用于稠合氨基二氫噻嗪衍生物的合成的方法和化合物與流程
相關申請
本申請要求在35U.S.C.§119(e)下的2011年1月21日提交的美國臨時專利申請第61/434,849號的權益。
技術領域
本發明涉及用于制備用作β-位點淀粉狀前體蛋白(APP)-裂解酶抑制劑的化合物的化合物和方法。
背景技術:
阿爾茲海默氏病是特征為神經元變性和喪失以及形成老年斑和神經纖維變性的疾病。目前,阿爾茲海默氏病僅用使用以乙酰膽堿酯酶抑制劑為代表的癥狀改善劑的癥狀療法來治療,和用于抑制疾病的進展的基本治療還未開發。需要開發用于控制病理學發作的原因的方法以創建對阿爾茲海默氏病的基礎治療。
據認為Aβ-蛋白,作為淀粉狀前體蛋白(APP)的分解產物,涉及神經元的變性和喪失和癡呆癥狀的發作(參見,例如,Klein等人,Alzheimer's disease-affected brain:Presence of oligomeric Aβ ligands(ADDLs)suggests a molecular basis for reversible memory loss,PNAS USA 2003,Sep 2;100(18),p.10417-10422;Nitsch等人,Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease,Neuron,2003,May22;38,p.547-554)。
Aβ-蛋白具有作為主要組分的由40個氨基酸組成的Aβ40和在C端添加了兩個氨基酸的Aβ42。已知Aβ40和Aβ42具有高聚集性并且是老年斑的主要組分(參見,例如,Jarrett等人,The carboxy terminus of the β amyloid protein is critical for the seeding of amyloid formation:Implications for the pathogenesis of Alzheimers'disease,Bi℃hemistry,1993,32(18),p.4693-4697;Glenner等人,Alzheimer's disease:initial report of the purification and characterization of a novel cerebrovascular amyloid protein,Biochemical and biophysical research communications,1984,May 16,120(3),p.885-890;Masters等人,Amyloid plaque core protein inAlzheimer disease and Down syndrome,PNAS USA,1985,Jun,82(12),p.4245-4249)。此外,已知Aβ40和Aβ42通過APP和早老素(presenilin)基因中的突變增加,其在家族性阿爾茲海默氏病中觀察到(參見,例如,Gouras等人,IntraneuronalAβ42accumulation in human brain,American Journal of Pathology,2000,Jan,156(1),p.15-20;Scheuner等人,Secreted amyloidβ-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1and 2and APP mutations linked to familial Alzheimer's disease,Nature Medicine,1996,Aug,2(8),p.864-870;Forman等人,Differential effects of the swedish mutant amyloid precursor protein onβ-amyloid accumulation and secretion in neurons and nonneuronal cells,The Journal of Biological Chemistry,1997,Dec 19,272(51),p.32247-32253)。因此,期望減少Aβ40和Aβ42的產生的化合物是阿爾茲海默氏病的進展抑制劑或預防劑。
Aβ通過β-分泌酶(BACE1)和隨后通過γ-分泌酶裂解APP來產生。因為這一原因,已經嘗試產生γ-分泌酶和β-分泌酶抑制劑以抑制Aβ產生。
技術實現要素:
本發明涉及用于制備β-位點淀粉狀前體蛋白(APP)-裂解酶(也稱為β-分泌酶或BACE1)的抑制劑的化合物和方法。BACE1抑制劑化合物和制備其的方法已經例如在WO2009/091016中描述,其內容通過參考以其全部并入本文。
本文提供式I化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6烷基)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式I遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
還提供了如上面所定義的式I化合物的制備方法。在一些實施方案中,所述方法包括加熱包含以下的混合物的步驟:
(i)式A的肟:
其中R1、R2和R3如上面為式I所示出;
(ii)氫醌;和
(iii)烴溶劑(例如,苯、甲苯、二甲苯等),
所述加熱達到90℃、95℃、100℃、105℃或110℃至115℃、120℃、125℃、130℃、135℃、140℃、145℃、150℃、155℃或160℃的溫度(例如,90℃至160℃,或110℃至140℃,或100℃至120℃,或120℃至140℃),
以制備所述式I的化合物或其鹽。
在一些實施方案中,所述方法包括加熱包含以下的混合物的步驟:
(i)式B化合物:
其中R1、R2和R3如上面所示出;
(ii)乙酸鹽;
(iii)羥基胺或其鹽;和
(iv)烴溶劑(例如,苯、甲苯、二甲苯等),
所述加熱達到90℃、95℃、100℃、105℃或110℃至115℃、120℃、125℃、130℃、135℃、140℃、145℃、150℃、155℃或160℃的溫度(例如,90℃至160℃,或110℃至140℃,或100℃至120℃,或120℃至140℃),
以制備所述式I的化合物或其鹽。
還提供了式II化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟);
在一些實施方案中,式II遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
還提供了制備式II化合物或其鹽的方法:
其中:
R1是C1-4烷基;和
R2和R3各自獨立地是氫或鹵素,
所述方法包括以下步驟:
(a)加熱包含以下的混合物:
(i)式B的酮:
其中R1、R2和R3如上面為式II所示出;
(ii)羥基胺或其鹽;
(iii)堿(例如,乙酸鹽、丙酸鹽、叔胺堿例如三乙胺或二乙基異丙基胺、吡啶或其鹽);和
(iv)選自以下的溶劑:水和醇(例如,C3-8醇),
所述加熱達到至少90或100攝氏度(例如,如通過回流達到沸騰)產生加熱的混合物,
(b)將加熱的混合物冷卻至60、50或45攝氏度或低于60、50或45攝氏度,并且然后
(c)向所述混合物加入有機溶劑(例如,四氫呋喃或醚溶劑如二乙基醚或甲基叔丁基醚),酸(例如,羧酸如乙酸或無機酸例如鹽酸或硫酸),和鋅(例如,鋅粉末或鋅粉),其中在所述加入過程中將所述混合物維持在60、50或45攝氏度或低于60、50或45攝氏度(例如,25、30或35至45、50或60攝氏度),
以制備所述式II化合物。
在一些實施方案中,將加熱進行至少30小時。
還提供制備式IIa的立體化學純的化合物或其鹽的方法:
其中R1、R2和R3如上面為式II所示出,
所述方法包括以下步驟:
(a)向溶劑(例如,醇如乙醇或異丙醇,或混合物如甲苯-乙腈混合物、甲苯-丙酮混合物、甲苯-四氫呋喃混合物或醇-水混合物)加入式II化合物的立體異構體混合物:
其中R1、R2和R3如上面所示出,
該溶劑包括手性羧酸化合物(例如,D-二苯甲酰基酒石酸、L-二苯甲酰基酒石酸、D-二甲苯甲酰基(ditoluyl)酒石酸和L-二甲苯甲酰基酒石酸等)以形成式II化合物的非對映體鹽的混合物;并且然后(b)使由式II化合物形成的單個非對映體鹽結晶以制備式IIa的立體化學純的化合物或其鹽。
在一些實施方案中,立體化學純的化合物具有大于約80wt%的化合物的一種立體異構體和少于約20wt%的化合物的其它立體異構體,更優選大于約90wt%的化合物的一種立體異構體和小于約10wt%的化合物的其它立體異構體,甚至更優選大于約95wt%的化合物的一種立體異構體和少于約5wt%的化合物的其它立體異構體,和最優選大于約97wt%的化合物的一種立體異構體和小于約3wt%的化合物的其它立體異構體,大于約90wt%的化合物的一種立體異構體和小于約10wt%的化合物的其它立體異構體。
在一些實施方案中,結晶步驟通過將式II化合物的非對映異構體鹽的混合物冷卻(例如,達到40、45、50、55或60℃至80、90、100或110℃的溫度,達到-10、-5、0、5或10℃至15、20、29或35℃的溫度)來執行。
進一步提供式III化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式III遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
還提供式IV化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式IV遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
進一步提供式V化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式V遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
還提供用于制備式V化合物或其鹽的方法:
其中:
R1是C3-4烷基、鹵-C1-4烷基、羥基-C1-4烷基或–C1-4烷基-OR',
其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素;
所述方法包括以下步驟:
(a)提供以下的混合物
(i)式III化合物,
其中R1、R2和R3如上面為式V所示出;
(ii)胺堿(例如,吡啶,或取代吡啶如三甲基吡啶);和
(iii)有機溶劑(例如,芳族烴溶劑,甲苯、二甲苯、苯、二氯甲烷等),
其中所述混合物在0攝氏度或低于0攝氏度的溫度下,
(b)使所述混合物與三氟甲基磺酸酐、4-甲苯磺酰氯、甲基磺酰氯或甲基磺酰酐反應,并且然后,
(c)加入:
(i)醇(例如,丙醇,如2-丙醇);和
(ii)堿,如選自以下的強堿:氫氧化物、醇鹽和氨,
以制備所述式V化合物。
在一些實施方案中,在-15、-10或-5至0攝氏度的溫度下進行反應步驟。
還提供式VI化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式VI遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
進一步提供了式VII化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,R1是甲基、三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
還提供了制備式VII化合物或其鹽的方法,其包括以下步驟:
還原式VI化合物以形成式VII化合物,如下面所示:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
在一些實施方案中,還原式VI化合物以形成式VII化合物可包括以下步驟中的一個或更多個:(a)將粉末狀鐵加到甲醇溶劑以形成其混合物;(b)將鹽酸(HCl)(例如,濃HCl)加到步驟(a)的混合物;(c)將步驟(b)的混合物加熱至31℃、40℃或50℃至65℃、70℃、75℃、80℃、85℃的溫度,或至步驟(b)的混合物的沸點(例如,持續0–4小時)以形成加熱的混合物;和(d)將式VI化合物或其鹽加到步驟(c)的加熱的混合物中。
在一些實施方案中,該方法還包括以下步驟:(e)將步驟(d)的混合物冷卻至10至30℃的溫度以形成冷卻的混合物;和(f)任選地,攪拌所述冷卻的混合物(例如,持續0-4小時)。
在一些實施方案中,以選自以下的量提供HCl:關于式VI化合物的量,化學計量的HCl量;和關于式VI化合物的量,摩爾過量的HCl量。
進一步提供制備式IX化合物或其鹽的方法:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素;和
R4選自:烷基、鹵烷基、烷氧基和用烷氧基取代的烷基;
其包括以下步驟:
使式VII化合物與式VIII的吡嗪羧酸反應:
其中R1、R2、R3和R4如上面所示出,以形成所述式IX化合物或其鹽。
