基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料及其合成方法與流程
本發明屬于熒光染料技術領域,具體涉及一種基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料及其合成方法。
背景技術:
雙光子吸收是一種三階非線性光學效應。具有大的雙光子吸收截面的有機材料在三維光存儲、雙光子熒光成像、雙光子光限幅材料以及光動力治療等領域有著具有廣闊的應用前景。在雙光子熒光染料研究領域,開發具有較大雙光子吸收截面的熒光染料是關鍵。
氟硼二吡咯是一類性能優良的熒光發色團,其摩爾消光系數大,熒光量子產率高,光穩定性好,對極性和PH耐受性好,且容易進行結構修飾,是一種應用廣泛的熒光染料,但其缺點是雙光子吸收截面較小(一般不高于10 GM),限制了其在雙光子熒光材料領域的應用。目前,人們對該類染料的雙光子熒光性能的研究還很少。例如,Wang等在氟硼二吡咯的2,6位引入了炔基給電子團,但對其其雙光子吸收截面的提高不明顯(29-60 GM)。Yilmaz及Sezen等在氟硼二吡咯的1及7位引入給電子基團,使氟硼二吡咯熒光團的雙光子吸收截面有所增加。
在雙光子吸收化合物的設計與合成中,利用分子內剛性D-π-A-π-D結構增加熒光基團的雙光子吸收截面,進而提高雙光子吸收及熒光性能,是一種重要的分子設計理念。此外,共軛橋的平面性和剛性具有非常重要的意義,芴環可以看作是具有嚴格剛性的聯苯結構,具有高平面性、高共軛性、良好的光化學與熱穩定性,可作為有效的分子內電荷轉移的π共軛橋引入到雙光子熒光基團中。再者,芳基胺上的氮原子主要采用sp2雜化方式與三個芳香環成鍵,所以往往具有較強的給電子能力,而且由于芳基的電子離域作用,使得分子即使在電荷分離形式的激發態也可以具有較好的穩定性,而且,眾多文獻報道采用二芳基胺為電子給體的熒光集團通常都具有較好的發光性質。因此,本發明正是基于以上文獻調研基礎,以及現有氟硼二吡咯雙光子熒光染料開發所存在的不足,設計并合成了一種基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料,使氟硼二吡咯的雙光子吸收截面有顯著提高,并具有較高的熒光量子產率。
技術實現要素:
本發明的目的是提供一種基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料及其合成方法,該染料具有相對較高的合成產率、良好的單光子及雙光子吸收及熒光性能,該合成方法為基于氟硼二吡咯的雙光子熒光染料的合成及應用提供了新的設計思路,并具有潛在的應用價值。
為實現上述目的,本發明采用以下技術方案:
一種基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料,該雙光子熒光染料具有以下結構通式:
其中,R為C1~C18的烷基。
所述的基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料的合成方法,步驟如下:
(1)2,6-碘代氟硼二吡咯熒光團的合成:以2,4-二甲基吡咯和苯甲醛為原料,以三氟乙酸為催化劑,合成中間產物聯二吡咯,再通過2,3-二氯-4,5-二氰基苯醌氧化脫氫,與三氟化硼乙醚配位成環,生成1,3,5,7-四甲基-8-苯基氟硼二吡咯,然后與碘代丁二酰亞胺反應實現2,6-位的碘化,得2,6-碘代氟硼二吡咯熒光團;
(2)二苯胺-芴-乙炔化合物的合成:以芴為原料,經過溴化、烷基化、Pd(0)催化胺化,Pd(0)和碘化亞銅催化Sonogashira交叉偶聯反應、脫掉三甲基硅基,得到二苯胺-芴-乙炔化合物;
(3)雙光子熒光染料的合成:2,6-位碘代氟硼二吡咯熒光團與二苯胺-芴-乙炔化合物進行Sonogashira偶聯反應,得到基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料。
所述步驟(1)的合成方法為:
A1:氬氣保護下,以無水二氯甲烷為溶劑,加入2,4-二甲基吡咯與苯甲醛,加入三氟乙酸作為催化劑,室溫下磁力攪拌6 h;
B1:將2,3-二氯-4,5-二氰基苯醌溶于干燥二氯甲烷中,加至步驟A1得到的反應液中,繼續室溫攪拌15 min;
C1:往步驟B1的反應液中加入二異丙基乙胺和三氟化硼乙醚溶液,繼續室溫攪拌30 min后,加入水淬滅反應,減壓濃縮,以二氯甲烷:石油醚 = 1:2為洗脫劑,經硅膠層析色譜柱分離,得到1,3,5,7-四甲基-8-苯基氟硼二吡咯
D1:以1,3,5,7-四甲基-8-苯基氟硼二吡咯為原料,二氯甲烷為溶劑,加入N-碘代丁二酰亞胺避光反應過夜,減壓濃縮,以二氯甲烷:石油醚 = 1:2為洗脫劑,經硅膠層析色譜柱分離,得到2,6-碘代氟硼二吡咯熒光團。
所述步驟A1中2,4-二甲基吡咯與苯甲醛的物質的量之比為1:0.4,以1mol 2,4-二甲基吡咯為基準,需要三氟乙酸0.1mL。
所述步驟B1中2,3-二氯-4,5-二氰基苯醌與步驟A1中2,4-二甲基吡咯的物質的量之比為0.4:1。
所述步驟C1中二異丙基乙胺和三氟化硼乙醚溶液的體積比為1:1,以18.9mmol 2,4-二甲基吡咯為基準,需要二異丙基乙胺10mL,三氟化硼乙醚溶液10mL。
所述步驟D1中1,3,5,7-四甲基-8-苯基氟硼二吡咯與N-碘代丁二酰亞胺的物質的量之比為1:5。
所述步驟(2)的合成方法為:
A2:芴與液溴按物質的量之比為1:2.75在干燥氯仿中避光反應過夜,飽和硫代硫酸鈉溶液淬滅反應,在乙醇中重結晶得到2,7-二溴-9H-芴;
B2:2,7-二溴-9H-芴與2.6倍量溴代烷烴(R-Br),3倍量氫氧化鉀,0.1倍量碘化鉀,以二甲基亞砜為溶劑,室溫攪拌過夜,反應完畢,以石油醚為洗脫劑,經硅膠層析色譜柱分離得到2,7-二溴-9,9-二烷基芴;其中2,7-二溴-9H-芴、溴代烷烴、氫氧化鉀和碘化鉀的物質的量之比為1:2.6: 5:0.1;
