一種核殼量子點/聚苯乙烯熒光微球的制備方法與流程
本發明涉及納米功能材料設計領域,特別是一種核殼量子點/聚苯乙烯熒光微球的制備方法,合成的量子點/聚苯乙烯熒光微球可用于高靈敏生物分析與檢測。
背景技術:
熒光微球是指熒光物質通過包埋法、物理吸附法、自組裝法、化學鍵合法、共聚法等方法吸附或者包覆在微球的內部而形成的納米至微米級負載熒光物質的微球。微球的外形一般為球形,所以被稱為熒光微球,熒光微球形態結構穩定、對熒光物質有保護作用及表面可修飾性,在標記、檢測、示蹤、免疫醫學、高通量藥物篩選等領域擁有巨大潛力。
量子點是三個維度的尺寸都在100納米以下,半徑小于或接近于激光波爾半徑,能夠接受激光產生熒光的一類半導體納米顆粒。新型的半導體熒光材料具有寬的吸收峰,窄而對稱的發射峰,且發射峰即發光顏色隨尺寸可調,以及較高的熒光強度、較強的抗光漂白能力等特點,能夠克服傳統熒光材料的熒光信號不穩定、制備條件苛刻、瞬時熒光干擾等問題,與傳統有機染料相比具有更優越的性能,是一種極具潛力的熒光探針制備物。其應用領域越來越廣泛,特別是其在免疫生物學和臨床檢驗學等研究中的潛在的應用價值,己引起了廣大科學工作者的極大關注,發光量子點作為熒光試劑探針標記生物大分子,正是近年來迅速發展的納米材料在生物分析領域的重要應用之一。
聚苯乙烯為載體的熒光微球是最常見的微球之一,聚苯乙烯微球由于比表面積大、粒徑均勻、表面易修飾官能團及官能團反應性強等優點,是非常理想的熒光物質載體,聚苯乙烯微球作為載體,量子點可以通過溶脹、吸附、包埋方式載到聚苯乙烯微球上,但是這些制備方法都存在某個或一些致命的缺點,導致這些微球無法大規模生產及商業化;如溶脹方法制備的熒光微球長時間量子點容易脫漏;物理吸附制備的熒光微球容易受到外界環境的的影響,如溶劑、酸、堿等容易造成吸附量子點的脫漏和熒光淬滅。普通包埋法雖然合成條件稍微苛刻,也有一些優點,如通過單體聚合方式將量子點包覆在微球的內部形成穩定的量子點熒光微球,大大降低了外界環境的影響,大大提高量子點熒光微球的穩定性,而且可以通過表面修飾官能團,與生物分子偶聯,用于高靈敏生物分析與檢測,但是通過多年發展也仍然存在包埋法制備的量子點/聚苯乙烯微球中包覆不均勻,包覆量不夠、量子點外漏、微球粒徑大等問題。
技術實現要素:
本發明的目的在于提供一種核殼量子點/聚苯乙烯熒光微球的制備方法,采用乳液聚合成功將大量量子點包覆在聚苯乙烯微球中,保留了包埋法的主要優點并克服了它包覆不均勻,包覆量不夠、量子點外漏等問題缺點,制備出了粒徑均勻、熒光穩定、熒光強度極高的量子點熒光微球,本發明合成方法簡單、微球粒徑可控,重復性好,可用在生物分子檢測。
為實現上述技術目的,達到上述技術效果,本發明公開了一種核殼量子點/聚苯乙烯熒光微球的制備方法,核殼量子點采用熱循環耦合法制備,并使用硫醇重新對量子點進行表面修飾,采用乳液聚合將量子點包埋在聚苯乙烯微球中,獲得核殼量子點/聚苯乙烯熒光微球,具體制備方法如下:
步驟1:稱量表面活性劑2-20份、碳酸氫鈉0.2-2份、分散劑2-30份,加入到玻璃瓶內,再加入去離子水超聲5-10分鐘,形成均勻混合水溶液A,以100份苯乙烯為參考標準;