在一些實施方案中,反應可包括以下步驟中的一個或更多個:(a)將亞硫酰氯加到包含在溶劑中的式VIII化合物的溶液以制備形成的混合物;和(b)將式VII化合物加到步驟(a)的形成的混合物,以形成所述式IX化合物或其鹽。
在一些實施方案中,在加入步驟(a)之前或過程中將包含在溶劑中的式VIII化合物的溶液冷卻(例如,到0至25℃的溫度)。
在一些實施方案中,溶劑可包括:N,N'-二甲基咪唑啉-2-酮、甲苯、二氯甲烷、二甲基甲酰胺、N-甲基吡咯烷或二甲基乙酰胺。
在一些實施方案中,反應可包括以下步驟中的一種或更多種:(a)將乙酸乙酯(EtOAc)加到式VIII化合物和式VII化合物以制備形成的混合物;和(b)將烷基膦酸酐(例如,2-丙基膦酸酐)加到步驟(a)的形成的混合物以形成所述式IX化合物或其鹽。
在一些實施方案中,可以在所述加入步驟(a)過程中將形成的混合物維持在0℃至35℃的溫度。
附圖說明
圖1A-1B表示根據實施例4,部分C的式II的四氫呋喃的非對映體拆分后來自手性HPLC的典型色譜。
圖2表示((2S,3R,4S)-4-氨基-4-(2-氟苯基)-2-((三芐基氧基)甲基)四氫呋喃-3-基)甲醇(2R,3R)-2,3-雙(苯甲酰基氧基)琥珀酸酯的x-射線晶體結構:
具體實施方式
A.定義
本發明的化合物包括上面一般性描述的那些,并且通過實施方案、亞實施方案和本文公開的形式進一步說明。如本文所使用,除非另外說明否則下述定義將適用。
如本文所描述,本發明的化合物可任選地用一種或更多種取代基(如上面一般說明的或如通過本發明的特別的類、亞類和形式例示的)取代。一般而言,術語“取代的指用特定的取代基取代給定結構中的氫。除非另外說明,否則被取代的基團可以具有在概基團的每個可取代位置的取代基,并且當任何給定結構中多于一個位置可以用選自特定基團的多于一個的取代基取代時,在每一位置取代基可以是相同的或不同的。本發明預期的取代基的組合優選是致使形成穩定的或化學上可行的化合物的那些。
“異構體“指具有相同數目和種類的原子并因此具有相同分子量,但關于原子的排列或構型不同的化合物。
“立體異構體”指僅在空間中原子的排列不同的異構體。
“非對映異構體”指相互不是鏡像的立體異構體。
“對映體”指相互不可重疊的鏡像的立體異構體。
對映體包括“對映體純”異構體,其包括基本上單一對映體,例如,大于或等于90%、92%、95%、98%或99%,或等于100%的單一對映體。
“對映體純”如本文所用指包括基本上單一對映體,例如大于或等于90%、92%、95%、98%或99%,或等于100%的單一對映體的化合物或化合物的組合物。
“立體異構體純”如本文所用指包括化合物的一種立體異構體并且基本上不含那種化合物的其它立體異構體的其化合物或其組合物。例如,具有一個手性中心的化合物的立體異構體純的組合物基本上不含該化合物的相對的對映體。具有兩個手性中心的化合物的立體異構體純的組合物基本上不含該化合物的非對映體,并且基本上不含該化合物的對映體。典型的立體異構體純的化合物包含大于約80wt%的該化合物的一種立體異構體和少于約20wt%的該化合物的其它立體異構體,更優選大于約90wt%的該化合物的一種立體異構體和少于約10wt%的該化合物的其它立體異構體,甚至更優選大于約95wt%的該化合物的一種立體異構體和少于5wt%的該化合物的其它立體異構體,和最優選大于約97wt%的該化合物的一種立體異構體和少于約3wt%的該化合物的其它立體異構體。參見,例如,美國專利第7,189,715號。
“R”和“S”作為描述異構體的術語是不對稱取代的碳原子上立體化學構型的描述符號。將不對稱取代的碳原子指定為"R"或"S"通過應用Cahn-Ingold-Prelog優選規則進行,這是本領域技術人員所熟知,并且描述在國際理論和應用化學聯合會(International Union of Pure and Applied Chemistry)(IUPAC)關于有機化學命名規則,第E節,立體化學中。
對映體的“對映體過量”(ee)是[(主要對映體的摩爾分數)減(次要對映體的摩爾分數)]x 100。
"穩定的"如本文所用,指當經受考慮它們的生產、檢測和優選它們的回收、純化和用于本文公開的一種或更多種目的的條件時基本上不改變的化合物。在一些實施方案中,穩定的化合物或化學上可行的化合物是當保持在40℃或更低溫度下,不存在濕氣或其它化學反應性條件時持續至少一周基本上不改變的化合物。
"回流"如本文所用指其中來自沸騰液體的蒸氣冷凝并返回到其所來自的混合物的技術,該技術典型地通過在連接冷凝器的容器中使液體沸騰。
"粉狀鐵"或"鐵粉末"是具有小于0.1、0.5、1、5、10、20、50、250、500或1000μm的平均粒徑的鐵。可使用本領域已知方法測定粒徑,例如篩分(mesh sizing)、激光衍射等。
"鋅粉"是具有小于0.001、0.05、0.1、0.5、1、5、10、15或20μm的平均粒徑的鋅。"鋅粉末"是具有小于200、175、150、125或100μm的平均粒徑的鋅。可使用本領域已知方法測定粒徑,例如篩分、激光衍射等。
“有機”化合物如本文所用是含碳化合物。類似地,“有機溶劑”是用作溶劑的含碳化合物。“無機”化合物是不含碳的化合物。
"無機酸"如本文所用是無機化合物的酸。實例包括但不限于鹽酸(HCl)、硝酸(HNO3)、磷酸(H3PO4)、硫酸(H2SO4)、硼酸(B(OH)3)、氫氟酸(HF)、氫溴酸(HBr)、高氯酸(HClO4)等。
"烴"是由碳和氫原子組成的有機化合物。用作“烴溶劑”的烴的實例包括但不限于“芳族烴溶劑”如苯、甲苯、二甲苯等,和“脂族烴溶劑”如戊烷、己烷、庚烷等。
“胺”或“胺堿”如本文所用指具有堿性氮原子的有機化合物(R-NR'R"),并且可以是伯(R-NH2)、仲(R-NHR')或叔(R-NR'R")胺。
“強堿”如本文所用是能夠使非常弱的酸去質子化的化合物。強堿的實例包括但不限于氫氧化物、醇鹽和氨。
“氫氧化物”是通常已知的二原子陰離子OH-或其鹽(典型地其堿金屬或堿土金屬鹽)。氫氧化物的實例包括但不限于:氫氧化鈉(NaCl)、氫氧化鉀(KOH)、氫氧化鋰(LiOH)和氫氧化鈣(CaOH)。
“醇鹽”是RO-,醇的共軛堿。實例包括但不限于甲醇鹽、乙醇鹽和丙醇鹽。
"芳(Ar)"或"芳基(aryl)"指具有一個或更多個閉合的環的芳族碳環部分。實例包括但不限于:苯基、萘基、蒽基、菲基、雙苯基和芘基。
"雜芳基"指具有一個或更多個閉合環的環部分,在至少一個環中具有一個或更多個雜原子(例如,氧、氮或硫),其中至少一個環是芳族的,并且其中所述環獨立地是稠合的,和/或橋連的。實例非限制性地包括:喹啉基、異喹啉基、吲哚基、呋喃基、噻吩基、吡唑基、喹喔啉基、吡咯基、吲唑基、噻吩并[2,3-c]吡唑基、苯并呋喃基、吡唑并[1,5-a]吡啶基、苯硫基吡唑基、苯并噻吩基、苯并噻唑基、噻唑基、2-苯基噻唑基和異噁唑基。
"烷基"或"烷基基團"如本文所用,指完全飽和的直鏈(即,非支鏈的)、支鏈的或環狀烴鏈。在一些實施方案中,烷基包含1、2或3至4、5或6個碳原子(例如,C1-4、C2-4、C3-4、C1-5、C2-5、C3-5、C1-6、C2-6、C3-6)。在一些實施方案中,烷基包含3-4個碳原子。在某些實施方案中,烷基包含1-3個碳原子。在其它實施方案中,烷基包含2-3個碳原子,并且在其它實施方案中烷基包含1-2個碳原子。在某些實施方案中,術語“烷基”或“烷基基團”指環烷基,也稱為碳環。示例性烷基的非限制性實例包括甲基、乙基、丙基、異丙基、丁基、環丙基和環己基。
"烯基"或"烯基基團"如本文所用指具有一個或更多個雙鍵的直鏈(即非支鏈)、支鏈或環狀烴鏈。在某些實施方案中,烯基包含2-6個碳原子。在某些實施方案中,烯基包含2-4個碳原子。在其它實施方案中,烯基包含3-4個碳原子,和在其它實施方案中烯基包含2-3碳原子。根據另一個方面,術語烯基指具有兩個雙鍵的直鏈烴,也稱為“二烯”。在其它實施方案中,術語“烯基”或“烯基基團”指環烯基基團。示例性烯基基團的非限制性實例包括:-CH=CH2、-CH2CH=CH2(也稱為烯丙基)、-CH=CHCH3、-CH2CH2CH=CH2、-CH2CH=CHCH3、-CH=CH2CH2CH3、-CH=CH2CH=CH2和環丁烯基。
如之前所定義,"烷氧基"或"烷基硫代"如本文所用指通過氧("烷氧基")或硫("烷基硫代")原子連接到主要碳鏈的烷基。
“亞甲基”、“亞乙基”、“亞丙基”如本文所用分別指二價部分-CH2-、-CH2CH2-和-CH2CH2CH2-。
“亞乙烯基”、“亞丙烯基”和“亞丁烯基”如本文所用指二價部分-CH=CH-、-CH=CHCH2-、-CH2CH=CH-、-CH=CHCH2CH2-、-CH2CH=CH2CH2-和-CH2CH2CH=CH-,其中每個亞乙烯基、亞丙烯基和亞丁烯基可以是順式或反式構型。在某些實施方案中,亞乙烯基、亞丙烯基或亞丁烯基可以是反式構型。
“烷叉”指由甲叉的單或二烷基取代形成的二價烴基團。在某些實施方案中,烷叉基團具有1-6個碳原子。在其它實施方案中,烷叉基團具有2-6、1-5、2-4或1-3個碳原子。這些基團包括丙叉(CH3CH2CH=)、乙叉(CH3CH=),和異丙叉(CH3(CH3)CH=)以及類似物。
“烯叉”指由甲叉的單或二烯基取代形成的具有一個或更多個雙鍵的二價烴基團。在某些實施方案中,烯叉基團具有2-6個碳原子。在其它實施方案中,烯叉基團具有2-6、2-5、2-4或2-3個碳原子。根據一方面,烯叉具有兩個雙鍵。示例性烯叉基團包括CH3CH=C=、CH2=CHCH=、CH2=CHCH2CH=和CH2=CHCH2CH=CHCH=。
“C1-6烷基酯或酰胺”指C1-6烷基酯或C1-6烷基酰胺,其中每個C1-6烷基如上面所定義。這些C1-6烷基酯基團是式(C1-6烷基)OC(=O)-或(C1-6烷基)C(=O)O-。這些C1-6烷基酰胺基團是式(C1-6烷基)NHC(=O)-或(C1-6烷基)C(=O)NH-。
“C2-6烯基酯或酰胺”指C2-6烯基酯或C2-6烯基酰胺,其中每個C2-6烯基如上面所定義。這些C2-6烯基酯基團是式(C2-6烯基)OC(=O)-或(C2-6烯基)C(=O)O-。這些C2-6烯基酰胺基團是式(C2-6烯基)NHC(=O)-或(C2-6烯基)C(=O)NH-。
“鹵”指氟、氯、溴或碘。
“鹵烷基”指用一個或更多個鹵原子取代的烷基。例如,“氟甲基”指用一個或更多個氟原子取代的甲基(例如,單氟甲基、二氟甲基、三氟甲基)。
“羥基烷基”指用羥基(-OH)取代的烷基。
“氟甲氧基”如本文所用,指通過氧原子連接主要碳鏈的氟甲基,氟甲基如之前所定義。
“保護基”如本文所用,指使特別的官能部分例如O、S或N被暫時阻塞使得在多官能化合物的其它反應部位選擇性地進行反應。例如,在某些實施方案中,如本文所詳述,使用某些示例性氧保護基。氧保護基包括但不限于連接到氧以形成醚的基團,如甲基、取代甲基(例如,Trt(三苯基甲基)、MOM(甲氧基甲基)、MTM(甲基硫代甲基)、BOM(芐氧基甲基)、PMBM或MPM(對甲氧基芐氧基甲基)),取代乙基(例如,2-(三甲基甲硅烷基)乙基)、芐基、取代芐基(例如,對甲氧基芐基)、甲硅烷基(例如,TMS(三甲基甲硅烷基)、TES(三乙基甲硅烷基)、TIPS(三異丙基甲硅烷基)、TBDMS(叔丁基二甲基甲硅烷基)、三芐基甲硅烷基、TBDPS(叔丁基二苯基甲硅烷基)、2-三甲基甲硅烷基丙-2-烯基、叔丁基、四氫吡喃基、烯丙基等。
在一些實施方案中,可作為鹽如藥學上可接受的鹽的形式提供化合物。藥學上可接受的鹽是保留母體化合物的期望的生物學活性并不賦予不期望的毒理學效果的鹽。藥學上可接受的鹽的特定實例包括無機酸鹽(如硫酸鹽、硝酸鹽、高氯酸鹽、磷酸鹽、碳酸鹽、碳酸氫鹽、氫氟酸鹽、氫氯酸鹽、氫溴酸鹽和氫碘酸鹽),有機羧酸鹽(如乙酸鹽、草酸鹽、馬來酸鹽、酒石酸鹽、延胡索酸鹽和檸檬酸鹽),有機磺酸鹽(如甲磺酸鹽、三氟甲基磺酸鹽、乙基磺酸鹽、苯磺酸鹽、甲苯磺酸鹽和樟腦磺酸鹽),氨基酸鹽(如天冬氨酸鹽和谷氨酸鹽),季胺鹽,堿金屬鹽(如鈉鹽和鉀鹽)以及堿土金屬鹽(如鎂鹽和鈣鹽)。
除非另外說明,否則用于描述如本文使用的化學基團或部分的術語遵循慣例,其中從左向右讀名字,連接到分子剩余部分的點在名字的右手側。例如,基團“(C1-3烷氧基)C1-3烷基”在烷基末端連接到分子的剩余部分。進一步的實例包括甲氧基乙基,其中連接點在乙基末端,和甲基氨基,其中連接點在氨基末端。
除非另外說明,否則其中通過其化學式描述單價或二價基團,包括由“-”表示的一種或兩種終端結合部分,應當理解連接從左向右讀。
除非另外說明,否則本文描述的結構也指包括結構的所有對映體、非對映體和幾何(或構象)形式;例如,對于每個不對稱中心的R和S構型,(Z)和(E)雙鍵異構體,以及(Z)和(E)構象異構體。因此,本發明化合物的單一立體化學異構體以及對映體、非對映體和幾何(或構象)混合物屬于本發明的范圍。除非另外說明,否則本發明化合物的所有互變異構形式屬于本發明的范圍。
此外,除非另外說明,否則本文描述的結構也指包括僅在存在一個或更多個同位素富集的原子時不同的化合物。例如,除通過氘或氚置換氫或通過13C-或14C富集的碳置換碳之外,具有本發明結構的化合物屬于本發明的范圍。這些化合物例如用作分析工具或生物學測試中的探針。
B.化合物和化學合成
