用CRISPR/Cas9技術易于篩選獲得目的基因敲除細胞系的方法及產品與流程
本發明屬于基因工程
技術領域:
中基因組編輯技術,涉及一種用CRISPR/Cas9技術敲除細胞中目的基因進行基因定向編輯并易于篩選獲得細胞系的方法與應用。
背景技術:
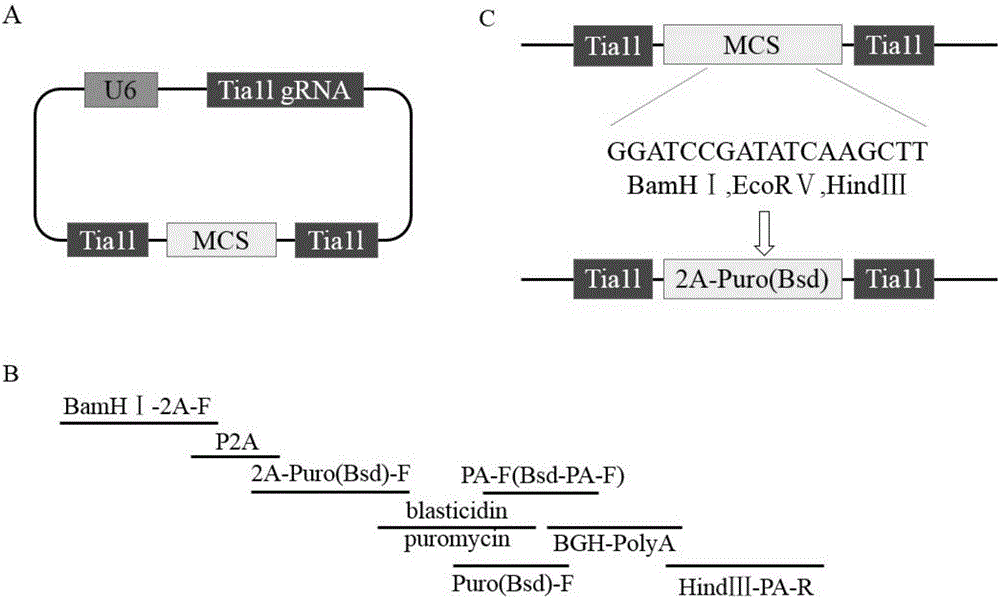
:CRISPR/Cas9基因組編輯技術具有“基因魔剪”之稱,是最近幾年興起的用于靶向基因編輯的一種重要工具,已被應用于多種模式生物,包括人、小鼠、大鼠、斑馬魚、秀麗隱桿線蟲、植物及細菌的基因編輯。該技術是利用人工設計特殊的向導RNA(single-guideRNA,sgRNA,又稱靶向RNA)介導外源表達的核酸內切酶cas9蛋白對基因組DNA上的靶點進行特異性剪切,隨后,細胞的非同源末端連接修復機制(NHEJ)重新連接斷裂處的基因組DNA,從而引入堿基插入或缺失突變,達到基因敲除實現基因定向編輯的目的。實驗室目前普遍使用的PX330載體質粒(Addgene/FengZhangLabPlasmids)具有Cas9表達盒和U6啟動子表達盒,可以將gRNA序列插入到U6啟動子之后。當把該質粒轉入細胞之后,質粒將表達特異性gRNA和Cas9,在gRNA的介導下,Cas9對基因進行切割。被剪切的基因在機體修復作用下有可能引入移碼造成目的基因缺失,然而轉染后的細胞中存在未被轉染的細胞和經過機體修復目的基因只是發生點突變而沒有發生移碼的細胞。PX330沒有篩選標記基因,從而造成篩選工作量非常巨大、獲得基因敲除的陽性細胞系的幾率比較低的問題,將給后續研究造成極大的不便和困難。技術實現要素:本發明的目的是提供一種用CRISPR/Cas9技術敲除細胞中目的基因易于篩選獲得目的基因敲除細胞系的方法。本發明用CRISPR/Cas9技術敲除細胞中目的基因易于篩選獲得目的基因敲除細胞系的方法,可包括以下步驟:1)構建攜帶篩選標記基因及2A自剪切肽編碼序列的Tia1l重組載體;所述篩選標記可選但不限于puromycin(嘌呤霉素)或blasticidin(殺稻瘟菌素);2)設計目的基因的sgRNA,合成其OligoDNA,構建攜帶目的基因的sgRNAOligoDNA和Cas9基因的重組載體(CRISPR/Cas9載體);3)將步驟1)和步驟2)構建的重組載體共轉染細胞;4)用篩選標志物(puromycin或blasticidin)進行篩選,快速篩選得到目的基因敲除的細胞系。在上述方法中,所述步驟1)中構建的Tia1l重組載體,具體為攜帶puromycin或blasticidin篩選標記及2A自剪切肽編碼序列的Tia1l重組載體,可包括以下過程:1.1)OverlappingPCR擴增BamHⅠ-2A-篩選標志基因-BGHpA-HindⅢ片段,具體可為BamHⅠ-2A-Puro-BGHpA-HindⅢ片段(對應puromycin)或BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段(對應blasticidin);1.2)用BamH1、HindⅢ雙酶切Tiall載體、BamHⅠ-2A-篩選標志基因-BGHpA-HindⅢ片段(BamHⅠ-2A-Puro-BGHpA-HindⅢ片段或BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段),回收酶切產物;1.3)將線性Tia1l載體與BamHⅠ-2A-篩選標志基因-BGHpA-HindⅢ酶切片段(BamHⅠ-2A-Puro-BGHpA-HindⅢ酶切片段或BamHⅠ-2A-Bsd-BGHpA-HindⅢ酶切片段)進行連接,得到Tia1l重組載(攜帶puromycin篩選標記的重組載體Tia1l-Puro或攜帶blasticidin篩選標記的重組載體Tia1l-Bsd)。所述步驟1.1)中的BamHⅠ-2A-篩選標志基因-BGHpA-HindⅢ片段自5’至3’端依次由BamHⅠ識別序列、2A自剪切肽編碼序列、篩選標志基因、BGHpA基因(BGH終止子)、HindⅢ識別序列連接而成。具體的:BamHⅠ-2A-Puro-BGHpA-HindⅢ片段自5’至3’端依次由BamHⅠ識別序列、2A自剪切肽編碼序列、puromycin基因、BGHpA基因(BGH終止子)、HindⅢ識別序列連接而成,其核苷酸序列如序列表中序列1所示;BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段自5’至3’端依次由BamHⅠ識別序列、2A自剪切肽編碼序列、blasticidin基因、BGHpA基因(BGH終止子)、HindⅢ識別序列連接而成,其核苷酸序列如序列表中序列2所示。