C2:在氮氣氛圍下,將2,7-二溴-9,9-二烷基芴、Pd(dba)2、1,1'-雙(二苯基膦)二茂鐵、二苯胺和叔丁醇鈉加入無水甲苯中,加熱回流,TLC板監測反應進程,反應完畢減壓濃縮,以石油醚:二氯甲烷=10:1為洗脫劑,經硅膠層析色譜柱分離,即可得到2-二苯胺-7-溴-9,9-二烷基芴;其中2-二苯胺-7-溴-9,9-二烷基芴、二苯胺、Pd(dba)2、1,1'-雙(二苯基膦)二茂鐵和叔丁醇鈉的物質的量之比為1:0.5:0.05:0.05:1;
D2:在氮氣保護下,將2-二苯胺-7-溴-9,9-二烷基芴與三甲基硅基乙炔、三苯基膦, Pd(dba)2和碘化亞銅加入無水三乙胺中,85 ℃攪拌過夜,TLC板監測反應進程,反應完畢后減壓濃縮,過硅膠柱除掉金屬鹽后,得到油狀化合物2-二苯胺-7-三甲基硅基乙炔基-9,9-二烷基芴;其中2-二苯胺-7-溴-9,9-二烷基芴、三甲基硅基乙炔、三苯基膦、Pd(dba)2和碘化亞銅的物質的量之比為1:1.5:0.04:0.02:0.02;
E2:2-二苯胺-7-三甲基硅基乙炔基-9,9-二烷基芴與四丁基氟化銨水合物按物質的量之比為1:1.25混合,以四氫呋喃和甲醇為溶劑,氮氣保護下,室溫反應8 h,減壓濃縮,以二氯甲烷:石油醚=1:2為洗脫劑,經硅膠層析色譜柱分離,得到二苯胺-芴-乙炔化合物。
所述步驟(3)的合成方法為:2,6-位碘代氟硼二吡咯熒光團與二苯胺-芴-乙炔化合物在Pd2(dba)3、三(2-呋喃基)膦和碘化亞銅條件下,以無水DMF和Et3N 按體積比 4:1作為混合溶劑,60℃反應半小時,發生Sonogashira交叉偶聯反應,反應完畢后減壓濃縮,以石油醚:二氯甲烷=2:1為洗脫劑,經硅膠層析色譜柱分離,得到目標產物。
所述2,6-碘代氟硼二吡咯熒光團、二苯胺-芴-乙炔化合物、Pd2(dba)3、三(2-呋喃基)膦和碘化亞銅的物質的量之比為1:2.6:0.05:0.1:0.1。
本發明的有益效果:本發明首次提出將二苯胺基芴通過炔基連接到氟硼二吡咯熒光團的2,6位,進而得到一系列具有分子內剛性結構的基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料,該染料與2,6位未取代的氟硼二吡咯熒光團相比,其雙光子吸收截面顯著提高,且具有較高的熒光量子產率。本發明所合成的目標化合物具有較強的雙光子熒光性能,在甲苯中,其最大雙光子吸收截面達到265 GM,熒光量子產率為0.35,本發明為基于氟硼二吡咯的雙光子熒光染料的合成及應用提供了新的設計思路,并具有潛在的應用價值。
附圖說明
圖1為雙光子熒光染料TPFD的核磁氫譜圖。
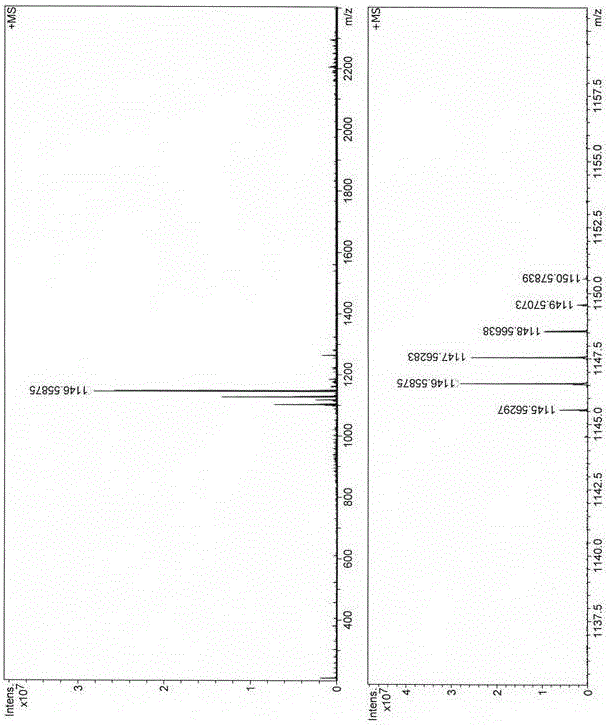
圖2為雙光子熒光染料TPFD的高分辨質譜圖。
圖3為雙光子熒光染料TPFD及1a在不同激發波長下在甲苯溶液(1×10-4mol/L)中的雙光子吸收截面(1a在810-910 nm處無雙光子吸收)。
圖4為雙光子熒光染料TPFD在甲苯溶液(1×10-4mol/L)中的雙光子誘導熒光強度與激發光功率關系圖。
具體實施方式