本文提供了式I化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6烷基)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式I遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式I化合物的實例包括:
還提供了制備如上面定義的式I化合物的方法。在一些實施方案中,該方法包括加熱包含以下的混合物的步驟:
(i)式A的肟:
其中R1、R2和R3如上面為式I所示出;
(ii)氫醌;和
(iii)烴溶劑(例如,苯、甲苯、二甲苯等),
所述加熱達到90℃、95℃、100℃、105℃或110℃至115℃、120℃、125℃、130℃、135℃、140℃、145℃、150℃、155℃或160℃的溫度(例如,90℃至160℃,或110℃至140℃,或100℃至120℃,或120℃至140℃),
以制備所述式I的化合物或其鹽。
在一些實施方案中,所述方法包括加熱包含以下的混合物的步驟:
(i)式B化合物:
其中R1、R2和R3如上面所示出;
(ii)乙酸鹽;
(iii)羥基胺或其鹽;和
(iv)烴溶劑(例如,苯、甲苯、二甲苯等),
所述加熱達到90℃、95℃、100℃、105℃或110℃至115℃、120℃、125℃、130℃、135℃、140℃、145℃、150℃、155℃或160℃的溫度(例如,90℃至160℃,或110℃至140℃,或100℃至120℃,或120℃至140℃),
以制備所述式I的化合物或其鹽。
還提供了式II化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟);
在一些實施方案中,式II遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式II化合物的實例包括:
還提供了制備式II化合物或其鹽的方法:
其中:
R1是C1-4烷基;和
R2和R3各自獨立地是氫或鹵素,
所述方法包括以下步驟:
(a)加熱包含以下的混合物:
(i)式B的酮:
其中R1、R2和R3如上面為式II所示出;
(ii)羥基胺或其鹽;
(iii)堿(例如,乙酸鹽、丙酸鹽、叔胺堿如三乙胺或二乙基異丙基胺、吡啶或其鹽);和
(iv)選自以下的溶劑:水和醇(例如,C3-8醇),
所述加熱達到至少90或100攝氏度(例如,如通過回流達到沸騰)產生加熱的混合物,
(b)將加熱的混合物冷卻至60、50或45攝氏度或低于60、50或45攝氏度,并且然后
(c)向所述混合物加入有機溶劑(例如,四氫呋喃或醚溶劑如二乙基醚或甲基叔丁基醚),酸(例如,羧酸如乙酸或無機酸例如鹽酸或硫酸),和鋅(例如,鋅粉末或鋅粉),其中在所述加入過程中將所述混合物維持在60、50或45攝氏度或低于60、50或45攝氏度(例如,25、30或35至45、50或60攝氏度),
以制備所述式II化合物。
在一些實施方案中,將加熱進行至少30小時。
還提供制備式IIa的立體化學純的化合物或其鹽的方法:
其中R1、R2和R3如上面為式II所示出,
其包括以下步驟:
(a)向溶劑(例如,醇如乙醇或異丙醇,或混合物如甲苯-乙腈混合物、甲苯-丙酮混合物、甲苯-四氫呋喃混合物或醇-水混合物)加入式II化合物的立體異構體的混合物:
其中R1、R2和R3如上面所示出,
該溶劑包括手性羧酸化合物(例如,D-二苯甲酰基酒石酸、L-二苯甲酰基酒石酸、D-二甲苯甲酰基酒石酸和L-二甲苯甲酰基酒石酸等)以形成式II化合物的非對映體鹽的混合物;并且然后
(b)使由式II化合物形成的單個非對映體鹽結晶以制備式IIa的立體化學純的化合物或其鹽。
在一些實施方案中,立體化學純的化合物具有大于約80wt%的化合物的一種立體異構體和少于約20wt%的化合物的其它立體異構體,更優選大于約90wt%的化合物的一種立體異構體和小于約10wt%的化合物的其它立體異構體,甚至更優選大于約95wt%的化合物的一種立體異構體和少于約5wt%的化合物的其它立體異構體,和最優選大于約97wt%的化合物的一種立體異構體和小于約3wt%的化合物的其它立體異構體,大于約90wt%的化合物的一種立體異構體和小于約10wt%的化合物的其它立體異構體。
在一些實施方案中,結晶步驟通過將式II化合物的非對映異構體鹽的混合物冷卻(例如,達到40、45、50、55或60℃至80、90、100或110℃的溫度,達到-10、-5、0、5或10℃至15、20、29或35℃的溫度)來執行。
進一步提供式III化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式III遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式III化合物的實例包括:
還提供式IV化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式IV遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式IV化合物的實例包括:
進一步提供式V化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式V遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式V化合物的實例包括:
還提供用于制備式V化合物或其鹽的方法:
其中:
R1是C3-4烷基、鹵-C1-4烷基、羥基-C1-4烷基或–C1-4烷基-OR',
其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素;
所述方法包括以下步驟:
(a)提供以下的混合物
(i)式III化合物,
其中R1、R2和R3如上面為式V所示出;
(ii)胺堿(例如,吡啶,或取代吡啶如三甲基吡啶);和
(iii)有機溶劑(例如,芳族烴溶劑,甲苯、二甲苯、苯、二氯甲烷等),
其中所述混合物在0攝氏度或低于0攝氏度的溫度下,
(b)使所述混合物與三氟甲基磺酸酐、4-甲苯磺酰氯、甲基磺酰氯或甲基磺酰酐反應,并且然后,
(c)加入:
(i)醇(例如,丙醇,如2-丙醇);和
(ii)堿,如選自以下的強堿:氫氧化物、醇鹽和氨,
以制備所述式V化合物。
在一些實施方案中,在-15、-10或-5至0攝氏度的溫度下進行所述反應步驟。
還提供式VI化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,式VI遵守這樣的條件:當R2或R3中的一個是鄰-氟,并且另一個是氫時,R1不是–CH2F。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式VI化合物的實例包括:
進一步提供了式VII化合物或其鹽:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素(例如,氟)。
在一些實施方案中,R1是甲基、三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
式VII化合物的實例包括:
還提供了制備式VII化合物或其鹽的方法,其包括以下步驟:
還原式VI化合物以形成式VII化合物,如下面所示:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素。
在一些實施方案中,R1是三氟甲基、羥甲基、單氟甲基或三苯基甲氧基甲基。
在一些實施方案中,R2是氟,和/或R3是氫或氟。
在一些實施方案中,還原式VI化合物以形成式VII化合物可包括以下步驟中的一個或更多個:
(a)將粉末狀鐵加到甲醇溶劑以形成其混合物;
(b)將鹽酸(HCl)(例如,濃HCl)加到步驟(a)的混合物中;
(c)將步驟(b)的混合物加熱至31℃、40℃或50℃至65℃、70℃、75℃、80℃、85℃的溫度,或至步驟(b)的混合物的沸點(例如,持續0–4小時)以形成加熱的混合物;和
(d)將式VI化合物或其鹽加到步驟(c)的加熱的混合物中。
在一些實施方案中,該方法還包括以下步驟:
(e)將步驟(d)的混合物冷卻至10至30℃的溫度以形成冷卻的混合物;和
(f)任選地,攪拌所述冷卻的混合物(例如,持續0-4小時)。
在一些實施方案中,以選自以下的量提供HCl:關于式VI化合物的量,化學計量的HCl量;和關于式VI化合物的量,摩爾過量的HCl量。
進一步提供制備式IX化合物或其鹽的方法:
其中:
R1是烷基、鹵烷基、羥基烷基或–(C1-6)-OR',其中R'是氧保護基;和
R2和R3各自獨立地是氫或鹵素;和
R4選自:烷基、鹵烷基、烷氧基和用烷氧基取代的烷基;
其包括以下步驟:
使式VII化合物與式VIII的吡嗪羧酸反應:
其中R1、R2、R3和R4如上面所示出,以形成所述式IX化合物或其鹽。
在一些實施方案中,反應可包括以下步驟中的一個或更多個:
(a)將亞硫酰氯加到包含在溶劑中的式VIII化合物的溶液以制備形成的混合物;和
(b)將式VII化合物加到步驟(a)的形成的混合物,以形成所述式IX化合物或其鹽。
在一些實施方案中,在加入步驟(a)之前或過程中將包含在溶劑中的式VIII化合物的溶液冷卻(例如,到0至25℃的溫度)。
在一些實施方案中,溶劑可包括:N,N'-二甲基咪唑啉-2-酮、甲苯、二氯甲烷、二甲基甲酰胺、N-甲基吡咯烷或二甲基乙酰胺。
在一些實施方案中,反應可包括以下步驟中的一種或更多種:
(a)將乙酸乙酯(EtOAc)加到式VIII化合物和式VII化合物以制備形成的混合物;和
(b)將丙烷膦酸酐(例如,Archimica,Germany)加到步驟(a)的形成的混合物以形成所述式IX化合物或其鹽。
在一些實施方案中,可以在所述加入步驟(a)過程中將形成的混合物維持在0℃至35℃的溫度。
這些化合物或其鹽以及制備方法用于稠合氨基二氫噻嗪衍生物的合成方法,例如下面方案1中提出的方法。
為了可以更充分地理解本文描述的發明,闡述以下實施例。應當理解這些實施例僅用于說明性目的并且不應被解釋為以任何方式限制本發明。
實施例
一般:
使用Biotage SP4進行柱色譜法。使用Büchii旋轉蒸發儀或Genevac離心蒸發儀進行溶劑除去。使用Water自動純化器和19x 100mm XTerra 5微米MS C18柱在酸性流動相條件下進行制備型LC/MS。使用Varian 400MHz分光計記錄NMR譜。
當術語“惰性的”用于描述反應器(例如,反應容器、燒瓶、玻璃反應器和類似物)時,指反應器中的空氣已經用基本上無水的或干燥的惰性氣體(如氮氣、氬氣和類似氣體)置換。術語“當量”(縮寫:當量)如本文所用描述了通過與預定的起始物質相比試劑或反應化合物的化學計量(摩爾比率)。術語“重量”(縮寫:wt)如本文所用相當通過與下面在實施例中具體引用的反應或純化的特別化學組分的質量相比物質或物質組的質量比。該比率計算為:g/g或Kg/Kg。術語“體積”(縮寫:vol)如本文所用相當給定物質或物質組與反應或純化的預定化學組分的質量或體積的體積比。方程中使用的單位涉及匹配數量級。例如,比率計算為:mL/mL、mL/g、L/L或L/Kg。
制備本發明的化合物的一般方法和實驗在下面闡述。在某些情況下,特別的化合物通過實施例描述。然而,應當理解在每種情況下一系列本發明的化合物根據下面描述的方案和實驗制備。
本文使用以下縮寫:
A.式I的六氫呋喃并異噁唑化合物的制備
實施例1:6a-(2-氟苯基)-4-(三氟甲基)六氫呋喃并-[3,4-c]異噁唑的制備
2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酸叔丁酯在rt在氮氣下向反應容器裝入甲苯(3.2L)、THF(0.60L)和丙烯醛(0.40L,5.985mol)。在17℃下加入(三氟甲基)三甲基硅烷(1.003kg,7.059mol)。將反應混合物冷卻至2.5℃并經2小時加入TBAF(0.01M在THF中,0.400L,0.004mol)。在加入TBAF過程中,將反應混合物的溫度升高至65℃。將反應混合物冷卻至0℃,并且2h之后,加入四正丁基銨硫酸氫鹽(0.171kg,0.503mol),隨后加入溴乙酸叔丁基酯(0.987kg,5.064mol)。經2h加入氫氧化鈉(50%wt在水中,4.2kg,52.6mol)同時維持溫度在10℃之下。在0-5℃2h之后,向反應混合物加入水(2.9L)和甲基叔丁基醚(6.0L)。將水相用甲基叔丁基醚(6.0L)再萃取一次。合并有機相并用14%NaCl水溶液洗滌(3x1.6L)。在真空下濃縮有機物以得到油狀標題化合物1(1.150kg,94.5%),其不經另外的純化用于隨后的階段。
2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酸叔丁酯:
1H NMR(500MHz,CDCl3)δ5.86-5.74(m,1H),5.59(d,J=17.5Hz,1H),5.56(d,J=10.9Hz,1H),4.37-4.30(m,1H),4.11(d,J=16.5Hz,1H),4.06(d,J=16.4Hz,1H),1.40(s,9H);13C NMR(125MHz,CDCl3)δ168.51,128.49(d,J=1.7Hz),123.86,123.71(q,J=281.8Hz),82.22,78.67(q,J=31.5Hz),66.60,28.02.
