一種α?氨基三乙氧基硅烷的制備方法與流程
本發明涉及一種胺烴基三乙氧基硅烷的制備方法,具體地說是一種α-位碳上連有氨基的三乙氧基硅烷交聯劑的制備方法,屬于有機硅合成技術領域。
背景技術:
單組分室溫硫化硅橡膠的交聯劑是含可水解性基團的多官能性硅烷化合物,通式為R4-nSiYn,其中n=3或n=4;R為碳官能基,Y為可水解性基團。在單組分室溫硫化硅橡膠中起固化交聯的作用。
按照硅橡膠交聯時脫除的小分子物不同可將其大致分為脫羧酸型,脫酮肟型,脫醇型,脫胺型,脫酰胺型,脫丙酮型及脫羥胺型。脫除的小分子物不同其水解速度不同,且不同型號硅橡膠性質和用途也不同。其中,脫醇型室溫硫化硅橡膠固化速度較慢,還需要加入催化劑加快其固化速度。常用的催化劑有有機錫化合物(如二月桂酸二丁基錫、辛酸亞錫)、氨基硅烷等。而有機錫類具有一定毒性,由于催化劑加入后難以在膠料中充分混合均勻而出現局部凝膠化,導致交聯反應不夠完全。
有機硅化合物結構中,取代基的位置會對有機硅化合物的穩定性產生不同程度的影響。α-官能基硅烷交聯劑由于官能團與硅原子間只隔一個碳原子,因而官能團的電子效應對硅原子的影響較大,α-官能基的硅烷交聯劑已被證實具有一定的自催化性能;雖然在堿性條件下容易斷裂硅碳鍵,但是其熱穩定性較高;且合成方法簡單,不需用貴金屬催化劑。因此開發和研究α-硅烷交聯劑有非常大的應用價值。
氨基碳官能基硅烷是用量最大的硅烷交聯劑之一,由其固化交聯的室溫硫化硅橡膠具有優異的粘結性,胺丙基烷氧基硅烷是最常見的氨基碳官能基硅烷。但α-氨基三乙氧基硅烷被證實具有自催化性能,能使固化時間大大縮短。可是由于合成工藝復雜并沒有得到工業化生產和廣泛應用。
有機硅單體是有機硅工業發展的基礎,甲基氯硅烷是合成有機硅材料的重要原料,二甲基二氯硅烷是用量最大、應用最廣的有機硅單體。在直接法合成甲基氯硅烷的過程中,除了主要產物二甲基二氯硅烷外,甲基三氯硅烷是主要副產物,約占總產物的5%~15%,比例相當大。我國有機硅產業發展迅猛異常,截止2015年底,國內甲基氯硅烷總產能已達800萬噸/年,甲基三氯硅烷也相應的大量積壓。研究甲基三氯硅烷新的利用方法,開發有機硅新材料,實現有機硅單體的循環利用,不僅具有重要的科學意義,而且具有重要的實用價值。
中國專利文件CN105131028A采用甲基三氯硅烷和無水乙醇進行醇解反應來制備甲基三乙氧基硅烷,反應要加入催化劑還生了大量腐蝕性氯化氫氣體,往往不能及時排干凈,氯化氫的存在腐蝕設備并且使醇解反應復雜化,出現了很多副反應,使產率降低。
中國專利文件CN101768180A采用氯丙基三乙氧基硅烷和氨氣在高壓釜中反應生成氨基丙基三乙氧基硅烷,此法耗能高,成本高。
尹以高、張藝等將有機胺滴加入氯甲基三乙氧基硅烷的方法來制備氨甲基三乙氧基硅烷,(參見:“自催化交聯型有機硅密封膠的合成和性能研究”,尹以高等,《中國交聯劑》,2007年第5期)。此法采用堿滴酸的方法,易產生多取代等副產物,且在反應體系中要加入大量的酸吸收劑,也操作變得復雜。
技術實現要素:
針對現有技術的不足,本發明提供一種α-氨基三乙氧基硅烷的制備方法。
本發明的技術方案如下:
一種α-氨基三乙氧基硅烷的制備方法,包括步驟如下:
(1)將乙醇鈉溶解于足量有機溶劑中,然后均勻滴加到氯甲基三氯硅烷中,滴加完后常溫反應一段時間,常壓蒸餾除去有機溶劑并濾去不溶物,減壓蒸餾,得到氯甲基三乙氧基硅烷;