所述步驟2)中設計目的基因的sgRNA,合成編碼目的基因sgRNA的雙鏈OligoDNA,在正義鏈模板的5’端添加CACC,反義鏈模板的5’端添加AAAC,使其可與BbsI酶切后形成的粘性末端互補。所述步驟2)中攜帶目的基因的sgRNAOligoDNA和Cas9基因的重組載體的構建方法,可包括以下步驟:2.1)根據設計的sgRNA序列,合成相應的單鏈OligoDNA及其互補鏈的OligoDNA單鏈,將合成的OligoDNA及其互補鏈按照分子數1:1比例混合,進行退火處理,以形成雙鏈OligoDNA;2.2)使用BbsI酶切PX330質粒和sgRNAOligoDNA,再將兩者進行連接,得到攜帶目的基因的sgRNAOligoDNA和Cas9基因的重組載體,命名為“PX330-目的基因-sgRNA”。所述步驟3)中將步驟1)構建的Tia1l重組載體(重組載體Tia1l-Puro或Tia1l-Bsd)與步驟2)構建的重組載體PX330-目的基因-sgRNA共轉染細胞的方法為:將細胞用DMEM高糖培養基(含10%滅活的胎牛血清)、雙抗(青霉素100unit/mL和鏈霉素100ug/mL,以防止細胞污染)、37℃、5%CO2恒溫培養,轉染前將4×106個細胞接種于100mm細胞培養皿中,當細胞密度達到80%-90%時使用胰酶消化,取出1/2的細胞,用PBS洗去培養基,用Invitrogen公司neon轉染系統并按說明書中的操作方法將Tia1l重組載體(Tia1l-Puro或Tia1l-Bsd)與PX330-目的基因-sgRNA共轉染細胞,將重組載體轉入細胞,載體使用量7μg,轉染體系為:PX330-Nedd4-sgRNA質粒3.5μg、Tia1l重組載體質粒(Tia1l-Puro或Tia1l-Bsd質粒)3.5μg,電轉條件為:1960V、10ms、2pulse,電轉完成后繼續培養細胞48-72h使細胞恢復。所述步驟4)中用篩選標記物(puromycin或blasticidin)篩選目的基因敲除的細胞的方法為:當細胞生長至正常狀態時,加入10μg/mL篩選藥物(puromycin或blasticidin),2天更換1次培養基,持續培養細胞2周,獲得穩定敲除目的基因的細胞系。用上述方法篩選獲得的敲除目的基因的細胞系也屬于本發明。上述方法步驟1)中擴增得到BamHⅠ-2A-篩選標記基因-BGHpA-HindⅢ片段,具體為BamHⅠ-2A-Puro-BGHpA-HindⅢ片段(對應puromycin)或BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段(對應blasticidin)也屬于本發明。上述方法步驟1)中構建的Tia1l重組載體,具體為攜帶puromycin篩選標記的重組載體Tia1l-Puro或攜帶blasticidin篩選標記的重組載體Tia1l-Bsd也屬于本發明。上述方法步驟2)獲得的重組載體PX330-目的基因-sgRNA也屬于本發明。具體來講,所述目的基因為Nedd4基因,在用CRISPR/Cas9技術敲除細胞中Nedd4基因中,所述步驟2)中設計并合成Nedd4基因的sgRNA,共設計3種sgRNA,其中兩種針對外顯子1(Exon1),一種針對外顯子4(Exon4),其RNA序列如下:(1)針對Nedd4基因外顯子1(Exon1)的sgRNA,序列表中序列3,命名為Nedd4-Exon1-sgRNA1(sgRNA1);(2)針對Nedd4基因外顯子1(Exon1)的sgRNA,序列表中序列4,命名為Nedd4-Exon1-sgRNA2(sgRNA2);(3)針對Nedd4基因外顯子4(Exon4)的sgRNA,序列表中序列5,命名為Nedd4-Exon4-sgRNA3(sgRNA3)。所述步驟2)中編碼Nedd4基因sgRNA的雙鏈OligoDNA,在正義鏈模板的5’端添加CACC,反義鏈模板的5’端添加AAAC,使其可與BbsI酶切后形成的粘性末端互補,可為下述雙鏈DNA序列之一:(1)sgRNA1的OligoDNA單鏈為序列表中序列6,其互補鏈為序列表中序列7;(2)sgRNA2的OligoDNA單鏈為序列表中序列8,其互補鏈為序列表中序列9;(3)sgRNA3的OligoDNA單鏈為序列表中序列10,其互補鏈為序列表中序列11。所述步驟2)中攜帶Nedd4基因的sgRNA1OligoDNA和Cas9基因的重組載體為PX330-Nedd4-sgRNA1,攜帶Nedd4基因的sgRNA2OligoDNA和Cas9基因的重組載體為PX330-Nedd4-sgRNA2,攜帶Nedd4基因的sgRNA3OligoDNA和Cas9基因的重組載體為PX330-Nedd4-sgRNA3,以上三種重組載體統稱為“PX330-Nedd4-sgRNA”。所述步驟3)中的細胞為BMDM細胞。用上述方法篩選獲得的敲除Nedd4基因的BMDM細胞系也屬于本發明。