N-甲氧基-N-甲基-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酰胺.在rt向包含2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酸叔丁酯1(1.150kg,4.788mol)的反應器加入甲酸(6.2kg)。將反應混合物加入55-60℃持續4-5小時。在真空下蒸發甲酸(T=40-45℃)并用甲苯(2x 3.0L)逐出(chased)。向殘余物加入CH2Cl2(2.0L)并在真空中進一步濃縮。向所得殘余物加入CH2Cl2(4.6L)并將溶液冷卻至0℃,隨后以五部分加入N,N-羰基二咪唑(1.05kg,6.49mol)。將混合物攪拌30分鐘并分幾部分加入N,O-二甲基羥基胺鹽酸鹽(0.67kg,6.72mol)同時維持溫度低于10℃。將反應混合物升溫至rt并經14小時攪拌。將反應混合物冷卻至3.2℃并以兩部分裝入咪唑(100.7g,1.48mol)。將反應混合物升溫至rt并加入水(1.4kg),隨后加入甲基叔丁基醚(14.0L)。將有機相用2.0N HCl水溶液(1.0L和0.7L)洗滌,隨后用NaHCO3飽和水溶液(1.2L)和NaCl飽和水溶液(1.20L)洗滌。將有機物濃縮以得到油狀標題化合物2(0.95kg,87.2%)。
N-甲氧基-N-甲基-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酰胺:
1H NMR(500MHz,CDCl3)δ5.85-5.76(m,1H),5.62(d,J=17.2Hz,1H),5.56(d,J=10.4Hz,1H),4.49-4.34(m,3H),3.68(s,3H),3.67(s,1H),3.18(s,3H),3.08(s,1H);13C NMR(126MHz,cdcl3)δ169.9*,163.4*,128.61,123.87(d,J=282.0Hz),123.82,78.54(q,J=31.3Hz),66.12,61.52,60.56,36.20,32.24.
HRMS計算C8H12F3NO3[M+H]+228.0848;實測228.0836。
1-(2-氟苯基)-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酮.在-75℃向1-溴-2-氟苯(0.967kg,5.527mol)在THF(6.2L)中的溶液加入正丁基鋰(己烷中2.50M,2.09L,5.22mol)同時維持溫度低于-65℃(ca.100min.)。15分鐘之后,加入N-甲氧基-N-甲基-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酰胺2(0.949kg,4.178mol)在THF(1.6L)中的溶液同時維持溫度低于-65℃(ca.70min.)。在-78℃2.5h之后,將反應通過加入NH4Cl飽和水溶液(3.0L)和甲基叔丁基醚(9.0L)猝滅。將反應混合物升溫至rt,將水相再次用甲基叔丁基醚(2.5L)萃取。將有機相合并,用NaCl飽和水溶液洗滌(2x 0.3L)并在真空下濃縮得到油狀標題化合物3(1.007kg,80.0%)。
1-(2-氟苯基)-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酮:
1H NMR(500MHz,CDCl3)δ7.96(td,J=7.6,1.8Hz,1H),7.62-7.54(m,1H),7.33-7.25(m,1H),7.20-7.12(m,1H),5.86(ddd,J=17.5,10.4,7.3Hz,1H),5.60(dd,J=20.5,13.8Hz,2H),4.91-4.76(m,2H),4.39(dq,J=12.8,6.4Hz,1H);13C NMR(125MHz,CDCl3)δ193.55,162.14(d,JCF=254.1Hz),135.36(d,JCF=9.2Hz),130.62(d,JCF=3.2Hz),128.49,124.85(d,JCF=3.3Hz),123.89,122.93,122.72(d,JCF=24.5Hz),116.50(d,JCF=23.7Hz),78.97(q,JCF=31.4Hz),74.56(d,JCF=12.4Hz).
HRMS計算C12H10F4O2[M+H]+263.0695;實測263.0709。
1-(2-氟苯基)-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酮肟.向反應器加入羥基胺鹽酸鹽(0.34kg,4.95mol)、乙酸鈉(0.47kg,5.70mol)和MeOH(2.68L)。向這種懸浮液中加入1-(2-氟苯基)-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酮3(0.998kg,3.806mol)在MeOH(1.8L)中的溶液并將反應混合物加熱至40-50℃。完成(ca.2h)之后,將反應混合物冷卻至rt,并濾過硅藻土(0.5wt)并用EtOAc(3.0L)沖洗。在真空下濃縮濾液并向所得殘余物加入甲基叔丁基醚(6.3L)、水(0.94L)和NaHCO3飽和水溶液(2.5L)。將有機相用水(1.6L)和NaCl飽和水溶液(0.1L)洗滌一次。在真空下濃縮有機相以提供油狀標題化合物4(1.03kg,95.0%)。
1-(2-氟苯基)-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酮肟:
1H NMR(500MHz,CDCl3)δ7.49-7.35(m,2H),7.24-7.06(m,2H),5.78-5.65(m,1H),5.54-5.40(m,2H),4.89-4.81(m,1H),4.53(d,J=12.6Hz,1H),4.47(d,J=12.6Hz,0.5H),4.27-4.18(m,1H),4.13-4.05(m,0.5H)
HRMS計算C12H11F4NO2[M+H]+278.0804;實測278.0780。
注意:肟4作為結構異構體的平衡存在,其解釋了低于總數的積分值。
6a-(2-氟苯基)-4-(三氟甲基)六氫呋喃并[3,4-c]異噁唑.在rt向1-(2-氟苯基)-2-(1,1,1-三氟丁-3-烯-2-基氧基)乙酮肟4(1.085kg,3.328mol)在二甲苯(6.9L)中的溶液加入氫醌(86.2g,0.8mol)。將溶液加熱至128℃(內溫)持續18h。將溶液冷卻至rt,加入己烷(7.0L),隨后加入4.0M HCl水溶液(2.4L)。將反應混合物攪拌1h并過濾。向固體加入水(2.0L)、甲基叔丁基醚(7.0L)和25wt.%NaOH水溶液(0.4L)。將水層用甲基叔丁基醚(7.0L)萃取一次,將有機物合并,用27%NaCl水溶液(2.0L)洗滌并在真空下濃縮至黑色油狀物5(512.0g,56%)。
6a-(2-氟苯基)-4-(三氟甲基)六氫呋喃并[3,4-c]異噁唑:
1H NMR(500MHz,CDCl3)δ7.64-7.52(m,1H),7.39-7.31(m,1H),7.19(td,J=7.7,1,2Hz,1H),7.11(ddd,J=11.9,8.2,1.0Hz,1H),4.54(d,J=10.1Hz,1H),4.34-4.23(m,1H),4.26-4.17(m,1II),4.16(d,J=10.2Hz,1H),4.10(d,J=8.5Hz,1H),3.71(d,J=20.2Hz,1H);13C NMR(125MHz,CDCl3)δ160.59(d,JCF=247.0Hz),130.50(d,JCF=8.7Hz),128.72,124.69(d,JCF=3.3Hz),124.45(q,JCF=281.8Hz),124.43(d,JCF=11.9Hz),116.66(d,JCF=22.7Hz),83.70(q,JCF=32.1Hz),78.17(d,JCF=3.1Hz),77.63.54.53.
HRMS計算C12H11F4NO2[M+H]+278.0804;實測278.0802。
實施例2:6a-(2-氟苯基)-4-甲基六氫呋喃并[3,4-c]異噁唑的制備
2-(丁-3-烯-2-基氧基)乙酸叔丁酯:向反應器裝入四丁基銨硫酸氫鹽(0.17Wt,0.10當量)和甲苯(2.6Wt,3.0V)。將混合物攪拌并冷卻至0-5℃。同時維持內部溫度低于10℃,加入50wt%氫氧化鈉水溶液(4.5wt,3.0V,10.5當量;由2.25wt的氫氧化鈉和2.25wt的水制備的50%wt氫氧化鈉),隨后加入3-丁烯-2-醇(0.45Wt,0.53V,1.20當量)。將混合物在0-10℃攪拌15分鐘。加入溴乙酸叔丁酯(1.0Wt,1.00當量)同時維持內部溫度在0-10℃。加入之后,將混合物在0-5℃攪拌1h并監測溴乙酸叔丁基酯的完全消耗(目標>98%轉化)。向反應器裝入水(3.0Wt,3.0V)和MTBE(4.4Wt,6.00V)并升溫至20-25℃。將雙相混合物劇烈攪拌15分鐘并然后允許分開。將有機層用水(1.5Wt,1.5V)洗滌。在減壓下濃縮有機層(T<40℃并不低于15torr或20mbar)。加入庚烷(1.0wts,1.5V)和MTBE(1.1wts,1.5V)。如為使產物澄清所需,將溶液過濾以除去顆粒,并用另外的庚烷:MTBE沖洗。將溶液返回到反應器中并在減壓下濃縮以得到油狀6(0.91Wt,95.0%)其帶有少于1體積的溶劑。
2-(丁-3-烯-2-基氧基)乙酸叔丁酯:
1H NMR(500MHz,CDCl3)δ5.78-5.66(m,1H),5.23-5.12(m,2H),4.00-3.85(m,3H),1.47(s,11H),1.31(d,J=6.4Hz,3H);13C NMR(125MHz,CDCl3)δ169.87,139.30,116.72,81.20,77.53,65.83,28.04,27.92,21.08.