(2)將有機胺在氮氣保護下加熱至沸騰,然后滴加步驟(1)所制得的氯甲基三乙氧基硅烷,滴加完后在70~200℃,反應1~8個小時;反應結束后過濾掉生成的鹽,常壓蒸餾除去低沸物,然后減壓蒸餾,即得α-氨基三乙氧基硅烷。
根據本發明,優選的,步驟(1)中所述的有機溶劑為乙醇,乙醇和乙醇鈉的摩爾比為:(4~5):1。
根據本發明,優選的,步驟(1)中所述乙醇鈉和氯甲基三氯硅烷的摩爾比為(2~4):1,進一步優選(3~4):1。
根據本發明,優選的,步驟(1)中乙醇鈉溶液滴加的終點為反應體系pH為中性的時候;
優選的,所述的反應溫度為室溫;反應時間為:5min-1h;
優選的,乙醇鈉溶液的滴加速度為:1滴/秒。
根據本發明,優選的,步驟(1)中減壓蒸餾的壓力范圍為5mmHg~20mmHg。
根據本發明,優選的,步驟(1)中還包括將蒸發出來的乙醇回收利用步驟,過濾除去的不溶物為氯化鈉和稍過量的乙醇鈉。
根據本發明,優選的,步驟(2)中氮氣的通入速率為:0.1~2L/min,有機胺與氯甲基三乙氧基硅烷的摩爾比為(2~8):1,進一步優選(4~6):1。
根據本發明,優選的,步驟(2)中所述的有機胺為RaNH(3-a)或RN2H4,R是含有1-6個碳的烷烴,a是1到2的整數;所述的氮氣為無水無氧的干燥氮氣。
根據本發明,優選的,步驟(2)中氯甲基三乙氧基硅烷的滴加速率為(1.3~1.7)g/min,所述的反應溫度為100-180℃,反應時間為3-6小時,減壓蒸餾的壓力范圍為5mmHg~20mmHg。
根據本發明,優選的,步驟(2)中所述的有機胺為乙二胺、二乙胺、二正丁胺、一正丁胺或環己胺。
本發明中步驟(2)反應結束后,過濾除去有機胺的鹽酸鹽,常壓蒸餾除去低沸物,然后減壓蒸餾收集產物,剩余的為高沸點的副產物。
本發明的反應路線及原理:
本發明中,氯甲基三氯硅烷與乙醇鈉反應生成氯甲基三乙氧基硅烷和氯化鈉為中性的,避免了額外加入酸吸收劑的過程;然后,氯甲基三乙氧基硅烷再與過量的有機胺反應,生成α-氨基三乙氧基硅烷,在反應的過程中由于將氯甲基三乙氧基硅烷滴加到有機胺中,就保證了有機胺的過量,這樣在反應中有機胺既作為反應物又作為酸吸收劑,并且氯甲基三乙氧基硅烷逐滴加入到過量的有機胺中,可以減少有機胺的多取代物等副產物的生成。
本發明制得的α-氨基三乙氧基硅烷可在無催化劑催化的條件下應用于室溫硫化硅橡膠生膠(107膠)的固化交聯。
本發明的有益效果如下:
1.本發明α-氨基三乙氧基硅烷的制備方法中,以大量的工業副產物氯甲基三氯硅烷為起始物,環保節能,既做到了變廢為寶使能源再生利用,又減少了有毒催化劑的使用,符合當今綠色環保的理念。
2.本發明α-氨基三乙氧基硅烷的制備方法中醇解這一步驟不需要加入催化劑也不會產生腐蝕性氣體氯化氫,使得反應變得簡單環保。氨解反應這一步驟不需要加壓,也不需要額外加入酸吸收劑,且胺的利用率高,沒有加入有機溶劑,后續分離簡單,反應體系易于控制。
3.本發明α-氨基三乙氧基硅烷的制備方法中采用沸點回流滴加氯甲基三乙氧基硅烷的方法,反應溫度易于控制,副反應較少。
4.本發明方法制得的α-氨基三乙氧基硅烷產品純度可達95%,產率在40%以上。
5.本發明方法制得的α-氨基三乙氧基硅烷產品可在無催化劑催化的條件下應用于室溫硫化硅橡膠生膠(107膠)的固化交聯,所固化交聯的室溫硫化硅橡膠具有良好的粘結性和熱穩定性。
附圖說明