上述方法中步驟2)構建的攜帶Nedd4基因的sgRNAOligoDNA和Cas9基因的重組載體PX330-Nedd4-sgRNA也屬于本發明。PX330載體(CRISPR/Cas9載體)缺少篩選標記,對后期細胞篩選帶來巨大困難;本發明選擇另外一種載體Tia1l,其含有Tia1l識別序列且兩段Tia1l序列中間具有多克隆位點,載體可表達Tia1lgRNA,識別Tia1l序列,cas9蛋白可對其進行特異性切割,但是該載體不具有篩選標記,因此對其進行改進,在多克隆位點處插入篩選標記puromycin(嘌呤霉素)或blasticidin(殺稻瘟菌素)及2A自剪切肽編碼序列,cas9蛋白特異性切割后,產生的片段兩端可與切割后的基因組切口粘性互補,然后通過細胞修復功能將篩選標記puromycin和blasticidin及2A自剪切肽編碼序列插入基因組切口處,細胞通過目的基因的啟動子,正常啟動puromycin或blasticidin轉錄后,在BGHpA處終止,細胞正常翻譯,并通過2A自剪切作用表達puromycin或blasticidin抗性蛋白,然后加入適量濃度的藥物puromycin和blasticidin篩選獲得目的基因敲除的細胞系。具體來講,本發明針對目的基因—Nedd4基因的外顯子,設計靶向外顯子的sgRNA,用CRISPR/Cas9技術并通過puromycin或blasticidin篩選及Westernblot鑒定,獲得了穩定敲除Nedd4基因并攜帶有puromycin或blasticidin篩選標記基因的細胞系。本發明將PX330重組載體(CRISPR/Cas9載體)和Tia1l重組載體聯合使用能快速篩選獲得了目的基因敲除的細胞系,為研究基因的功能及作用機制提供了有效工具,同時插入篩選基因,在特異性插入目的基因方面進行了嘗試并取得成功,這就意味著不僅可以敲除目的基因并且可以在特定的位點上特異性地插入想要插入的基因(Tia1l質粒中兩段Tia1l序列之間具有多克隆位點,可以插入想要插入的基因,同時利用cas9蛋白對Tia1l序列和目的DNA序列的切割作用,切割出的Tia1l之間的序列,該序列可以和基因組間的切口互補,通過DNA修復,修復切口,這樣就達到了在特定位點上插入目的基因的目的),這為基因編輯方面奠定了堅實的基礎。下面結合具體實施例對本發明做進一步詳細說明。附圖說明圖1為Tia1l載體結構及用其構建攜帶puromycin(Puro)或blasticidin(Bsd)篩選標記及2A自剪切肽編碼序列的Tia1l重組載體的示意圖;圖2為PX330載體結構(A)及Exon1/4sgRNA的靶向識別序列位置及其周圍DNA序列(B);圖3為使用neon系統將重組載體Tia1l-Puro或Tia1l-Bsd與PX330-Nedd4-sgRNA共轉染BMDM細胞的結果;圖4為用puromycin或blasticidin篩選Nedd4基因敲除的BMDM細胞的結果。具體實施方式下述實施例中所用方法如無特別說明均為常規方法,具體步驟可參見:《MolecularCloning:ALaboratoryManual》(Sambrook,J.,Russell,DavidW.,MolecularCloning:ALaboratoryManual,3rdedition,2001,NY,ColdSpringHarbor)。所述百分比濃度如無特別說明均為質量/質量(W/W,單位g/100g)百分比濃度、質量/體積(W/V,單位g/100mL)百分比濃度或體積/體積(V/V,單位mL/100mL)百分比濃度。實施例中描述到的各種生物材料的取得途徑僅是提供一種實驗獲取的途徑以達到具體公開的目的,不應成為對本發明生物材料來源的限制。事實上,所用到的生物材料的來源是廣泛的,任何不違反法律和道德倫理能夠獲取的生物材料都可以按照實施例中的提示替換使用。所用引物由Invitrogen公司合成,使用超純水溶解引物為100μm母液,再稀釋成濃度為10μm的工作液。實施例在以本發明技術方案為前提下進行實施,給出了詳細的實施方式和具體的操作過程,實施例將有助于理解本發明,但是本發明的保護范圍不限于下述的實施例。實施例、用CRISPR/Cas9技術敲除BMDM細胞中Nedd4基因并快速獲得細胞系實施例用CRISPR/Cas9技術敲除BMDM細胞中Nedd4基因(實施例以Nedd4基因作為目的基因,其它目的基因與本例方法類似)并快速篩選獲得細胞系,包括以下過程:1)構建攜帶篩選標記及2A自剪切肽編碼序列的Tia1l重組載體。實施例以puromycin(嘌呤霉素)或blasticidin(殺稻瘟菌素)作為篩選標記實例,使用其它篩選標記與本例方法類似。