2-(丁-3-烯-2-基氧基)-N-甲氧基-N-甲基乙酰胺:向反應器裝入6的甲苯溶液(1.0Wt,1.0V)并冷卻至0-5℃。加入甲酸(4.84Wt,4.0V,19.2當量)同時維持內溫低于10℃。加入甲酸后,將混合物加熱至35-40℃持續三小時。關于6的消耗監測反應(>90%轉化)。完成后在45℃在真空下除去甲酸。將殘余的甲酸通過在真空下在45℃下與甲苯(2.0-2.50Wt,2.3-3.0V)兩次共沸蒸餾除去以得到粗制酸(0.67Wt,95%)。
將粗制酸溶解在二氯甲烷(5.3Wt,4.0V)中并在0℃冷卻。向溶液以5-7部分加入N,N-羰基二咪唑(1.15Wt,1.28當量)并維持反應溫度在0-10℃。注意:在反應過程中存在氣體放出。完成加入N,N-羰基二咪唑后,將混合物升溫至15-20℃并攪拌30分鐘。然后將反應混合物冷卻回到0-5℃。以幾部分加入N,O-二甲基羥基胺鹽酸鹽(0.71Wt,1.33當量),維持反應在0-10℃。完成加入之后,將反應混合物升溫至15-20℃。關于中間體酸的完全消耗監測反應(>98%轉化)。在0-10℃向反應器裝入水(1.2Wt,1.2V)并且然后裝入MTBE(9.1Wt,12.3V)。將雙相混合物劇烈攪拌15分鐘并然后允許分開。將水層分離,然后將合并的有機層用2N HCl(1.8Wt,1.8V)洗滌(檢查pH以保證其在1-3之間或通過TLC檢查有機層以保證除去產物以下的極性物質。若需要裝入更多的2N HCl)。分離水層,將有機物用飽和碳酸氫鈉T(NaHCO3,0.1Wt和水1.1Wt,1.2V)和然后在水(1.2Wt,1.2V)中的20%NaCl洗滌。在減壓下濃縮有機相。加入無水THF(1.33Wt,1.50V),然后將溶液過濾以除去沉淀,若需要用另外的無水THF沖洗以使產物澄清。將THF溶液返回到反應器并在減壓下濃縮以得到油狀物7(0.73Wt,75.0%)。
2-(丁-3-烯-2-基氧基)-N-甲氧基-N-甲基乙酰胺:
1H NMR(500MHz,CDCl3)δ5.80-5.70(m,1H),5.21(d,J=17.3Hz,1H),5.17(d,J=10.3Hz,1H),4.28(d,J=15.6Hz,1H),4.18(d,J=15.6Hz,1H),4.03-3.96(m,1H),3.68(s,3H),3.18(s,3H),1.32(d,J=6.4Hz,3H);13C NMR(125MHz,CDCl3)δ139.62,116.73,77.49,65.34,61.44,32.36,21.25.
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮.向反應器裝入1-溴-2-氟苯(1.31Wt,1.30當量)和無水THF(7.60Wt,8.6V)。將反應混合物冷卻至低于-70℃.加入己烷中的2.5M正丁基鋰(1.94Wt,2.89V,1.25當量)同時維持內溫低于-60℃。完成加入之后,將混合物在低于-60℃的溫度下攪拌15分鐘并且然后加入酰胺7(1.0Wt,1.0當量)在THF(1.9Wt,2.2V)中的溶液同時維持內溫低于-60℃。攪拌反應60分鐘之后就,關于酰胺7的完全消耗監測反應(>90%轉化)。向反應器裝入20wt%氯化銨水溶液(1.0Wt,1.0V)并升溫至20-25℃。將混合物轉移到含有另外的20wt%氯化銨水溶液(3.0Wt,3.0V)的后處理容器中。將混合物攪拌15分鐘,然后停止攪拌并相分離。將下面的水層返回到反應器中。加入MTBE(4.45Wt,6.0V)。將雙相混合物劇烈攪拌15分鐘,并且然后允許分開。將有機層合并并用25wt%氯化鈉水溶液(4.75Wt,4.75V)洗滌。在減壓下濃縮有機相(T<40℃)。加入無水THF(1.78Wt,2.0V),然后將混合物過濾以除去顆粒,若需要用另外的無水THF沖洗以使產物澄清。將THF溶液返回到反應器并在減壓下濃縮以得到油狀物酮8(0.8Wt,73.0%)。
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮:
1H NMR(500MHz,CDCl3)δ7.93(td,J=7.6,1.8Hz,1H),7.57-7.49(m,1H),7.24(dt,J=14.6,3.8Hz,1H),7.12(ddd,J=11.0,8.3,0.8Hz,1H),5.21(dd,J=17.3,1.3Hz,1H),5.17(dd,J=10.3,0.8Hz,1H),4.69(dd,J=18.1,3.2Hz,1H),4.60(dd,J=18.1,3,4IIz,1II);4.00(dq,J=12.9,6.4Hz,1H),1.36(d,J=6.4Hz,3H);13C NMR(125MHz,CDCl3)δ195.24(d,JCF=5.3Hz),162.03(d,JCF=253.9Hz),139.42,134.86(d,JCF=8.9Hz),130.64(d,JCF=3.4Hz),124.67(d,JCF=3.2Hz),123.57(d,JCF=15.3Hz),116.89,116.45(d,JCF=23.7Hz),77.93,74.11(d,JCF=11.2Hz),21.19.
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮肟.向反應器裝入粗制酮8(1.0Wt,1.0當量)、羥基胺鹽酸鹽(0.43Wt,1.30當量)、乙酸鈉(0.59Wt,1.50當量)和甲醇(4.6Wt,5.8V)。將混合物升溫至50-55℃,并且然后繼續攪拌1-2hr。關于酮8的消耗(>95%轉化)監測反應。將混合物冷卻至15-20℃,然后將反應混合物過濾以除去固體。將固體用乙酸乙酯沖洗直到產物澄清(3.6Wt,4.0V)。將濾液返回到反應器并在減壓下濃縮(T<40℃)。加入MTBE(5.9Wt,8.0V)和水(1.0Wt,1.0V)。將雙相混合物劇烈攪拌15分鐘并然后分開。將有機相用10wt%碳酸氫鈉水溶液(3.2Wt,3.2V)、水(2.0Wt,2.0V)和最后25wt%氯化鈉水溶液(1.0Wt,1.0V)順序洗滌。在減壓下濃縮有機相(T<40℃)。加入無水THF(1.78Wt,2.0V),然后將混合物過濾以除去顆粒,若需要用另外的無水THF沖洗以使產物澄清。將THF溶液返回到反應器并在減壓下濃縮以得到油狀物肟9(0.8-1.2Wt,90.0%)。
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮肟:
1H NMR(500MHz,cdcl3)δ7.45(ddd,J=9.2,6.2,2.5Hz,1H),7.38-7.30(m,1H),7.20-7.03(m,2H),5.73-5.58(m,1H),5.21-5.05(m,2H),4.67(s,2H),4.38(d,J=13.0Hz,1H),4.26(d,J=13.0Hz,1H),3.97-3.88(m,1H),3.79-3.72(m,1H),1.18(d,J=6.4Hz,1H),1.05(d,J=6.4Hz,2H);13C NMR(126MHz,cdcl3)δ160.68(d,JCF=249.9Hz),159.13(d,JCF=249.1Hz),156.42,151.80,139.39,130.78(d,JCF=8.2Hz),130.67(d,JCF=8.4Hz),130.27(d,JCF=3.7Hz),129.77(d,JCF=4.2Hz),123.96(d,JCF=3.5Hz),123.86(d,JCF=3.4Hz),122.51(d,JCF=14.1Hz),120.08(d,JCF=17.1Hz),116.59,116.29,115.72(d,JCF=7.4Hz),115.55(d,JCF=7.7Hz),77.45,76.53,68.48(d,JCF=1.7Hz),77.45,76.53,68.48(d,JCF=1.7Hz),62.01(d,JCF=2.4Hz),20.99,20.81.
注意:存在許多1H和13C信號,因為這一化合物是四種可能的異構體的混合物。
6a-(2-氟苯基)-4-甲基六氫呋喃并[3,4-c]異噁唑.向反應器裝入肟9(1.0Wt,1.0當量)、二甲苯(6.8Wt,7.9V)和然后氫醌(0.10Wt,0.2當量.)。將混合物加熱至125-130℃并關于肟9的消耗(>90%轉化)監測反應。將混合物冷卻至20-25℃。加入2N鹽酸水溶液(4.0Wt,4.0V)。將雙相混合物劇烈攪拌15分鐘并然后分離相。將有機相用2N鹽酸水溶液(3.0Wt,3.0V)萃取。注意;產物存在于水層。將水相返回到反應器中。加入叔丁基甲基醚(5.8Wt,7.9V)并且然后加入氫氧化鈉(0.55Wt)以將pH調節至9-10。將雙相混合物劇烈攪拌15分鐘并然后分離相。將水相用MTBE(5.8Wt,8.0vol)萃取第二次。將合并的有機相用25wt%氯化鈉水溶液(1.0Wt,1.0V)洗滌。在減壓下濃縮有機相(T<40℃)。加入無水THF(1.0Wt,1.1V),并過濾以除去顆粒,若需要用另外的無水THF浸洗以使產物澄清。將THF溶液返回到反應器并在減壓下濃縮以得到油狀物四氫異噁唑10(0.6-0.8Wt,65.0%)。
6a-(2-氟苯基)-4-甲基六氫呋喃并[3,4-c]異噁唑:
1H NMR(400MHz,CDCl3)δ7.61(t,J=8.0Hz,1H),7.30(ddd,J=15.0,5.2,1.8Hz,1H),7.19-7.03(m,2H),4.36(d,J=10.2Hz,1H),4.23-4.16(m,1H),4.05(dt,J=12.2,6.2Hz,1H),3.98-3.87(m,2H),3.19-3.09(m,1H),1.40(d,J=6.4Hz,3H).
注意:對于這一化合物未獲得13C-NMR數據。
用于制備酮8的可選過程#1:
2-(丁-3-烯-2-基氧基)-3-(2-氟苯基)-3-氧代丙酸叔丁酯.在氮氣氣氛下向反應器加入酯6的甲苯溶液(1.00Wt,1.0當量)、2-氟苯甲酸甲酯(0.91Wt,0.75Vol 1.10當量)和THF(0.53Wt,1.8Vol)。將溶液冷卻至低于-70℃。以這樣的速率加入六甲基二硅基氨基鋰(LiHMDS,1.0M在THF中,7.6Wt,8.6Vol,1.6當量)使得內部溫度不超過-60℃。在完成LiHMDS加入之后,將混合物升溫(10-20℃/h的速率)并在-40℃攪拌1h。通過GC監測反應(轉化目標≥95%)。如果4h之后轉化仍然在目標量以下,加入另外的THF中LiHMDS 1M(基于未反應的原料1.1當量)。達到目標轉化之后,使內部溫度升高(20-30℃/h的速率)至-15至-20℃。將25%氯化銨水溶液(1.7Wt,1.6Vol)加入反應器,并維持內部溫度低于0℃。加入完成后,將混合物升溫至>0℃并轉移含有25wt%氯化銨水溶液(4.4Wt,4.1Vol)和水(1.1Wt,1.1Vol)的容器中。在20-25℃將混合物攪拌至少30分鐘。除去下面的水層并且有機層用20wt%氯化鈉水溶液(3.0Wt,1.4Vol)洗滌。將混合物攪拌至少15分鐘,然后使其沉淀。除去下面的水層。在減壓下濃縮有機相(夾套<25℃)直到蒸餾停止。加入甲苯(6.6Wt,6.7Vol)并且所得的混合物用水洗滌(3.0Wt,3.0Vol),攪拌15分鐘,然后使其沉淀。有機物用水洗滌(3.0Wt,3.0Vol)直到pH恒定在pH=7-8。有機相20wt%氯化鈉水溶液(3.0Wt,1.4Vol)洗滌。在減壓下濃縮有機相(夾套<25℃)以提供橙色油狀物的酮酯11(2.1Wt)。
2-(丁-3-烯-2-基氧基)-3-(2-氟苯基)-3-氧代丙酸叔丁酯:
1H NMR(500MHz,CDCl3)δ7.88-7.78(m,1H),7.57-7.48(m,1H),7.29-7.19(m,1H),7.17-7.07(m,1H),5.85-5.64(m,1H),5.28-5.13(m,2H),5.09(s,1H),5.05(s,1H),4.18-4.10(m,1H),4.02-3.94(m,1H),1.39(s,9H),1.37(s,9H),1.35(d,J=6.3Hz,3H),1.28(d,J=6.4Hz,3H);13C NMR(125MHz,CDCl3)δ193.17,193.14,192.33,192.30,166.57,166.16,162.44,162.36,160.42,160.33,138.93,138.80,134.76,134.68,134.61,131.17,131.15,131.05,131.02,124.50,124.47,124.40,124.37,117.86,117.60,116.37,116.34,116.19,116.16,82.95,82.91,82.76,82.73,82.38,82.34,78.75,78.48,27.82,27.76,21.21,21.13.
