一種雙核嗎啉磷鎢酸鹽催化氧化苯甲醇制備苯甲醛的方法與流程
本發明涉及一種雙核嗎啉磷鎢酸鹽催化氧化苯甲醇制備苯甲醛的方法,屬于催化氧化苯甲醇技術領域。
背景技術:
苯甲醛是制造染料的常用中間體,同時也是制備造醫藥、香料、涂料等精細化學品的重要原料。傳統上,苯甲醛的生產方法主要是甲苯氧化法或甲苯氯化水解法。然而他們存在工藝復雜、鹵素殘留、選擇性低等問題。從原子經濟性和環境友好的角度考慮,選擇性催化氧化苯甲醇合成苯甲醛的方法有極大的前景,得到了廣泛研究。選擇性催化氧化苯甲醇制備苯甲醛是利用空氣、氧氣或H2O2作為氧化劑對苯甲醇進行催化氧化生成苯甲醛,常用的催化劑包括含有Ru、Pd等負載型貴金屬催化劑、含Mn、Cr等的非貴金屬氧化物催化劑,但存在環境污染、催化劑成本過高或轉化率低等問題。日益嚴格的環保要求和不斷增長的高品質苯甲醛市場需求,要求研究者研制備高效“綠色”液相氧化苯甲醇制備苯甲醛的催化劑。
近年來,雜多酸作為一種性能優異、環境友好的催化新材料,在有機合成反應中應用發展迅速。盡管雜多酸鹽能夠得到較好的催化效果,但卻存在催化劑不易回收等問題。
離子液體是一種綠色溶劑和催化劑,其結構和酸、堿性等指標可調控。在苯甲醇氧化過程中主要作為溶劑使用,但存在成本高、反應產物與離子液體分離困難等問題。
若將離子液體的有機陽離子部分與雜多酸陰離子結合,則可得到一種新型的有機-雜多酸雜化材料。通過對有機陽離子的結構和雜多酸陰離子的功能化進行設計,調節該類催化劑的綜合性能,在保持催化活性的前提下,緩解傳統催化劑以及固體酸堿催化劑存在的腐蝕性強、難分離以及成本較高等問題。
技術實現要素:
針對現有技術存在的問題,本發明提供一種雙核嗎啉磷鎢酸鹽催化苯甲醇氧化制備苯甲醛的方法,該方法具有催化活性高、工藝簡單、催化劑熱穩定性好、容易回收以及可多次重復使用等優點。
為解決上述技術問題,本發明是這樣實現的:
以雙核嗎啉磷鎢酸鹽作為催化劑,將所述雙核嗎啉磷鎢酸鹽、過氧化氫和苯甲醇混合,攪拌條件下反應一段時間,離心分離,固體催化劑回收待用,將液體倒入分液漏斗中靜置分層,分離出的下層油相即為苯甲醛。
作為一種優選方案,本發明所述雙核嗎啉磷鎢酸鹽具有如下結構:
進一步地,本發明所述的反應溫度為60~100℃。
進一步地,本發明所述雙核嗎啉磷鎢酸鹽的加入量為苯甲醇質量的1.0~5.0%。
進一步地,本發明所述催化氧化反應時間為1.0~8.0h。
進一步地,本發明所述苯甲醇和過氧化氫的質量比為1.048~2.096:1。
進一步地,本發明所述過氧化氫為30%過氧化氫。
所述的雙核嗎啉磷鎢酸鹽的制備方法包括以下步驟:
將1,4-二溴丁烷溶于丙酮中,通過恒壓滴液漏斗加入嗎啡啉,1,4-二溴丁烷和嗎啡啉摩爾比為1:2,1,4-二溴丁烷與丙酮的質量比為1:1~2,冰浴條件下反應5h,反應結束后離心分離除去未反應的原料,剩余固體分三次用50mL丙酮洗滌,得到白色固體。取一定量的磷鎢酸,加入50mL去離子水,配成磷鎢酸水溶液。然后在冰浴條件下向三口燒瓶中滴加磷鎢酸水溶液,反應一定時間,靜置24h,然后離心分離除去水,將固體放入烘箱干燥至恒重,最后得到灰色固體即為最終產物。
本發明的優點在于:
(1)催化劑活性高,用量少;
(2)催化氧化反應條件溫和,反應時間短;
(3)無溶劑條件下進行催化氧化反應;
(4)雙核嗎啉磷鎢酸鹽性能穩定,可重復使用,分離工藝簡單,后處理成本低,無污染,不腐蝕設備,環境友好,可望成為極具競爭力的清潔工藝路線。
附圖說明
下面結合附圖和具體實施方式對本發明作進一步說明。本發明的保護范圍不僅局限于下列內容的表述。
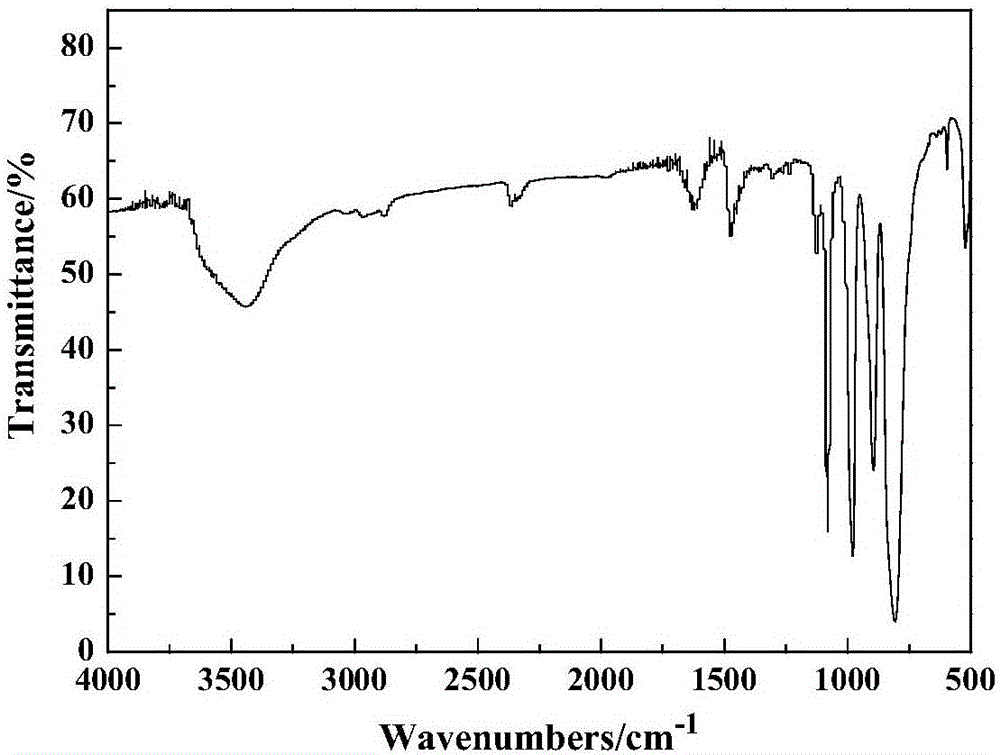
圖1是本發明實施例1制備的雙核嗎啉磷鎢酸鹽的紅外光譜圖;
圖2是本發明實施例1制備的雙核嗎啉磷鎢酸鹽的TGA曲線;
圖3是本發明實施例1制備的雙核嗎啉磷鎢酸鹽的XRD曲線;