步驟2:稱量核殼量子點10-150份,再加入100份苯乙烯和4-30份丙烯酸叔丁酯,在冰浴中超聲5分鐘,形成均勻的油相液B;
步驟3:將水溶液A和油相液B兩者混合后磁力攪拌10分鐘,再在冰浴中超聲5-20分鐘,形成穩定的微乳液;
步驟4:將微乳液轉移到三口瓶中,通氮氣30分鐘去除三口瓶中的氧氣,升溫至60-80℃,加入引發劑1-30份,在磁力攪拌下,反應6-12小時,將產物離心純化,獲得聚苯乙烯熒光微球。
其中,核殼量子點通過熱循環耦合法在量子點核上外延生長量子點殼層為1-16層,核殼量子點的熒光光譜發射波長為548nm-750nm。
其中,核殼量子點表面修飾的硫醇配體為烷基硫醇,核殼量子點配體交換所用量子點與烷基硫醇的質量之比為1:(0.5-2)。
其中,步驟1中表面活性劑是十二烷基硫酸胺、十二烷基硫酸鈉、十二烷基磺酸鈉或者十二烷基乙氧基磺基甜菜堿中的一種,分散劑為各分子量的聚乙烯吡咯烷酮。
其中,步驟4中引發劑為過硫酸鉀或過硫酸銨中的一種。
本發明具有以下有益效果:
1.本發明通過量子點的配體交換、加入無機鹽、酯的微乳聚合,合成了粒徑均勻、熒光穩定、熒光強度極高的量子點熒光微球,通過后續官能團修飾可用于高靈敏生物分析及熒光免疫層析快速檢測。
2.通過采用量子點熒光微球與傳統熒光微球相比,能夠克服傳統熒光材料的熒光信號不穩定、制備條件苛刻、瞬時熒光干擾等問題。
3.量子點成功包埋在聚苯乙烯微球中,相對于溶脹法、物理吸附法等,穩定性更好,量子點不會從微球內脫漏,同時在微球的保護下耐苛刻生理條件效果更好。
附圖說明
圖1為本發明的流程示意圖。
圖2為本發明的不同殼層厚度核殼量子點熒光發射圖。
圖3為本發明的不同殼層厚度核殼量子點熒光量子產率圖和熒光發射峰半高寬圖。
圖4為本發明的不同殼層厚度核殼量子點透射電鏡圖。
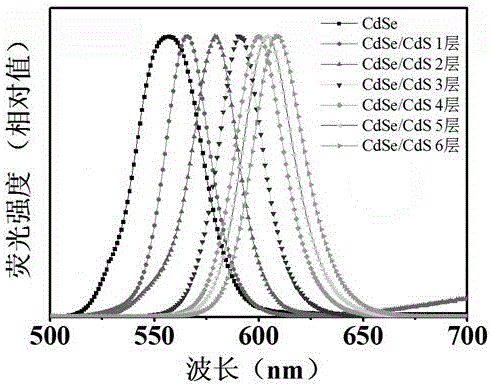
圖5為本發明的核殼量子點/聚苯乙烯熒光微球熒光發射光譜。
圖6為本發明的不同聚苯乙烯用量合成熒光微球透射電鏡圖。
具體實施方式
為了使本發明的目的、技術方案及優點更加清楚明白,以下結合附圖及實施例,對本發明進行進一步詳細說明。
實施例1
本發明公開了一種核殼量子點/聚苯乙烯熒光微球的制備方法,其中核殼量子點以閃鋅礦CdSe/CdS量子點為例,但不限于該種量子點。
閃鋅礦型CdSe量子點的合成及提純:稱取0.2712g的Cd(St)2于三口燒瓶中,再移入7mL的十八烯酸;在室溫下攪拌、抽真空,通氮氣,再升高溫度至100℃,以促使融化,并繼續通氮氣10分鐘,快速升溫,待體系溫度升到250℃時,快速注入2mL 0.1M的Se-十八烯酸懸濁液,保持此溫度進行反應8分鐘后,再次注入0.05mL 0.1M的Se-十八烯酸懸濁液,3-4分鐘后,第三次注入0.05mL 0.1M的Se-十八烯酸懸濁液。此后每3-4分鐘,再次注入0.1M的Se-十八烯酸懸濁液直到量子點的尺寸達預期尺寸;在反應進行過程中,可用注射器吸取少量溶液于比色皿中,通過UV-Vis測定光譜來監測反應進度。當反應獲得預期尺寸的納米晶體,即刻停止加熱,反應溶液快速冷卻至50℃時,進行原位提純。