注意:該化合物是非對映體的混合物。
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮.在氮氣氣氛下向反應器加入酮酯11(1.00Wt,1.0當量)和甲酸(10.0Wt,8.0Vol)。將所得混合物在25-30℃攪拌12h,然后取樣測定轉化(目標>97%)。達到目標轉化之后,在減壓下濃縮混合物并維持內部溫度低于30℃。加入甲苯(1.4Wt,1.6Vol)并在減壓下將混合物濃縮,維持內部溫度低于30℃以使殘余的甲酸共沸。加入甲苯(1.4Wt,1.6Vol)并在減壓下將混合物濃縮,維持內部溫度低于30℃。粗制酮8(0.6Wt,77%)直接用于下一階段。
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮:
1H NMR(500MHz,CDCl3)δ7.93(td,J=7.6,1.8Hz,1H),7.57-7.49(m,1H),7.24(dt,J=14.6,3.8Hz,1H),7.12(ddd,J=11.0,8.3,0.8Hz,1H),5.21(dd,J=17.3,1.3Hz,1H),5.17(dd,J=10.3,0.8Hz,1H),4.69(dd,J=18.1,3.2IIz,1II),4.60(dd,J=18.1,3.4Hz,1H);4.00(dq,J=12.9,6.4Hz,1H),1.36(d,J=6.4Hz,3H);13C NMR(125MHz,CDCl3)δ195.24(d,JCF=5.3Hz),162.03(d,JCF=253.9Hz),139.42,134.86(d,JCF=8.9Hz),130.64(d,JCF=3.4Hz),124.67(d,JCF=3.2Hz),123.57(d,JCF=15.3Hz),116.89,116.45(d,JCF=23.7Hz),77.93,74.11(d,JCF=11.2Hz),21.19.
用于制備酮8的可選過程#2:
2-(丁-3-烯-2-基氧基)-1-嗎啉代乙酮.向反應器裝入四丁基銨硫酸氫鹽(0.10當量)和甲苯(4.0V)。將混合物冷卻至10-20℃。當維持內部溫度低于20℃時,加入50wt%氫氧化鈉水溶液(2.2V,9.7當量)并且然后加入3-丁烯-2-醇(0.66V,1.25當量)。將混合物在10-20℃攪拌15分鐘。加入4-(氯乙酰基)嗎啉(1.0Wt,1.00當量)同時維持內部溫度在10-20℃。加入之后,將混合物在10-20℃攪拌1h并關于4-(氯乙酰基)嗎啉的完全消耗(目標>98%轉化)監測反應。向反應器裝入水(3.0V)和MTBE(6.00V)同時維持內部溫度在10-20℃。將雙相混合物劇烈攪拌15分鐘并然后允許分開。將水層用MTBE(3.0V)萃取。將合并的有機層用水洗滌兩次(每次1.5V)。將水洗物用MTBE萃取兩次(每次1.6V)。將有機層合并并在減壓下濃縮(T<40℃)。加入庚烷(1.0V)和MTBE(1.0V)。如為使產物澄清所需,將溶液過濾以除去顆粒,用另外的庚烷:MTBE沖洗。將溶液返回到反應器并在減壓下濃縮以得到油狀酰胺12(77%)。
2-(丁-3-烯-2-基氧基)-1-(2-氟苯基)乙酮.向反應器裝入THF中1.3M的異丙基氯化鎂-氯化鋰(5.15V,1.33當量)并冷卻至0-5℃。加入1-溴-2-氟苯(0.726V,1.33當量)同時維持內部溫度在0-10℃。完成加入之后,將混合物攪拌0-10℃持續1h。加入酰胺12(1.0Wt,1.0當量.)在THF(1.0V)中的溶液同時維持內部溫度在0-10℃。攪拌反應3h之后,關于酰胺12的完全消耗(>97%轉化)監測反應。將反應混合物轉移到1M HCl(11V,.2.2當量)中同時維持內部溫度低于20℃。將混合物攪拌15分鐘并然后分離相。將下面的水層用MTBE(5.0V)萃取。將所有有機層合并并用25wt%氯化鈉水溶液(6.0V)洗滌。將有機相在減壓下濃縮(T<40℃)以得到油狀酮8(93%)。
實施例3:6a-(2,3-二氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑的制備
1-嗎啉代-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮。向具有1-(三苯甲基氧基)丁-3-烯-2-醇13(41.6g,0.111mol,1.0當量)的反應器加入甲苯(146mL)。將所得溶液冷卻至0-5℃并加入四正丁基銨硫酸氫鹽(7.52g,0.0222mol,0.20當量)。在0-5℃下加入4-(氯乙酰基)嗎啉(18.1g,0.111mol,1.00當量)。將氫氧化鈉(50%wt.在水中;88.6g,1.10mol,10當量)冷卻至15℃并加入反應混合物,同時T<10℃。在T<15℃將反應混合物攪拌1hr并監測1-(三苯甲基氧基)丁-3-烯-2-醇的消耗(目標>99%)。向反應混合物加入2-甲氧基-2-甲基丙烷(146mL)和水(146mL),同時T<20℃。將有機物用18%NaCl水溶液(73mL)和NH4Cl飽和水溶液(21mL)洗滌。將有機物濾過硅藻土(10.4g)以除去顆粒并用2-甲氧基-2-甲基丙烷(83mL)沖洗。在真空下在T<30℃蒸發溶劑以經靜置得到白色固體14(55.0g,98.7%收率,是殘余溶劑的緣故)。
1-嗎啉代-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮:
1H NMR(500MHz,CDCl3)δ7.46-7.41(m,6H),7.32-7.25(m,6H),7.25-7.18(m,3H),5.79-5.68(m,1H),5.28(d,J=17.0Hz,1H),5.26(d,J=10.1Hz,1H),4.20(d,J=12.7Hz,1H),4.10(d,J=12.7Hz,1H),3.97-3.88(m,1H),3.69-3.47(m,8H),3.25(dd,J=9.9,6.5Hz,1H),3.17(dd,J=9.9,4.3Hz,1H);13C NMR(125MHz,CDCl3)δ167.84,143.84,134.84,128.67,127.78,127.04,119.06,86.73,81.07,68.78,66.78,66.34,45.94,42.16.
1-(2,3-二氟苯基)-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮.在氮氣下將異丙基氯化鎂-LiCl復合物(1.30M于THF中,155.0mL,0.0715mol)冷卻至0-5℃并加入2,3-二氟溴苯(13.8g,0.0715mol,1.50當量)同時T<10℃。在0-5℃1h后,加入1-嗎啉代-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮14(21.8g,0.048mol,1.0當量)在THF(2.0vols)中的溶液,同時T<10℃。將反應混合物攪拌2.5小時并監測1-嗎啉代-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮的消耗(目標>97%)。將反應混合物通過加入冷的NH4Cl飽和水溶液(110mL)和水(33mL)猝滅同時T<20℃。加入2-甲氧基-2-甲基丙烷(218mL)并分層。將有機物用NH4Cl飽和水溶液(65mL)和18%NaCl水溶液(44mL.)洗滌。將有機物在T<25℃真空下濃縮至淺黃色油狀酮15(21.6g)。
1-(2,3-二氟苯基)-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮:
1H NMR(500MHz,CDCl3)δ7.70-7.64(m,1H),7.47-7.42(m,6H),7.37-7.30(m,1H),7.30-7.25(m,6H),7.24-7.19(m,3H),7.19-7.13(m,1H),5.84-5.71(m,1H),5.30(d,J=17.3Hz,1H),5.25(d,J=10.4Hz,1H),4.78(dd,J=17.7,3.1Hz,1H),4.70(dd,J=17.7,3.1Hz,lH),4.03(dd,J=11.8,6.5Hz,1H),3.35(dd,J=9.8,6.4Hz,1H),3.18(dd,J=9.8,4.6Hz,1H);13C NMR(125MHz,CDCl3)δ194.15(dd,JCF=4.9,2.4Hz),150.77(dd,JCF=250.3,14.1Hz),150.29(dd,JCF=256.4,13.9Hz),143.98,135.34,128.75,127.76,126.95,125.70(d,JCF=12.0Hz),125.16(d,JCF=3.5Hz),124.55(dd,JCF=6.4,4.2Hz),121.62(d,JCF=17.6Hz),118.83,86.78,81.35,74.98(d,JCF=9.5Hz),66.71.
1-(2,3-二氟苯基)-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮肟.在環境溫度下向1-(2,3-二氟苯基)-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮15(21.1g,0.0435mol,1.0當量)在MeOH(106mL)中的溶液加入NaOAc(5.36g,0.0653mol,1.50當量)。5min.之后,以幾部分加入NH2OH HCl(3.33g,0.048mol,1.10當量)。將懸浮液升溫至35℃并攪拌1小時。將反應混合物冷卻至17.8℃并攪拌15分鐘,然后過濾并用乙酸乙酯(84mL)沖洗。在真空(T<30℃)下濃縮濾液。向所得殘余物加入2-甲氧基-2-甲基丙烷(169mL)和水(21.1mL)。將有機物用NaHCO3飽和水溶液(53mL)和18%NaCl水溶液(21mL)洗滌。在真空下濃縮有機物以得到油狀標題化合物16(21.0g)。
1-(2,3-二氟苯基)-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮肟:
1H NMR(500MHz,CDCl3)δ7.42-7.32(m,6H),7.30-7.15(m,10H),7.16-7.06(m,1H),7.03-6.95(m,1H),5.72-5.58(m,1H),5.24-5.13(m,2H),4.80(d,J=14.6Hz,1H),4.73(d,J=14.6Hz,1H),4.45(d,J=12.4Hz,0.3H),4.35(d,J=12,4Hz,0.3H),3.99-3.91(m,0.3H),3.91-3.83(m,1H),3.20(dd,J=9.9,6.8Hz,0.3II),3.09(dd,J=9.8,6.6Hz,1H),3.02(dd,J-9.9,4.2Hz,0.3H),2.95(dd,J=9.8,4.0Hz,1H);13C NMR(125MHz,CDCl3)δ155.40,150.59(dd,JCF=248.1,13.0Hz),148.86(dd,JCF=252.6,13.5Hz),143.96,135.10,128.73,128.69,127.72,127.68,126.92,126.85,124.96,124.56(d,JCF=10.6Hz),124.03-123.86(m),118.64(d,JCF=15.1Hz),117.92(d,JCF=17.2Hz),86.66,86.50,81.30,80.32,69.20,66.54,66.48,62.57.
