芥酸金屬配合物及其制備方法與流程
本發明涉及一種芥酸金屬配合物及其制備方法,屬于有機材料制備技術領域。
背景技術:
近年來,隨著納米顆粒制備技術的不斷發展,尤其是伴隨著高溫熱分解法在單分散納米晶體制備方面的重大突破,選擇安全、簡單、廉價、通用、環境友好的前驅物來實現納米顆粒尺寸、形貌、組分的可控制備已成為當前納米化學合成的發展趨勢與技術挑戰。當前,納米晶體合成所依賴的前驅物包括乙酰丙酮類配合物(乙酰丙酮鐵等)、羰基類配合物(五羰基鐵等)、金屬有機類配合物(二甲基鋅、二甲基鉻等)等,這些前驅物往往毒性比較大、昂貴、不穩定、易爆炸,使用此類前驅物必須配備像手套箱這樣的復雜設備,而且所制備的納米晶體在質量與性質上并沒有顯示出別樣的優勢。因此,選擇安全、簡單、廉價、通用、環境友好的前驅物制備納米顆粒已成為當前納米化學合成技術的必然趨勢,并且為以后納米晶體工業化生產奠定良好基礎。
芥酸中文別名水芥油酸,蕪酸,瓢兒菜酸,順二十二碳-13-烯酸,(z)-13-二十二烯酸,順13-二十二烯酸,13-二十二碳烯酸,油菜酸,順式-13-二十二碳烯酸,為無色針狀結晶,分子式為c9h18=c13h24o2,屬脂肪酸類化合物,不溶于水,溶于乙醇、甲醇,易溶于乙醚。
技術實現要素:
本發明的目的在于提供一種芥酸金屬配合物及其制備方法和應用。
本發明的實現過程如下:
結構式(1)所示的芥酸金屬配合物,
m(eruciate)x(1),
其中m=fe、mn、co、ni、zn和cu,eruciate為脫去質子的芥酸根,x=2或3。
上述芥酸金屬配合物的制備方法,包含如下步驟:
(1)將金屬鹽與芥酸溶于質子性溶劑中得到溶液a,所述金屬鹽中的金屬為fe、mn、co、ni、zn和cu;
(2)將強堿溶于質子性溶劑中得到溶液b;
(3)將溶液b加至溶液a中,待反應結束,分離沉淀用去離子水與質子性溶劑洗滌數次后真空干燥。
所述金屬鹽為金屬氯化物、金屬硫酸鹽、金屬硝酸鹽和金屬醋酸鹽等。
所述質子性溶劑為水、甲醇、乙醇、甲酸、乙酸或乙胺等。
所述強堿為氫氧化鈉、氫氧化鉀等。
所述金屬鹽、芥酸和強堿溶解/分散于質子性溶劑中的濃度為0.01-100mmol/ml。
所述的金屬鹽與芥酸的摩爾比為1:100~100:1,強堿和芥酸的摩爾比為1:100~100:1。
制備過程中步驟(1)、(2)和(3)溶解、分散與反應所需溫度為20-80℃。制備結束后產物進行真空干燥溫度為20-80℃,真空干燥時間為1-24h。
所述芥酸金屬配合物在制備無機納米晶中作為前驅物的應用,特別是用作溶液法制備無機納米晶的前驅物。發明人發現使用該前驅物可以制備出超小納米晶。
本發明的優點及積極效果:該方法通過使用金屬與芥酸在質子溶劑中的配位反應來實現配合物的制備,工藝流程簡單,操作簡便快捷,反應條件溫和,制備成本低,穩定性好,無毒,在制備此類芥酸金屬配合物中具有普適性,可以大規模制備,該類配合物克服了現有前驅物的不足,可作為無毒、環保、成本低廉的前驅物制備納米晶,為以后工業化制備納米晶奠定基礎。
附圖說明
圖1是本發明所制備的芥酸鐵實物圖;
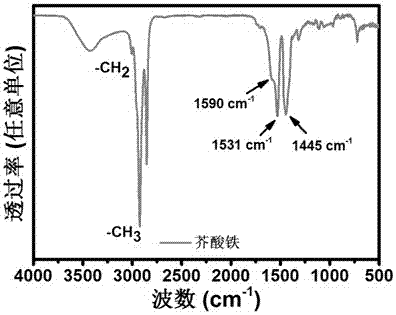
圖2是本發明所制備的芥酸鐵紅外光譜圖(ftir);
圖3是本發明所制備的芥酸鐵的核磁共振氫譜圖1hnmr;
圖4是本發明所制備的芥酸鐵質譜圖;
圖5(a)和(d)是2nmmnfe2o4納米晶的透射電鏡(tem)圖,圖5(b)和(e)是3nmmnfe2o4納米晶的tem圖,圖5(c)和(f)是3.9nmmnfe2o4納米晶的tem圖;
圖6本發明實施例中所制備的2nm、3nm和3.9nmmnfe2o4納米晶的x射線粉末衍射(xrd)圖。
具體實施方式
為使發明的目的、技術方案及優點更加清楚明白,以下參照附圖并舉實施例對發明進一步詳細說明。
實施例1:芥酸鐵的制備
5g六水合三氯化鐵和30g芥酸溶于200ml甲醇,把混合溶液充分攪拌后,向其中滴加含4gkoh的300ml甲醇溶液,生成的沉淀用甲醇和超純水反復洗劑,放入真空干燥箱中30℃干燥4小時,如圖1所示。
將所制備的芥酸鐵蠟狀產物與溴化鉀研磨均勻后壓片處理,然后測試紅外光譜。圖2是所制備的芥酸鐵的紅外光譜圖,從圖中可以看出,所制備的芥酸鐵顯示出羧酸配合物的特征振動峰,芥酸鐵的特征振動峰在1590cm-1,1531cm-1和1445cm-1,其中1590cm-1和1531cm-1是芥酸鐵中羧基官能團的非對稱振動峰;1445cm-1是芥酸鐵的中羧基官能團的對稱振動峰。