圖4-1、圖4-2、圖4-3和圖4-4是本發明實施例1制備的雙核嗎啉磷鎢酸鹽作為催化劑催化氧化苯甲醇的工藝條件考察示意圖。
具體實施方式
實施例1
將1,4-二溴丁烷溶于丙酮中,通過恒壓滴液漏斗加入嗎啡啉(1,4-二溴丁烷和嗎啡啉摩爾比為1:2),冰浴條件下反應5h,反應結束后離心分離除去未反應的原料,剩余固體分三次用50mL丙酮洗滌,得到白色固體。取一定量的磷鎢酸(磷鎢酸與上述剩余固體的摩爾比為3:2),加入50mL去離子水,配成磷鎢酸水溶液。然后在冰浴條件下向三口燒瓶中滴加磷鎢酸水溶液,反應24h,靜置24h,然后離心分離除去水,將固體放入烘箱干燥至恒重,最后得到固體即為最終產物雙核嗎啉磷鎢酸鹽。
將0.20g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入15.72g的30%H2O2溶液,常壓下在100℃反應8h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為100%,苯甲醛的選擇性為91.8%。
實施例2
將0.20g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入20.96g的30%H2O2溶液,常壓下在90℃反應8h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為85.0%,苯甲醛的選擇性為95.0%。
實施例3
將0.50g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入15.72g的30%H2O2溶液,常壓下在90℃反應8h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為92.4%,苯甲醛的選擇性為93.3%。
實施例4
將0.20g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入10.48g的30%H2O2溶液,常壓下在90℃反應8h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為34.9%,苯甲醛的選擇性為74.3%。
實施例5
將0.10g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入15.72g的30%H2O2溶液,常壓下在90℃反應8h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為53.1%,苯甲醛的選擇性為100%。
實施例6
將0.20g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入10.48g的30%H2O2溶液,常壓下在90℃反應4h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為15.4%,苯甲醛的選擇性為73.9%。
實施例7
將0.20g雙核嗎啉磷鎢酸鹽和10.00g苯甲醇分別加入到100mL帶回流冷凝裝置的三口燒瓶中,然后加入15.72g的30%H2O2溶液,常壓下在70℃反應8h。冷卻后經離心分離除去底部的剩余催化劑,傾倒出上層的液體,放入分液漏斗,靜置分層,上層為水相,下層為油相。取下層的油相進行氣相色譜分析,計算得到苯甲醇的轉化率為35.1%,苯甲醛的選擇性為100%。
以上關于本發明的具體描述,僅用于說明本發明而并非受限于本發明實施例所描述的技術方案。本領域的普通技術人員應當理解,仍然可以對本發明進行修改或等同替換,以達到相同的技術效果。只要滿足使用需要,都在本發明的保護范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!