下面結合具體實施例,對本發明做進一步說明。應理解,以下實施例僅用于說明本發明而非用于限制本發明的范圍,該領域的技術熟練人員可以根據上述發明的內容作出一些非本質的改進和調整。
實施例1
本實施例的基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料的合成路線如下:
其中,R為C2H5。
本實施例的基于苯基取代氟硼二吡咯及二苯胺基芴的雙光子熒光染料的合成方法如下:
(1)2,6-碘代氟硼二吡咯熒光團1a的合成
A1:氬氣保護下,在1000 mL三口瓶中加入500 mL二氯甲烷,18.9 mmol 2,4-二甲基吡咯,7.6mmol苯甲醛和0.1 mL三氟乙酸作為催化劑,室溫下磁力攪拌6 h;
B1:將7.6 mmol 2,3-二氯-5,6-二氰基苯醌(DDQ)溶于150 mL二氯甲烷中,加至步驟A1得到的反應液中,繼續室溫攪拌15 min;
C1:然后往步驟B1得到反應液中加入10 mL二異丙基乙胺(DIEA)和10 mL三氟化硼乙醚溶液,繼續室溫攪拌30 min后,加入200 mL水淬滅反應,二氯甲烷萃取,分液,無水硫酸鈉干燥,真空蒸掉溶劑二氯甲烷,以二氯甲烷:石油醚= 1:2為洗脫劑,經硅膠層析色譜柱分離,得到橘紅色固體1,3,5,7-四甲基-8-苯基氟硼二吡咯,產率為30%;
D1:在100 mL圓底燒瓶中把0.6 mmol 1,3,5,7-四甲基-8-苯基氟硼二吡咯溶于35 mL二氯甲烷中,加入3 mmol NIS,避光條件室溫攪拌12 h,真空蒸發濃縮,經硅膠層析色譜柱分離(洗脫劑:二氯甲烷:石油醚 = 1:2),即得到1a,產率為60%。
(2)二苯胺-芴-乙炔化合物2e的合成(R = C2H5)
A2:2,7-二溴-9H-芴2a的合成
在250 mL圓底燒瓶中,將5 g(30.1mmol)芴溶解到50 mL干燥的氯仿中,把13.25 g 83 mmol (2.75倍)液溴溶于30 mL干燥氯仿溶液中,在恒壓滴液漏斗中在0 °C下將上述混合液滴加到反應瓶中,避光條件下攪拌過夜,將飽和Na2S2O3溶液加入反應液中中和過量的液溴,淬滅反應,分液,有機層用水洗滌幾次后,在有機相中加入無水MgSO4干燥半小時,過濾除掉無水硫酸鎂,真空旋蒸除掉溶劑,在乙醇中重結晶,得到白色固體2,7-二溴-9H-芴2a,產率95%。
B2:2,7-二溴-9,9-二烷基芴2b的合成
在50 mL圓底燒瓶中加入2,7-二溴-9H-芴(5 g, 15 mmol),氫氧化鉀(4.2 g, 75mmol),碘化鉀(0.26 g, 1.5mmol),然后加入12 mL二甲基亞砜后放入10°C的冷肼中,磁力攪拌,逐滴加入溴乙烷(4.25 g, 39 mmol)(五分鐘的時間),可以看到反應混合液由紅色變為淺紫色。滴加完畢,將反應溫度升溫至20 °C,攪拌過夜,反應完全后加入水以除掉二甲基亞砜并用二氯甲烷萃取幾次,靜置分層,在收集的有機相中加入無水Na2SO4干燥2 h后,過濾除掉無水硫酸鈉,真空蒸發濃縮,然后將所得樣品吸附至硅膠上,用石油醚為洗脫劑,經硅膠層析色譜柱分離,得到白色固體2b,產率30%。