6a-(2,3-二氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑.向1-(2,3-二氟苯基)-2-(1-(三苯甲基氧基)丁-3-烯-2-基氧基)乙酮肟16(20.0g,0.0400mol,1.0當量)在甲苯(140mL)中的溶液加入氫醌(0.0882g,0.008mol,0.2當量)。將反應混合物加熱至回流(110-115℃)并保持13小時。將反應混合物冷卻至20-25℃并然后在真空(T<45℃)下濃縮。將溶劑用2-丙醇(100mL)逐出。向粗的殘余物加入異丙醇(140mL)并將混合物加熱至65-70℃直到形成澄清溶液。以10℃/hr將溶液冷卻至0℃并且然后保持1小時。將所得懸浮液過濾并將固體用冷的2-丙醇(40.0)沖洗。干燥后獲得粉末狀標題化合物17(13.2g)。
6a-(2,3-二氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑:
1H NMR(500MHz,DMSO)δ7.42-7.33(m,8H),7.31(t,J=7.6Hz,6H),7.28-7.21(m,3H),7.20-7.12(m,1H),6.39(s,1H),4.13(d,J=.8.2Hz,1H),4.03(s,1H),3.97(s,1H),3.89(d,J=9.2IIz,1H),3.81-3.70(m,1H),3.36-3.29(m,1H),3.26(dd,J=10.1,6.0Hz,1H),3.15(dd,J=10.0,3.5Hz,1H);13C NMR(125MHz,DMSO)δ150.74(dd,JCF=246.4,14.4Hz),148.66(dd,JCF=248.3,13.5Hz),144.09,130.58,128.67,128.32,127.48,124.75,124.45,117.18(d,JCF=17.0Hz),86.57,84.91,78.16,76.81,64.82,56.46.
實施例4:6a-(2-氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑的制備
6a-(2-氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑.向反應器裝入1-(2-氟苯基)-2-((1-(三苯甲基氧基)丁-3-烯-2-基)氧基)乙酮(61.2g,1.0當量)、甲苯(428mL)和乙酸鈉(16.1g,1.5當量)。完成加入之后,將反應混合物在環境溫度下攪拌5分鐘。加入羥基胺鹽酸鹽(10.0g,1.1當量)并將懸浮液加熱至111℃。在111℃攪拌>20小時后,允許反應混合物冷卻至環境溫度,并將懸浮液過濾并將固體用甲苯(245mL)沖洗并然后用THF(10體積)沖洗。在減壓下將濾液濃縮(浴溫≤40℃)并用2-丙醇(275mL)逐出。將2-丙醇(428mL)加到固體中并將懸浮液加熱至83℃。在83℃攪拌1小時后,允許混合物冷卻到環境溫度。將漿液經1小時期間冷卻至0℃并在0℃下攪拌1小時。將懸浮液過濾并將固體分兩部分用冷的2-丙醇(490mL)沖洗以得到固體狀6a-(2-氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑(42.3g)。
1H NMR(400MHz,CDCl3)δ7.62(t,1H),7.41(d,J=7.2Hz,6H),7.31-7.21(m,10H),7.12(m,1H),7.03(dd,J=12.0,8.0Hz,1H),4.29-4.04(m,3H),3.91(dd,J=8.8,1.2Hz,1H),3.88(m,1H),3.42(dd,J=9.2,6.8Hz,1H),3.29(m,1H),3.22(dd,J=10.0,4.8Hz,1H).
B.式II的四氫呋喃的制備
4-氨基-4-(2-氟苯基)-2-(三氟甲基)四氫呋喃-3-基)甲醇.將鋅(389.2g,5.95mol)放入反應容器中,并加入水(893mL)。加入乙酸同時維持溫度低于10℃。15分鐘后,作為在THF(665mL)中的溶液加入6a-(2-氟苯基)-4-((三氟甲基)六氫呋喃并[3,4-c]異噁唑5(550.0g,1.98mol)。反應混合物在rt下攪拌16小時。加入二氯甲烷(1.89L),隨后加入28%NH4OH水溶液(552mL),同時使溫度維持在低于30℃。將混合物攪拌30分鐘,然后濾過硅藻土(80g),用二氯甲烷(378mL)沖洗。水層用二氯甲烷(1.89L)萃取。合并有機物,用飽和NaCl水溶液(1.0L)洗滌并且在真空下濃縮以提供氨基醇18(502g,90.6%)。粗制的殘留物不經附加的純化在下一步驟中使用。
HRMS計算C12H13F4NO2[M+H]+280.0961;實測280.0972.
4-氨基-4-(2-氟苯基)-2-甲基四氫呋喃-3-基)甲醇.向反應器裝入四氫異噁唑10(1.0Wt,1.0當量)和冰乙酸(4.1Wt,6.0V)。將混合物在20℃攪拌并且以3部分逐漸地加入鋅粉(1.17Wt,4.0當量),保持反應溫度低于60℃。一旦放熱減退,將混合物在40℃攪拌2h并且然后監測四氫異噁唑10的完全消耗(>99%轉化)。將混合物過濾以除去殘余的鋅。將反應器和固體用乙酸乙酯(4.5Wt,5V)沖洗。將濾液返回到反應器并在減壓(T<40℃)下濃縮。加入甲苯(1.73wt,2.0V)并在減壓下將混合物濃縮。加入甲苯(1.73wt,2.0V)并在減壓下將混合物濃縮。加入二氯甲烷(6.81wt,5V)和水(5.0wt,5.0vol)。加入28%氫氧化銨直到pH到達8-9。將混合物攪拌15分鐘,然后允許相分離并將較低的有機層除去。加入二氯甲烷(5.4wt,4.0V)。將混合物攪拌15分鐘然后分相。將有機相合并然后用25%氯化鈉水溶液(2.0Wt,2.0V)洗滌。在減壓下濃縮有機相(T<40℃)。如為使產物澄清所需,加入無水THF(2.0Wt),并將溶液過濾以除去顆粒,用另外的無水THF沖洗。將THF溶液返回到反應器并在減壓下濃縮以得到油狀氨基醇19(0.93Wt,94.0%)。
4-氨基-4-(2-氟苯基)-2-甲基四氫呋喃-3-基)甲醇:
1H NMR(500MHz,DMSO)δ7.63(td,J=8.6,1.7Hz,1H),7.32-7.24(m,1H),7.20-7.08(m,2H),4.43(s,1H),4.07(dd,J=8.7,1.3Hz,1H),4.02(dq,J=8.5,6.1Hz,1H),3.68-3.57(m,2H),3.55-3.46(m,1H),2.23(dd,J=14.4,6.7Hz,1H),1.24(d,J=6.1Hz,3II);13C NMR(125MHz,DMSO)δ160.69(d,JCF=244.7Hz),133.57(d,JCF=11.3Hz),128.95(d,JCF=8.8Hz),128.81(d,JCF=4.8Hz),124.33(d,JCF=3.2Hz),116.32(d,JCF=23.5Hz),79.58(d,JCF=4.9Hz),78.52,63.99(d,JCF=3.5Hz),59.39,56.91(d,JCF=2.2Hz),21.37.
4-氨基-4-(2,3-二氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇.向6a-(2,3-二氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑17(87.5g,0.175mol,1.0當量)中加入乙酸(385mL)。將懸浮液冷卻至15℃,并經2-3min以小部分加入鋅(88.4g,1.35mol,7.71當量)。允許反應經3-4小時升溫至18℃并攪拌9h。將反應濾過硅藻土(40g)并用甲苯(210mL)沖洗。將收集的濾液在減壓下濃縮,其中浴溫低于35℃并用三部分的甲苯(3x 130mL)逐出。將所得油狀物溶解在CH2Cl2(320mL)中。加入水(195mL),隨后加入28%NH4OH(43.4mL)。將有機層分離并將剩余的水相用CH2Cl2(130mL)萃取。將合并的有機相用27%NaCl水溶液(87.5mL)洗滌。將溶液在減壓下濃縮,溶解在THF(270mL)中并濾過硅藻土。將固體用THF(130mL)沖洗并將合并的濾液在真空下濃縮以產生泡沫狀標題化合物20(69.1g)。
4-氨基-4-(2,3-二氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇:
1H NMR(500MHz,DMSO)δ7.47(t,J=7.5Hz,1H),7.41(d,J=7.5Hz,6H),7.34-7.27(m,7H),7.28-7.21(m,3H),7.19-7.10(m,1H),4.14(d,J=8.7Hz,1H),4.15-4.09(m,1H),3.77(dd,J=8.6,2.6Hz,1H),3.59(dd,J=11.1,6.4Hz,1H),3.46(dd,J=11.1,6.6Hz,1H),3.18(dd,J=9.9,3.0Hz,1H),3.07(dd,J=9.9,5.8Hz,1H),2.62(dd,J=14.6,6.6Hz,1H);13C NMR(125MHz,DMSO)δ150.68(dd,JCF=244.4,13.9Hz),148.46(dd,JCF=246.7,13.0Hz),144.36,135.44(d,JCF=8.1Hz),128.76,128.23,127.37,124.31,124.23,116.12(d,JCF=17.1Hz),86.29,81.48,78.92(d,JCF=4.3Hz),66.03,63.93,59.49,51.27
HRMS計算C31H29F2NO3[M+Na]+524.2013;實測524.2039。
制備式II化合物的可選過程
4-氨基-4-(2-氟苯基)-2-甲基四氫呋喃-3-基)甲醇(19)
向反應器裝入酮8(1.0Wt,1.0當量)和水(10Wt,10V),加入羥基胺鹽酸鹽(0.350Wt,1.05當量)和乙酸鈉(0.433Wt,1.10當量)。將所得混合物攪拌并升溫至101-102℃持續至少40h并監測向四氫噁唑10的完全轉化(>95%)。將混合物冷卻到25℃以下并且然后裝入THF(1.8wt,2.0V)和乙酸(0.86Wt,0.82V,3.0當量)。加入鋅粉(0.942Wt,3.00當量)同時維持內部溫度低于45℃。將所得混合物在35-45℃攪拌1h并關于向氨基醇19的完全轉化(>99%)監測反應。將混合物冷卻到20℃以下并加入二氯甲烷(2.6Wt,2.0V)。加入氫氧化銨水溶液(28%;1.3wt,1.5V,2.2當量)直到pH達到10-12。將所得混合物攪拌15分鐘并入然后將固體過濾,用二氯甲烷(4.6Wt,3.5V)沖洗。允許濾液層分開之后,留出較低的有機層。將上面水層用二氯甲烷萃取兩次(2.6wt,2.0V每次)。將合并的有機層在減壓下濃縮(夾套T<45℃)以得到氨基醇19(0.88wt,81%)。
C.式II四氫呋喃的非對映體拆分
((2S,3R,4S)-4-氨基-4-(2-氟苯基)-2-(三氟甲基)四氫呋喃-3-基)甲醇(2S,3S)-2,3-雙(苯甲酰基氧基)琥珀酸酯.向4-氨基-4-(2-氟苯基)-2-(三氟甲基)四氫呋喃-3-基)甲醇18(0.502kg,1.798mol)在乙醇(4.865L)中的溶液加入二苯甲酰基-D-酒石酸(0.642kg,1.798mol)。將所得懸浮液加熱至67℃。經15min加入水(94.0mL,5.2mol)同時維持溫度>66℃。將所得溶液冷卻至45℃同時發生沉淀。將漿液再加熱至60℃,并且然后以5℃/小時冷卻至環境溫度。將漿液過濾,并將固體用乙醇(950mL)和水(20mL)的預混合并冷卻的溶液沖洗。將固體氨基醇二苯甲酰基酒石酸鹽21在真空下干燥至恒重(370g,97.6%ee)。
((2S,3R,4S)-4-氨基-4-(2-氟苯基)-2-(三氟甲基)四氫呋喃-3-基)甲醇(2S,3S)-2,3-雙(苯甲酰基氧基)琥珀酸酯:
1H NMR(500MHz,CD3OD)δ8.13(d,J=7.2Hz,4H),7.66-7.58(m,3H),7.54-7.45(m,5H),7.36-7.20(m,2H),5.92(s,2H),4.79-4.66(m,1H),4,40-4.28(m,1H),4.04(dd,J=12.1,3.4Hz,1H),3.92(dd,J=12.1,5.4Hz,1H),3.30-3.24(m,1H);13C NMR(125MHz,DMSO)δ169.61,165.81,160.23(d,J=246.1Hz),133.00,131.34(d,J=9.1Hz),129.65,129.55,128.08,127.97(d,J=3.5Hz),124.95(d,J=3.3Hz),116.56(d,J=23.5Hz),77.48(q,JCF=31.0Hz),76.33,73.20,65.61(d,J=3.1Hz),57.11.