如圖1(A:Tia1l載體結構;B:OverlappingPCR擴增BamH1-2A-Puro(Bsd)-BGHpA-HindⅢ片段;C:Tial載體多克隆位點結構:多克隆位點包括BamHⅠ、EcoRⅤ、HindⅢ酶切位點,在多克隆位點處插入篩選基因)所示,Tia1l載體具有一個U6啟動子、Tia1lgRNA片段及其兩個特異性識別序列Tia1l,其識別序列之間具有多克隆位點,但是其不具有篩選標記。本發明通過Overlapping的方法合成具有puromycin(Puro)或blasticidin(Bsd)篩選標記的片段,然后將該片段插入Tia1l質粒的多克隆位點,通過轉化,挑取單克隆,提取質粒,將質粒進行測序鑒定,獲得陽性質粒。1.1)OverlappingPCR擴增BamHⅠ-2A-Puro-BGHpA-HindⅢ片段和BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段BamHⅠ-2A-Puro-BGHpA-HindⅢ片段對應puromycin(嘌呤霉素),自5’至3’端依次由BamHⅠ識別序列、2A自剪切肽編碼序列、puromycin基因、BGHpA基因(BGH終止子)、HindⅢ識別序列連接而成,其核苷酸序列如序列表中序列1所示(BamHⅠ:1-6bp,P2A:7-72bp,puromycin:73-669bp,BGHpolyA:670-894bp,HindⅢ:895-900bp)。引物序列如下(5’-3’):BamHI-2A-F:CGGGATCCggaagcggagctacta,P2A:gaagcggagctactaacttcagcctgctgaagcaggctggagacgtggaggagaaccctggacct,2A-Puro-F:gagaaccctggacctaccgagtacaagccca,Puro-R:AACTAGAAGGCACAGTCAGGCACCGGGCTT,Puro-pA-F:aagcccggtgcctgactgtgccttctagtt,HindIII-pA-R:cccAAGCTTccatagagcccaccg。以PcDNA3.1(+)(購自Addgene)為模板,OverlappingPCR擴增BamHⅠ-2A-Puro-BGHpA-HindⅢ片段,PCR反應體系為:模板PcDNA3.1(+)1μL(20ng),5×reactionbuffer10μL,dNTPs4μL,PrimeSTARHSDNAPolymerase0.5μL,BamHI-2A-F1μL,HindIII-pA-R1μL,P2A0.3μL,2A-Puro-F0.2μL,Puro-pA-F0.1μL,Puro-R:0.3μL,用H2O補至50μL。PCR反應條件為:先94℃5min;然后98℃10s,56℃5s,72℃1min,共40個循環;最后72℃:10min。反應結束后,對PCR產物進行2%瓊脂糖凝膠電泳,使用膠回收試劑盒(購自博邁德生物技術有限公司)進行膠回收900bp的BamHⅠ-2A-Puro-BGHpA-HindⅢ片段。BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段對應blasticidin(殺稻瘟菌素),自5’至3’端依次由BamHⅠ識別序列、2A自剪切肽編碼序列、blasticidin基因、BGHpA基因(BGH終止子)、HindⅢ識別序列連接而成,其核苷酸序列如序列表中序列2所示(BamH1:1-6bp,P2A:7-72bp,blasticidin:73-468bp,BGHpolyA:469-693bp,HindⅢ:694-699bp)。引物序列如下(5’-3’):BamHI-2A-F:CGGGATCCggaagcggagctactaP2A:gaagcggagctactaacttcagcctgctgaagcaggctggagacgtggaggagaaccctggacct2A-Bsd-F:gagaaccctggacctgccaagcctttgtctBsd-pA-F:tgtgtgggagggctaactgtgccttctagttBsd-R:AACTAGAAGGCACAGTTAGCCCTCCCACACApA-F:aagcccggtgcctgactgtgccttctagttHindIII-pA-R:cccAAGCTTccatagagcccaccg以PcDNA3.1(+)為模板,OverlappingPCR擴增BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段,PCR反應體系為:模板PcDNA3.1(+)1μL(20ng),5×reactionbuffer10μL,dNTPs4μL,PrimeSTARHSDNAPolymerase0.5μL,BamHI-2A-F1μL,HindIII-pA-R1μL,P2A0.3μL,2A-Puro-F0.2μL,Bsd-pA-F0.1μL,Bsd-R0.3μL,用H2O補至50μL。