C2:2-二苯胺-7-溴-9,9-二烷基芴2c的合成
在50 mL干燥的史萊克瓶中加入2b (3.8 g, 10 mmol),二苯胺(0.85 g, 5 mmol),Pd(dba)2 (0.14 g, 0.25 mmol),DPPF (0.14 g, 0.25 mmol)和叔丁醇鈉(0.95 g,10 mmol),在雙排管上抽充換氣后,氮氣保護下加入18 mL剛處理過的無水甲苯,密封反應瓶,在80 °C下加熱回流18 h后將溫度升至100 °C繼續攪拌5 h,TLC板監測反應完全后,冷卻至室溫,加入水淬滅反應,將反應液轉移至分液漏斗中,用乙醚萃取幾次,分液收集有機相,用硫酸鎂干燥半個小時,過濾除掉硫酸鎂,真空蒸發濃縮,將所得油狀物吸附至硅膠上,以石油醚:二氯甲烷=10:1為洗脫劑,干法上樣,經硅膠層析色譜柱分離,得到黃色固體2c,產率60%。
D2:2-二苯胺-7-三甲基硅基乙炔基-9,9-二烷基芴的合成
權利要求中的記載為“2-二苯胺-7-溴-9,9-二烷基芴、三甲基硅基乙炔、三苯基膦、Pd(dba)2和碘化亞銅的物質的量之比為1:1.5:0.04:0.02:0.02”
在50 mL史萊克瓶中加入化合物2c (1.17 g, 2.5 mmol),PPh3 (26.2 mg, 0.1 mmol), Pd(dba)2 (28.7 mg, 0.05 mmol) 和CuI (10 mg, 0.05 mmol)在雙排管上抽充換氣后,氮氣保護下加入12 mL無水三乙胺,逐滴加入三甲基硅乙炔(0.53 mL, 3.75 mmol),密封反應體系,在85 °C溫度下攪拌過夜,TLC監測反應完全進行后,停止反應,真空蒸發濃縮溶劑三乙胺,將所得油狀物吸附至硅膠上,干法上樣,以二氯甲烷:石油醚=1:2為洗脫劑,經硅膠層析色譜柱分離,得到油狀化合物2d,產率65%。
E2:二苯胺-芴-乙炔化合物2e的合成
在50 mL史萊克瓶中加入化合物2d (0.97 g, 2 mmol),氮氣保護下加入17 mL 四氫呋喃、3 mL甲醇和0.65 g (2.5 mmol)四丁基氟化銨水合物,密封反應管,室溫攪拌8 h,TLC板檢測反應進度,反應結束后,真空蒸發濃縮,除掉溶劑后將所得油狀物吸附至硅膠上,干法上樣,以二氯甲烷:石油醚=1:2為洗脫劑,經硅膠層析色譜柱分離,得到黃色固體化合物2e,產率96%。1H NMR (300 MHz, CDCl3) δ7.58 (d, J= 8.0 Hz, 2H), 7.50 (dd, J= 7.8, 1.5Hz, 1H), 7.46 (br, s, 1H), 7.32-7.27 (m, 4H), 7.18-7.12 (m, 5H), 7.09-7.03 (m, 3H), 3.15 (s, 1H), 1.95 (two sets of diastereotopic quarters, J= 12.8, 7.5 Hz, 4H), 0.38 (t, J= 7.5Hz, 4H).