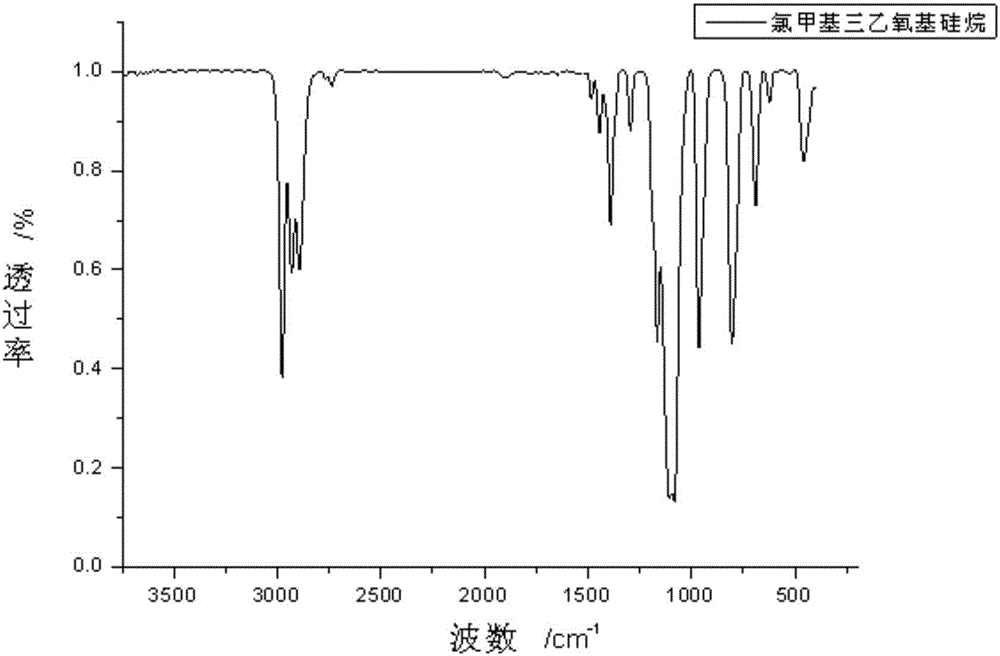
圖1為本發明實施例1制得的氯甲基三乙氧基硅烷的紅外譜圖。
圖2為本發明實施例1制得的氯甲基三乙氧基硅烷的核磁共振氫譜。
圖3為本發明實施例2制得的α-(N,N-二乙基)氨甲基三乙氧基硅烷的紅外譜圖。
圖4為本發明實施例2制得的α-(N,N-二乙基)氨甲基三乙氧基硅烷的核磁共振氫譜。
圖5為本發明實施例3制得的α-(N-正丁基)氨甲基三乙氧基硅烷的紅外譜圖。
圖6為本發明實施例3制得的α-(N-正丁基)氨甲基三乙氧基硅烷的核磁共振氫譜。
圖7為本發明實施例4制得的α-(N,N-二正丁基)氨甲基三乙氧基硅烷的紅外譜圖。
圖8為本發明實施例4制得的α-(N,N-二正丁基)氨甲基三乙氧基硅烷的核磁共振氫譜。
圖9為本發明實施例5制得的α-(N-環己基)氨甲基三乙氧基硅烷的紅外譜圖。
圖10為本發明實施例5制得的α-(N-環己基)氨甲基三乙氧基硅烷的核磁共振氫譜。
圖11為本發明實施例6制得的α-(β-氨乙基)氨甲基三乙氧基硅烷的紅外譜圖。
圖12為本發明實施例6制得的α-(β-氨乙基)氨甲基三乙氧基硅烷的核磁共振氫譜。
具體實施方式
下面通過具體實施例對本發明做進一步說明,但不限于此。
實施例中所用原料均為常規原料,市購產品。
實施例1
一種氯甲基三乙氧基硅烷的制備方法,包括步驟如下:
將19mL氯甲基三氯硅烷加入到250mL干燥的三口瓶中,將31g乙醇鈉溶于110mL乙醇中,在氮氣保護下通過恒壓滴液漏斗將其慢慢滴加到三口瓶中,滴加速度為1滴/秒,當體系的PH由酸性變為中性的時候停止滴加。在室溫下反應30min后進行常壓蒸餾,蒸出乙醇,然后過濾除去氯化鈉和少過量的乙醇鈉,減壓蒸餾,得氯甲基三乙氧基硅烷,留作后用。
本實施例制得的氯甲基三乙氧基硅烷的紅外譜圖如圖1所示,核磁共振氫譜如圖2所示。由圖1、2可知,本實施例制得的產物即為氯甲基三乙氧基硅烷。
實施例2
一種α-(N,N-二乙基)氨甲基三乙氧基硅烷的制備方法,包括步驟如下:
將52.4mL的二乙胺加入到200mL裝有冷凝管的四口瓶中,在氮氣保護下加熱至沸騰,通過恒壓滴液漏斗在35min內均勻滴加18.3mL氯甲基三乙氧基硅烷。滴加完成后,在62℃下反應6h,反應結束后,將混合物冷卻至室溫。進行減壓抽濾,濾去二乙胺鹽酸鹽。常壓蒸餾除去過量的二乙胺,再進行減壓蒸餾,真空度為13mmHg,取99~107℃的餾份,即得α-(N,N-二乙基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N,N-二乙基)氨甲基三乙氧基硅烷的紅外譜圖如圖3所示,核磁共振氫譜如圖4所示。由圖3、4可知本實施例制得的產物即為α-(N,N-二乙基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N,N-二乙基)氨甲基三乙氧基硅烷的純度為96%,收率為63.5%。
實施例3
一種α-(N-正丁基)氨甲基三乙氧基硅烷的制備方法,包括步驟如下:
將40mL的一正丁胺加入到200mL裝有冷凝管的四口瓶中,在氮氣保護下加熱至沸騰,通過恒壓滴液漏斗在40min內均勻滴加20.3mL氯甲基三乙氧基硅烷。滴加完成后,在86℃下反應260min,反應結束后,將混合物冷卻至室溫。進行減壓抽濾,濾去一正丁胺鹽酸鹽。常壓蒸餾除去過量的一正丁胺,再進行減壓蒸餾,真空度為23mmHg,取114~122℃的餾份,即得α-(N-正丁基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N-正丁基)氨甲基三乙氧基硅烷的紅外譜圖如圖5所示,核磁共振氫譜如圖6所示。由圖5、6可知本實施例制得的產物即為α-(N-正丁基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N-正丁基)氨甲基三乙氧基硅烷的純度為97%,收率為43.5%。
實施例4
一種α-(N,N-二正丁基)氨甲基三乙氧基硅烷的制備方法,包括步驟如下:
將84.3mL的二正丁胺加入到200mL裝有冷凝管的四口瓶中,在氮氣保護下加熱至沸騰,通過恒壓滴液漏斗在43min內均勻滴加20.3mL氯甲基三乙氧基硅烷。滴加完成后,在155℃下反應265min,反應結束后,將混合物冷卻至室溫。進行減壓抽濾,濾去二正丁胺鹽酸鹽。直接進行減壓蒸餾,真空度為8mmHg,除去二正丁胺等低沸物,再取122~130℃的餾份,即得α-(N,N-二正丁基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N,N-二正丁基)氨甲基三乙氧基硅烷的紅外譜圖如圖7所示,核磁共振氫譜如圖8所示。由圖7、8可知本實施例制得的產物即為α-(N,N-二正丁基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N,N-二正丁基)氨甲基三乙氧基硅烷的純度為96%,收率為70.1%。
實施例5
一種α-(N-環己基)氨甲基三乙氧基硅烷的制備方法,包括步驟如下:
將45.9mL的二正丁胺加入到200mL裝有冷凝管的四口瓶中,在氮氣保護下加熱至沸騰,通過恒壓滴液漏斗在55min內均勻滴加20.3mL氯甲基三乙氧基硅烷。滴加完成后,在140℃下反應5h,反應結束后,將混合物冷卻至室溫。進行減壓抽濾,濾去環己胺鹽酸鹽。直接進行減壓蒸餾,真空度為7mmHg,除去環己胺等低沸物,再取124~130℃的餾份,即得α-(N-環己基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N-環己基)氨甲基三乙氧基硅烷的紅外譜圖如圖9所示,核磁共振氫譜如圖10所示。由圖9、10可知本實施例制得的產物即為α-(N-環己基)氨甲基三乙氧基硅烷。
本實施例制得的α-(N-環己基)氨甲基三乙氧基硅烷的純度為98%,收率為71.3%。
實施例6
一種α-(β-氨乙基)氨甲基三乙氧基硅烷的制備方法,包括步驟如下:
將54.0mL的乙二胺胺加入到200mL裝有冷凝管的四口瓶中,在氮氣保護下加熱至沸騰,通過恒壓滴液漏斗在60min內均勻滴加20.0mL氯甲基三乙氧基硅烷。滴加完成后,在117℃下反應3h,反應結束后,將混合物冷卻至室溫。進行減壓抽濾,濾去乙二胺鹽酸鹽。常壓蒸餾除去過量的乙二胺,再進行減壓蒸餾,真空度為15mmHg,取120~132℃的餾份,即得α-(β-氨乙基)氨甲基三乙氧基硅烷。
本實施例制得的α-(β-氨乙基)氨甲基三乙氧基硅烷的紅外譜圖如圖11所示,核磁共振氫譜如圖12所示。由圖11、12可知本實施例制得的產物即為α-(β-氨乙基)氨甲基三乙氧基硅烷。
本實施例制得的α-(β-氨乙基)氨甲基三乙氧基硅烷的純度為95%,收率為57.3%。
- 還沒有人留言評論。精彩留言會獲得點贊!