PCR反應條件為:先94℃5min;然后98℃10s,56℃5s,72℃1min,共40個循環;最后72℃:10min。反應結束后,對PCR產物進行2%瓊脂糖凝膠電泳,使用膠回收試劑盒進行膠回收699bp的BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段。1.2)使用BamHⅠ、HindⅢ雙酶切Tia1l質粒、BamH1-2A-Puro-BGHpA-HindⅢ片段或BamH1-2A-Bsd-BGHpA-HindⅢ片段酶切體系為:Tai1l質粒(DHLackner,ACarre,PMGuzzardo,CBanning,RMangena,THenley,SOberndorfer,BVGapp,SMNijman,TRBrummelkamp,andTBurckstummer,AgenericstrategyforCRISPR-Cas9-mediatedgenetagging,NatCommun,2015,6(10237))10μL,BamH1-2A-Puro-BGHpA-HindⅢ片段或BamH1-2A-Bsd-BGHpA-HindⅢ片段20μL,BamHⅠ2μL,HindⅢ2μL,10×reactionbuffer5μL,用H2O補至50μL。酶切條件為:37℃酶切4小時。酶切結束后,對酶切產物進行2%瓊脂糖凝膠電泳,回收2786bp的線性Tia1l質粒與900bp的BamH1-2A-Puro-BGHpA-HindⅢ酶切片段或699bp的BamH1-2A-Bsd-BGHpA-HindⅢ酶切片段,與期望序列相同。1.3)將線性Tia1l載體與BamHⅠ-2A-Puro---BGHpA-HindⅢ酶切片段或BamHⅠ-2A-Bsd-BGHpA-HindⅢ酶切片段進行連接,得到攜帶puromycin篩選標記的重組載體Tia1l-Puro或攜帶blasticidin篩選標記的重組載體Tia1l-Bsd。連接體系為:線性Tia1l載體5ul,BamH1-2A-Puro-BGHpA-HindⅢ酶切片段或BamH1-2A-Bsd-BGHpA-HindⅢ酶切片段10ul,10×reactionbuffer2ul,T4DNA連接酶1ul,H2O1ul。連接條件為:16℃連接過夜(16小時)。將連接產物轉化DH5α大腸桿菌感受態細胞,挑取單克隆,小量提質粒并進行測序鑒定,測序結果表明獲得了序列及結構正確的攜帶puromycin或blasticidin篩選標記及2A自剪切肽編碼序列的Tia1l重組載體,分別命名為Tia1l-Puro和Tia1l-Bsd,選取陽性克隆,大量提質粒,備用。2)設計Nedd4基因的sgRNA及其OligoDNA,構建攜帶Nedd4基因的sgRNAOligoDNA和Cas9基因的重組載體2.1)設計Nedd4基因的sgRNA及其OligoDNA如圖2中B幅(B:Exon1/4sgRNA的靶向識別序列位置及其周圍DNA序列)所示,用哈佛大學的網上在線軟件(http://crispr.mit.edu)設計Nedd4基因的向導RNA(sgRNA)。共設計3種sgRNA,其中兩種針對外顯子1(Exon1),一種針對外顯子4(Exon4),其RNA序列如下:(1)針對Nedd4基因外顯子1(Exon1)的靶標序列(自5‘端第233-252位堿基)設計的sgRNA:ACGAGTCGGAGGCCCCAGTA,序列表中序列3,命名為Nedd4-Exon1-sgRNA1(sgRNA1);(2)針對Nedd4基因外顯子1(Exon1)的靶標序列(自5‘端第219-238位堿基)設計的sgRNA:GGACATGGCAGCCGACGAGT,序列表中序列4,命名為Nedd4-Exon1-sgRNA2(sgRNA2);(3)針對Nedd4基因外顯子4(Exon4)的靶標序列(自5‘端第14988-15007位堿基)設計的sgRNA:ACATTGTATGACCCGATGAG,序列表中序列5,命名為Nedd4-Exon4-sgRNA3(sgRNA3)。編碼Nedd4基因的sgRNA的雙鏈OligoDNA,在正義鏈模板的5’端添加CACC,反義鏈模板的5’端添加AAAC,使其可與BbsI酶切后形成的粘性末端互補,如表1所示,可為下述雙鏈DNA序列之一:(1)sgRNA1的OligoDNA單鏈為序列表中序列6,其互補鏈為序列表中序列7;(2)sgRNA2的OligoDNA單鏈為序列表中序列8,其互補鏈為序列表中序列9;(3)sgRNA3的OligoDNA單鏈為序列表中序列10,其互補鏈為序列表中序列11。