(3)雙光子熒光染料TPFD的合成(R = C2H5)
在10 mL史萊克反應管中,加入化合物2e (0.11 g, 0.26 mmol),Pd2(dba)3 (4.58 mg, 0.005 mmol),三(2-呋喃基)膦(2.32 mg, 0.01 mmol)和CuI (1.9 mg, 0.01 mmol),然后加入0.8 mL無水N,N-二甲基甲酰胺(DMF)和0.2 mL剛處理過的三乙胺的混合溶劑,氮氣保護下加入化合物1a ( 0.1 mmol),密封反應瓶,在60 °C下反應0.5 h后,TLC板檢測原料氟硼二吡咯反應完全后,停止反應,將反應液倒入乙醚中,轉移至分液漏斗,用水洗滌幾次,靜置分層,將收集到的上層有機相用無水MgSO4干燥,布氏漏斗抽濾除去無水硫酸鎂,真空蒸發濃縮,將所得藍色固體溶解在二氯甲烷中,吸附至硅膠上,以石油醚:二氯甲烷=2:1為洗脫劑,經硅膠層析色譜柱分離,以88%的產率得到藍色固體TPFD。mp 296 °C; 1H NMR (400 MHz, CDCl3) δ 7.56-7.53 (m, 7H), 7.42-7.40 (m, 2H), 7.35-7.31 (m, 4H), 7.27-7.23 (m, 8H), 7.12-7.08 (m, 10H), 7.03-6.99 (m, 6H), 2.77 (s, 6H), 1.97-1.84 (m, 8H), 1.56 (s, 6H), 0.37-0.33 (t,J= 8.0 Hz, 12H). 13C NMR (100 MHz, CDCl3) δ 158.5, 151.6, 150.0, 147.9, 147.7, 143.7, 142.2, 141.6, 135.8, 134.6, 131.3, 130.5, 129.3, 129.2, 128.0, 125.5, 124.0, 123.5, 122.7, 120.8, 120.6, 119.1, 119.0, 116.5, 97.8, 81.5, 56.1, 32.6, 13.8, 13.5, 8.5. 19F NMR (564 MHz, CDCl3)δ-146.4 (q,J(11B, F) = 32.3 Hz). m/z (FT-ICR-MS MALDI): calculated M+ for C81H69BF2N4 as 1146.5590; found as 1146.5587.
實施例1中步驟B2中的溴代烷烴為溴乙烷,需要說明的是,當溴代烷烴的碳原子數為除正溴丁烷的其他C1、C3~C18的溴代烷烴時,同樣能夠實現本發明。
(一)雙光子熒光染料TPFD的單光子吸收及熒光性能(R = C2H5)
將目標化合物溶解在甲苯中,配置成濃度為1×10-5 mol/L的溶液,并對其紫外吸收光譜和熒光發射光譜進行了測試,結果發現,其最大吸收波長為597 nm,最大熒光發射波長為670 nm,熒光量子產率為0.35。
(二)雙光子熒光染料TPFD的雙光子熒光性能(R = C2H5)
雙光子吸收截面測定:采用Ti:sapphire的激光作為激發光源,測定化合物TPFD在甲苯溶液中的雙光子熒光性能。所測定溶液的濃度為1×10-4mol/L,激發波長從810 nm到1010 nm,每相隔10 nm作為一個激發波長,并采用熒光參比法,以熒光素為參比,得到了化合物從810 nm到1010 nm的雙光子吸收截面(圖3)。可以看出,化合物TPFD的雙光子吸收截面最高可達到265 GM (1 GM = 1×10-50 cm4 s per photon-molecule)。TPFD的雙光子熒光強度歲激發光功率增加未變化的情況如圖4所示。由圖可知,當用940 nm波長的激光激發時,化合物的熒光強度隨激發光強度的增強而增強,且熒光強度與激發光功率的對數成正比關系(斜率約等于2),這證明了TPFD在940 nm波長激發下所發射的熒光確實為雙光子誘導熒光機制。
以上顯示和描述了本發明的基本原理和主要特征以及本發明的優點。本行業的技術人員應該了解,本發明不受上述實施例的限制,上述實施例和說明書中描述的只是說明本發明的原理,在不脫離本發明精神和范圍的前提下,本發明還會有各種變化和改進,這些變化和改進都落入要求保護的本發明范圍內。本發明要求保護范圍由所附的權利要求書及其等效物界定。
- 還沒有人留言評論。精彩留言會獲得點贊!