圖3是所制備的芥酸鐵的核磁共振氫譜圖。將少許芥酸鐵與油酸錳溶解在氘代氯仿中,裝入核磁管進行測試。從圖3測試結果可以看出芥酸鐵不同化學環境下氫原子的化學位移與模擬結果都能匹配。
圖4是所制備的芥酸鐵的質譜圖,將少許芥酸鐵與油酸錳分散在甲醇或乙腈中進行質譜分析。
實施例2:芥酸鋅的制備
7g醋酸鋅和30g芥酸溶于200ml乙醇,把混合溶液充分攪拌后,向其中滴加含4gkoh的300ml乙醇溶液,生成的沉淀用甲醇和超純水反復洗劑,放入真空干燥箱中30℃干燥4小時即可。
實施例3:芥酸鎳的制備
6g硝酸鎳和30g芥酸溶于200ml乙醇,把混合溶液充分攪拌后,向其中滴加含5gkoh的300ml乙醇溶液,生成的沉淀用甲醇和超純水反復洗劑,放入真空干燥箱中30℃干燥4小時即可。
實施例4:2nmmnfe2o4納米晶的制備
(1)前驅體的合成
芥酸鐵:5g六水合三氯化鐵和18g芥酸溶于200ml甲醇,把混合溶液充分攪拌后,向其中滴加200ml甲醇含4gnaoh的溶液。生成的沉淀用甲醇和超純水反復洗劑,放入真空干燥箱中30℃干燥4小時。
油酸錳:4g四水合氯化錳和14g油酸溶于150ml甲醇,把混合溶液充分攪拌后,向其中滴加200ml甲醇含4gnaoh的溶液。生成的沉淀用甲醇和超純水反復洗劑,放入真空干燥箱中30℃干燥4小時。
(2)2nmmnfe2o4納米晶的制備
分別稱取3g芥酸鐵、2g油酸錳和0.5g油醇,將其溶解到8g二芐醚中,氮氣氛圍下攪拌充分混合均勻;
混合物以15℃/min的速率升溫至245℃,回流45min;
混合物降溫到室溫,10ml丙酮加到混合物,離心分離沉淀物,將沉淀物分散于10ml甲苯,重復3次。
實施例5:3nmmnfe2o4納米晶的制備
前驅物芥酸鐵和油酸錳的合成參考實施例4。
分別稱取4g芥酸鐵、3g油酸錳、0.5g油醇和1g油酸,將其溶解到8g二辛醚中,氮氣氛圍下攪拌充分混合均勻;
混合物以15℃/min的速率升溫至245℃,回流45min;
混合物降溫到室溫,10ml甲醇加到混合物,離心分離沉淀物,將沉淀物分散于10ml正己烷,重復3次。
實施例6:3.9nmmnfe2o4納米晶的制備
前驅物芥酸鐵和油酸錳的合成參考實施例4。
分別稱取5g芥酸鐵、3g油酸錳、1g油醇和1g油酸,將其溶解到8g十八烯中,氮氣氛圍下攪拌充分混合均勻;
混合物以15℃/min的速率升溫至255℃,回流45min;
混合物降溫到室溫,10ml乙醇加到混合物,離心分離沉淀物,將沉淀物分散于10ml氯仿,重復3次。
將大約2μl所制備的2nm、3nm和3.9nmmnfe2o4納米晶滴在鍍有碳膜的cu網上,自然干燥后,做透射電鏡。圖5分別是實施例4、5、6所制備的2nm、3nm和3.9nmmnfe2o4納米晶tem圖。從圖中可以看出,所制備的2nm、3nm和4nmmnfe2o4納米晶粒徑均勻,對納米晶進行尺寸分布統計,結果顯示平均尺寸分別為2nm、3nm和3.9nm。
圖6是實施例4、5、6所制備的2nm、3nm和3.9nmmnfe2o4納米晶的xrd圖(cu/kα靶,λ=0.15418nm)。從圖6中可以看到反尖晶石結構的mnfe2o4納米晶的(111)、(220)、(311)、(400)、(511)、(440)和(622)衍射峰,根據最強的(311)衍射峰的2θ=34.980°,由謝樂公式計算得到晶粒尺寸分別為2.07nm、2.98nm和4.29nmmnfe2o4納米晶。
以上實施案例只用于對本發明作進一步說明,不能理解為對本發明保護范圍的限制,本領域的技術人員根據本發明的上述內容做出的一些非本質的改進和調整均屬于本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種羰基锝-99m標記的美法侖配合物及其制備方法與用圖
- 一種2-羰基-3-苯基丙酸苯甲酰腙二苯基錫配合物及其制備方法和應用
- 一種新型金屬有機配合物纖維及其衍生多孔碳纖維的制備方法
- 包含新的配體結構的金屬配合物的制作方法
- 一種2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯芐基)錫配合物及其制備方法和應用
- 一種2-羰基-2-苯基乙酸苯甲酰腙二丁基錫配合物及其制備方法和應用
- 一種2-羰基-3-苯基丙酸水楊酰腙二苯基錫配合物及其制備方法和應用
- 一種2-羰基-3-苯基丙酸苯甲酰腙二(2,4-二氯芐基)錫配合物及其制備方法和應用
- 一種2-羰基-2-苯基乙酸苯甲酰腙二丁基錫配合物及其制備方法和應用
- 一種2-羰基-3-苯基丙酸水楊酰腙二苯基錫配合物及其制備方法和應用