表1編碼Nedd4基因的sgRNA的雙鏈OligoDNA序列向導RNAOligoDNA序列sgRNAOligonucleotidesequenceNedd4-Exon1-sgRNA1F:5'-CACCGTACTGGGGCCTCCGACTCGT-3'(序列表中序列6)R:5'-AAACACGAGTCGGAGGCCCCAGTAC-3'(序列表中序列7)Nedd4-Exon1-sgRNA2F:5'-CACCGGACATGGCAGCCGACGAGT-3'(序列表中序列8)R:5'-AAACACTCGTCGGCTGCCATGTCC-3'(序列表中序列9)Nedd4-Exon4-sgRNA3F:5'-CACCGACATTGTATGACCCGATGAG-3'(序列表中序列10)R:5'-AAACCTCATCGGGTCATACAATGTC-3'(序列表中序列11)2.2)構建攜帶Nedd4基因的sgRNAOligoDNA和Cas9基因的的重組載體如圖2中A幅(A:PX330載體結構:攜帶Case9基因和2個BbsI酶切位點)所示,攜帶Nedd4基因的sgRNAOligoDNA和Cas9基因的重組載體的構建方法,包括以下步驟:2.2.1)合成Nedd4基因的sgRNAOligoDNA的單鏈及其互補鏈,將合成的OligoDNA單鏈及其互補鏈按照分子數1:1比例混合,進行退火處理,以形成雙鏈OligoDNA兩條sgRNA寡核苷酸單鏈退火形成雙鏈OligoDNA:兩條單鏈DNA使用超純水溶解使其濃度為100μm/L,退火體系為:sgRNA-F1μL,sgRNA-R1μL,10×PNKbuffer1μL,T4PNK1μL,H2O6μL。退火條件為:37℃30min,95℃5min,-1℃/s降溫至25℃。將退火產物使用高純水1:200稀釋。2.2.2)使用BbsI酶切PX330載體(攜帶Cas9基因)和sgRNAOligoDNA,再將兩者進行連接,得到攜帶Nedd4基因的sgRNAOligoDNA和Cas9基因的重組載體用限制性內切酶BbsI切割PX330載體,酶切體系為:PX330載體(購自Addgene)10μL(共計1μg),BbsI(購自Thrmo公司)1μL,10×reactionbuffer2μL,H2O7μL。酶切條件為:37℃酶切1h。用瓊脂糖凝膠回收試劑盒切膠回收8500bp的線性PX330載體將線性PX330載體和sgRNAOligoDNA進行連接,連接體系為:線性化PX330載體4μL(100ng),sgRNAOligoDNA2μL,10×T4DNALigaseBuffer2μL,T4DNA連接酶1μL,H2O11μL。連接條件為:16℃連接過夜(16小時)。將連接產物轉化DH5α大腸桿菌感受態細胞,氨芐抗性涂板篩選,挑取單克隆,小量提質粒并進行測序鑒定,測序結果表明獲得了序列及結構正確的攜帶Nedd4基因的sgRNAOligoDNA和Cas9基因的重組載體,統稱為PX330-Nedd4-sgRNA,其中,攜帶Nedd4基因的sgRNA1OligoDNA的重組載體為PX330-Nedd4-sgRNA1,攜帶Nedd4基因的sgRNA2OligoDNA的重組載體為PX330-Nedd4-sgRNA2,攜帶Nedd4基因的sgRNA3OligoDNA的重組載體為PX330-Nedd4-sgRNA3。選取陽性克隆,大量提質粒,備用。3)將步驟1)和步驟2)構建的重組載體共轉染BMDM細胞將步驟1)構建的重組載體Tia1l-Puro或Tia1l-Bsd與步驟2)構建的PX330-Nedd4-sgRNA共轉染BMDM細胞的方法為:將BMDM細胞用DMEM高糖培養基(購自gibco公司)(含10%滅活的胎牛血清,購自ExCellBiology.Inc公司)、雙抗(青霉素100unit/mL,鏈霉素100ug/mL,以防止細胞污染)、37℃、5%CO2恒溫培養,轉染前將4×106個BMDM細胞接種于100mm細胞培養皿中,當細胞密度達到80%-90%時使用胰酶消化,取出1/2的細胞,用PBS洗去培養基,用Invitrogen公司neon轉染系統并按說明書中的操作方法將重組載體Tia1l-Puro或Tia1l-Bsd與PX330-Nedd4-sgRNA(PX330-Nedd4-sgRNA1、PX330-Nedd4-sgRNA2或PX330-Nedd4-sgRNA3)共轉染BMDM細胞,以PX330-Nedd4-sgRNA(PX330-Nedd4-sgRNA1、PX330-Nedd4-sgRNA2或PX330-Nedd4-sgRNA3)單獨轉染BMDM細胞為對照,載體使用量7μg,轉染體系為:PX330-Nedd4-sgRNA質粒3.5ug,Tia1l-Puro或Tia1l-Bsd質粒3.5ug,電轉條件為:1960V、10ms、2pulse,電轉完成后繼續培養細胞48-72h使細胞恢復。BMDM細胞轉染結果如圖3(A:明場下視野(40×);B:熒光下視野(40×))所示,細胞轉染效率(達80%)及細胞存活率(達90%)均達到實驗要求,說明細胞轉染成功。