將制備的CdSe核純化后,測定熒光或者紫外可見光吸收測量吸光度,通過消光系數定量對于測定的粒徑為3.0nm的CdSe核物質的量為2*10-7mol的體系,包覆6層殼層,每層殼層需要加入的0.1mol/L的Cd(DDTC)2單前體體積分別為0.1,0.16,0.23,0.31,0.385和0.48mL。
取2mL十二烷、3mL油胺和原位提純過的1mL CdSe溶液加入到三口瓶中。通氮氣10分鐘,升溫至80℃;注入相應量的殼前體,在第1層注入前軀體時,溶液溫度設定為80℃,前驅體溶液用注射器注入到三口瓶中。在80℃下維持10分鐘,然后升溫至160℃,在保持20分鐘后停止加熱,使反應溶液降溫至80℃,第2層到第6層殼層的生長方式同第1層一樣,只是把生長溫度改為150℃。完成殼層的外延生長。
CdSe/CdS量子點的配體交換:
CdSe/CdS的配體交換具體步驟是將純化后的0.12g CdSe/CdS量子點溶解在8mL甲苯中,再加入0.8g十二烷基硫醇。轉移到50mL單口瓶中,升溫至75℃。保溫5小時。加入丙酮沉淀,5000rmp離心。
核殼量子點/聚苯乙烯熒光微球的合成:
聚苯乙烯熒光微球的合成:稱量0.017g十二烷基硫酸鈉、0.005g碳酸氫鈉、0.02g聚乙烯吡咯烷酮加入到50mL玻璃瓶內,再加入15mL去離子水超聲5分鐘,形成均勻混合水溶液。稱量十二烷基硫配體的0.04g CdSe/CdS量子點,再加入0.5mL苯乙烯和100L丙烯酸叔丁酯。在冰浴中超聲5分鐘,形成均勻的油相液。將兩者混合后磁力攪拌10分鐘,再在冰浴中超聲10分鐘,形成穩定的微乳液。將微乳液轉移到100mL三口瓶中,通氮氣20分鐘去除三口瓶中的氧氣。升溫至75℃。加入0.01g過硫酸鉀。反應12小時。將產物離心純化。
實施例2
實驗目的及方法:為了表征合成的核殼量子點,本實施例以實施例1合成的CdSe/CdS量子點和核殼量子點/聚苯乙烯熒光微球為表征對象,表征內容包括不同殼層數的CdSe/CdS量子點熒光發射光譜、熒光量子產率、熒光發射峰半高寬和透射電鏡圖,核殼量子點/聚苯乙烯熒光微球熒光發射光譜和不同聚苯乙烯用量合成熒光微球透射電鏡圖,具體實驗操作為標準實驗操作此處不再贅述。
實驗結果:
如圖2、3、4所示,通過控制熱循環耦合法在量子點核上外延生長量子點殼層數,可以獲得熒光發射峰從548nm-750nm規則變化的核殼量子點,并且通過熒光量子產率表征可以看出,得到的核殼量子點隨著外延生長殼層的增多,熒光量子產率逐漸接近100%,具有極高的量子產率。而從透射電鏡圖中可以看出,隨著外延生長的繼續,核殼量子點的平均粒徑有明顯的增加。
如圖5所述,在聚苯乙烯包裹的核殼量子點后,合成的熒光微球熒光強度較等當量的核殼量子點有所降低,熒光強度損失約15%,但是該量級的熒光強度仍然滿足現有儀器檢測的需求。反應過程中通過聚苯乙烯用量,可以合成出不同尺寸大小的熒光微球,如圖6所示,圖6中A-D小圖聚苯乙烯用量分別為0.5mL、0.8mL、1mL、1.2mL,其他實施步驟與實施例1相同,此處不再贅述。
以上所述,僅為本發明較佳的具體實施方式,但本發明的保護范圍并不局限于此,任何熟悉本技術領域的技術人員在本發明揭露的技術范圍內,可輕易想到的變化或替換,都應涵蓋在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!