4)用puromycin或blasticidin進行篩選,得到Nedd4基因敲除的BMDM細胞用puromycin或blasticidin篩選Nedd4基因敲除的BMDM細胞的方法為:當細胞生長至正常狀態時,加入10μg/mL篩選藥物(puromycin或blasticidin),2天更換1次培養基,持續培養細胞2周。單轉PX330-Nedd4-sgRNA載體時,cas9蛋白靶向切割目的基因,同時利用DNA修復機制使基因發生移碼、替換、插入或者缺失突變(圖4中A幅),使用無限稀釋法獲得單克隆細胞系,以期獲得Nedd4基因敲除的單克隆細胞系,然后提取轉染BMDM細胞總蛋白,用WesternBlot(抗體為Nedd4,購自santacruz)方法檢測Nedd4蛋白表達水平,結果如圖4中C幅所示,gRNA-1.1為完全敲除細胞系,同時提取其基因組對其測序,結果如圖4中D幅(帶底紋字體表示相同堿基,-表示缺失堿基)所示,Nedd4基因發生了替換以及16bp的缺失突變,這說明獲得一株Nedd4基因敲除的單克隆細胞系。PX330-Nedd4-sgRNA載體和Tia1l-Puro(或Tai1l-Bsd)載體共轉染BMDM細胞時,利用cas9蛋白的剪切作用,剪切后2A-Puro(或2A-Bsd)片段和基因組切口互補,通過連接作用將2A-Puro(或2A-Bsd)片段插入基因組(圖4中B幅),通過Nedd4基因的啟動子,正常啟動puromycin或blasticidin轉錄后,在BGHpA處終止,細胞正常翻譯,通過2A自剪切作用使細胞表達puromycin或blasticidin抗性蛋白,然后使用puromycin或blasticidin進行篩選,持續處理2周,提取篩選細胞總蛋白,Westernblot檢測Nedd4蛋白表達水平。結果如圖4中E幅所示,兩種篩選方式均獲得了Nedd4基因完全敲除的BMDM細胞株,這表明本方法通過簡單操作就能篩選獲得基因敲除的細胞系,與只用PX330載體通過有限稀釋法篩選獲得基因敲除的細胞系的方法相比大大提高了篩選效率。序列表<110>中國人民解放軍軍事醫學科學院野戰輸血研究所<120>用CRISPR/Cas9技術易于篩選獲得目的基因敲除細胞系的方法及產品<130>CGCNB165182W<160>11<210>1<211>900<212>DNA<213>BamHⅠ-2A-Puro-BGHpA-HindⅢ片段<400>1ggatccggaagcggagctactaacttcagcctgctgaagcaggctggagacgtggaggag60aaccctggacctaccgagtacaagcccacggtgcgcctcgccacccgcgacgacgtcccc120agggccgtacgcaccctcgccgccgcgttcgccgactaccccgccacgcgccacaccgtc180gatccggaccgccacatcgagcgggtcaccgagctgcaagaactcttcctcacgcgcgtc240gggctcgacatcggcaaggtgtgggtcgcggacgacggcgccgcggtggcggtctggacc300acgccggagagcgtcgaagcgggggcggtgttcgccgagatcggcccgcgcatggccgag360ttgagcggttcccggctggccgcgcagcaacagatggaaggcctcctggcgccgcaccgg420cccaaggagcccgcgtggttcctggccaccgtcggcgtctcgcccgaccaccagggcaag480ggtctgggcagcgccgtcgtgctccccggagtggaggcggccgagcgcgccggggtgccc540gccttcctggagacctccgcgccccgcaacctccccttctacgagcggctcggcttcacc600gtcaccgccgacgtcgaggtgcccgaaggaccgcgcacctggtgcatgacccgcaagccc660ggtgcctgactgtgccttctagttgccagccatctgttgtttgcccctcccccgtgcctt720ccttgaccctggaaggtgccactcccactgtcctttcctaataaaatgaggaaattgcat780cgcattgtctgagtaggtgtcattctattctggggggtggggtggggcaggacagcaagg840gggaggattgggaagacaatagcaggcatgctggggatgcggtgggctctatggaagctt900<210>2<211>699<212>DNA<213>BamHⅠ-2A-Bsd-BGHpA-HindⅢ片段<400>2ggatccggaagcggagctactaacttcagcctgctgaagcaggctggagacgtggaggag60aaccctggacctgccaagcctttgtctcaagaagaatccaccctcattgaaagagcaacg120gctacaatcaacagcatccccatctctgaagactacagcgtcgccagcgcagctctctct180agcgacggccgcatcttcactggtgtcaatgtatatcattttactgggggaccttgtgca240gaactcgtggtgctgggcactgctgctgctgcggcagctggcaacctgacttgtatcgtc300gcgatcggaaatgagaacaggggcatcttgagcccctgcggacggtgccgacaggtgctt360ctcgatctgcatcctgggatcaaagccatagtgaaggacagtgatggacagccgacggca420gttgggattcgtgaattgctgccctctggttatgtgtgggagggctaactgtgccttcta480gttgccagccatctgttgtttgcccctcccccgtgccttccttgaccctggaaggtgcca540ctcccactgtcctttcctaataaaatgaggaaattgcatcgcattgtctgagtaggtgtc600attctattctggggggtggggtggggcaggacagcaagggggaggattgggaagacaata660gcaggcatgctggggatgcggtgggctctatggaagctt699<210>3<211>20<212>RNA<213>Nedd4-Exon1-sgRNA1(sgRNA1)<400>3acgagucggaggccccagua20<210>4<211>20<212>RNA<213>Nedd4-Exon1-sgRNA2(sgRNA2)<400>4ggacauggcagccgacgagu20<210>5<211>20<212>RNA<213>Nedd4-Exon4-sgRNA3(sgRNA3)<400>5acauuguaugacccgaugag20<210>6<211>25<212>DNA<213>Nedd4-Exon1-sgRNA1(sgRNA1)的OligoDNA單鏈(F)<400>6caccgtactggggcctccgactcgt25<210>7<211>25<212>DNA<213>Nedd4-Exon1-sgRNA1(sgRNA1)的OligoDNA單鏈的互補鏈(R)<400>7aaacacgagtcggaggccccagtac25<210>8<211>24<212>DNA<213>Nedd4-Exon1-sgRNA2(sgRNA2)的OligoDNA單鏈(F)<400>8caccggacatggcagccgacgagt24<210>9<211>24<212>DNA<213>Nedd4-Exon1-sgRNA2(sgRNA2)的OligoDNA單鏈的互補鏈(R)<400>9aaacactcgtcggctgccatgtcc24<210>10<211>25<212>DNA<213>Nedd4-Exon4-sgRNA3(sgRNA3)的OligoDNA單鏈(F)<400>10caccgacattgtatgacccgatgag25<210>11<211>25<212>DNA<213>Nedd4-Exon4-sgRNA3(sgRNA3)的OligoDNA單鏈的互補鏈(R)<400>11aaacctcatcgggtcatacaatgtc25當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 用CRISPR/Cas9技術易于篩選獲得目的基因敲除細胞系的方法及產品與制造工藝
- 一種Candida amazonensis的FLP/FRT基因敲除方法與制造工藝
- 玉米低鎂條件下特異性轉運鎂離子基因ZmMGT10及其應用的制造方法與工藝
- CRISPR?Cas9特異性敲除豬SLA?3基因的方法及用于特異性靶向SLA?3基因的sgRNA與制造工藝
- 利用CRISPR/Cas9敲除人PD?1基因構建靶向CD19CAR?T細胞的方法與制造工藝
- 一種構建基因替代小鼠研究野生型和突變體非肌性肌球蛋白重鏈II功能的方法與制造工藝
- 構建在海馬體區域特異性敲除IKKα基因的小鼠模型的方法及打靶載體和試劑盒與制造工藝
- 豬ApoE基因敲除打靶載體及其構建方法和應用與制造工藝
- 一種血小板減少癥的斑馬魚模型的制造方法與工藝
- 基于CRISPR技術敲除FUT8基因的方法與制造工藝