HRMS計算C12H13F4NO2[M+H]+280.0961;實測280.0967(對于氨基醇)。
通過與從對映體富集的(S)-2-(三氟甲基)環氧乙烷開始制備的樣品比較,指定氨基醇21的絕對立體化學。
手性HPLC參數:
裝置、試劑和流動相:
裝置:
試劑:
流動相:
向適合的燒瓶加入70mL 2-丙醇和930mL庚烷(分別用100mL和1000-mL有刻度的量筒測定)和1.0mL三乙胺(用測體積用玻璃移液管測定)并混合。在使用過程中線內脫氣。
稀釋溶液:2-丙醇
HPLC參數:
分析物和雜質的保留時間:
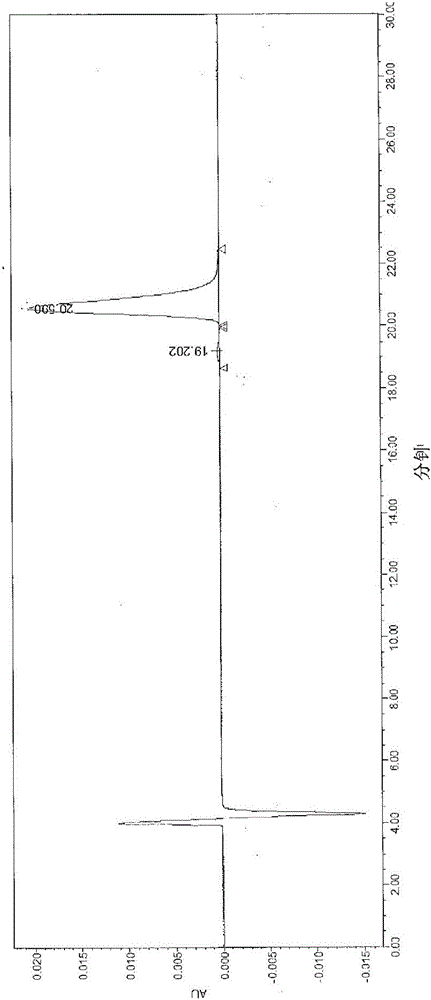
典型色譜圖呈現在圖1A中。
((2R,3R,4S)-4-氨基-4-(2-氟苯基)-2-甲基四氫呋喃-3-基)甲醇(2S,3S)-2,3-雙(苯甲酰基氧基)琥珀酸酯.向反應器裝入氨基醇19(1.0Wt,1.0當量)和乙醇(5.5Wt,7.0V)。將混合物加熱至內部溫度65-70℃。經30-60分鐘向混合物加入二苯甲酰基-D-酒石酸(1.0當量,1.59wt)在乙醇(1.97Wt,2.50V)和水(0.25Wt,0.25V)中的溶液,同時維持內部溫度高于60℃。將含有二苯甲酰基酒石酸的容器用最小量乙醇沖洗。然后將混合物以3℃/小時的速率冷卻至40℃。結晶之后將混合物冷卻至10℃(以10℃/小時)并在10℃攪拌至少2小時。然后將晶體過濾并用預冷卻的乙醇(1.4Wt,1.8V)沖洗并在真空下干燥以得到固體狀氨基醇二苯甲酰基酒石酸鹽22(0.86Wt,33.0%)。
((2R,3R,4S)-4-氨基-4-(2-氟苯基)-2-甲基四氫呋喃-3-基)甲醇(2S,3S)-2,3-雙(苯甲酰基氧基)琥珀酸酯:
1H NMR(500MHz,CD3OD)δ8.14-8.10(m,4H),7.60(t,J=7.4Hz,2H),7.55(td,J=8.1,1.3Hz,1H),7.47(t,J=7.8Hz,4H),7.46-7.42(m,1H),7.30-7.19(m,2H),5.90(s,2H),4.39-4.26(m,2H),4.15(dd,J=10.4,2.4Hz,1H),3.90(ddd,J=28.3,11.9,5.0Hz,2H),2.52(dt,J=8.4,5.0Hz,1H),1.31(d,J=6.1Hz,3H);13C NMR(126MHz,CD3OD)δ169.96,165.90,160.35(d,J=245.9Hz),132.95,131.05(d,JCF=9.1Hz),129.75,129.57,128.06,127.57(d,JCF=3.5Hz),124.82(d,JCF=3.2Hz),124.24(d,JCF=11.5Hz),116.45(d,JCF=23.1Hz),76.34,74.32(d,JCF=5.4Hz),73.50,65.60(d,JCF=2.8Hz),57.43,54.88,19.00.
(2S,3R,4S)-4-氨基-4-(2,3-二氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇(-)二苯甲酰基-L-酒石酸.將消旋的混合物6a-(2,3-二氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑20(69.1g,0.138mol,1.0當量)和(-)-二苯甲酰基-L-酒石酸(49.4g,0.138mol,1.0當量)溶解在丙酮(346mL)中。然后經15min將混合物加熱至61.9℃。加入甲苯(415mL)。然后允許溶液冷卻至室溫。然后將反應混合物冷卻至-5℃并在這一溫度下攪拌另外的一小時。將懸浮液過濾并用甲苯/丙酮(V/V 6/5,138mL,冷卻至0℃)溶液洗滌。在減壓下將固體干燥,并分離標題化合物23(44.5g)。
(2S,3R,4S)-4-氨基-4-(2,3-二氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇(-)二苯甲酰基-L-酒石酸:
1H NMR(500MHz,DMSO)δ8.00-7.95(m,4H),7.66(t,J=7.4Hz,2H),7.53(t,J=7.8Hz,4H),7.47-7.39(m,2H),7.40-7.35(m,6H),7.35-7.28(m,6H),7.28-7.23(m,3H),7.23-7.15(m,1H),5.75(s,1H),4.19(d,J=9.5Hz,2H),4.02(dd,J=9.5,2.1Hz,1H),3.61(dd,J=11.4,6.4Hz,1H),3.52(dd,J=11.4,6.2Hz,1H),3.21-3.14(m,1H),3.12-3.05(m,1H),2.77(dd,J=14.3,6.4Hz,1H);13C NMR(125MHz,DMSO)δ168.07,165.23,150.68(dd,JCF=245.2,13.4Hz),148.27(dd,JCF=248.0,13.6Hz),144.18,134.09,129.75,129.60,129.24,128.70,128.28,127.45,124.88,124.20,117.40(d,JCF=16.8Hz),86.40,80.73,76.73,72.22,65.42,64.58,58.63,50.51.
HRMS計算C31H29F2NO3[M+Na]+524.2013;實測524.2047.
手性HPLC參數:
裝置、試劑和流動相:
裝置:
試劑:
流動相:
向適合的燒瓶加入100mL 2-丙醇和900mL庚烷(分別用100mL和1000-mL有刻度的量筒測定)以及0.5mL三乙胺(用測體積用玻璃移液管測定)并混合。在使用過程中線內脫氣。
稀釋溶液:2-丙醇
HPLC參數:
分析物和雜質的保留時間:
典型色譜法呈現在圖1B中。
D.式III的氨基硫代甲酰基(Carbamothioyl)苯甲酰胺的合成
N-((3S,4R,5S)-3-(2-氟苯基)-4-(羥甲基)-5-(三氟甲基)-四氫呋喃-3-基氨基硫代甲酰基)苯甲酰胺.向手性鹽21(0.361kg,0.556mol)加入乙酸乙酯(1.08L)并將懸浮液冷卻至-3℃。經20分鐘加入1.0N NaOH水溶液(1.30L)同時維持T<5℃。5分鐘之后,經8分鐘加入苯甲酰基異硫代氰酸酯(80.0mL,594mmol)同時維持T<5℃。1hr之后,加入乙酸乙酯(722mL)。除去水層,并將有機物用飽和NaHCO3水溶液(361mL)和飽和NaCl水溶液(361mL)洗滌。將有機物濾過硅藻土(90g)并用乙酸乙酯(360mL)沖洗。在真空下濃縮有機物以得到殘余物,將殘余物再溶解到CH2Cl2(1.1L)中并濃縮以得到黃色泡沫狀標題化合物24(261g,99%收率,由于存在殘余溶劑的緣故),其用于下一步驟。
N-((3S,4R,5S)-3-(2-氟苯基)-4-(羥甲基)-5-(三氟甲基)-四氫呋喃-3-基氨基硫代甲酰基)苯甲酰胺:
1H NMR(500MHz,DMSO)δ12.04(s,2H),11.20(s,2H),7.95(d,J=7.4Hz,2H),7.69-7.60(m,1H),7.56-7.42(m,2H),7.37-7.28(m,1H),7.24-7.12(m,2H).5.59(t,J=4.5Hz,1H),5.03(d,J=9.7Hz,1H),4.92(d,J=9.7Hz,1H),4.75-4.63(m,1H).3.92-3.74(m,2H),2.77-2.66(m,1H);13C NMR(125MHz,DMSO)δ179.98,167.85,159.75(d,JCF=245.0Hz).133.44,132.58,129.88,129.81,129.04,128.85,126.31(d,JCF=9.8Hz),124.36,116.83(d,JCF=23.4Hz),76.11(q,JCF=31.0Hz).74.37(d,JCF=6.1Hz),68.77(d,JCF=3.4Hz),57.03,52.23.
HRMS計算C20H18F4N2O3S[M+H]+441.0896;實測441.0818.
N-((3S,4R,5R)-3-(2-氟苯基)-4-(羥甲基)-5-甲基四氫呋喃-3-基氨基硫代甲酰基)苯甲酰胺.向反應器裝入22(1Wt,1當量)和乙酸乙酯(2.7Wt,3.0V)。將混合物冷卻至0至10℃。加入1.00M NaOH水溶液(4.2Wt,4.0V,2.3當量)。加入苯甲酰基異硫氰酸酯(0.314Wt,0.259V,1.10當量),伴隨劇烈攪拌,同時維持T在0至10℃。完成加入之后,在0-5℃繼續攪拌2h并監測22的完全消耗(目標>98%轉化)。將反應混合物過濾并將濾餅用:1)水(2.0Wt,2.0V);2)庚烷(0.91Wt,1.3V)和乙酸乙酯(1.2Wt,1.3V)的混合物沖洗。在減壓下干燥固體(T<40℃)以得到25(0.57Wt,85.4%收率)。
N-((3S,4R,5R)-3-(2-氟苯基)-4-(羥甲基)-5-甲基四氫呋喃-3-基氨基硫代甲酰基)苯甲酰胺:
1H NMR(500MHz,CDCl3)δ11.78(s,1H),8.88(s,1H),7.88-7.82(m,2H),7.67(td,J=8.1,1.6Hz,1H),7.65-7.59(m,1H),7.50(dd,J=10.8,4.8Hz,2H),7.30-7.23(m,1H),7.15(td,J=7.8,1.2Hz,1H),7,01(ddd,J=12.3,8.2,1.1Hz,1H),4.73(d,J=10.1Hz,1H),4.42(dd,J=10.1,1.7Hz,1H),4.10-4.03(m,1H),4.03-3.91(m,2H),2.80(t,J=5.3Hz,1H),2.63-2.52(m,1H),1.35(d,J=6.1Hz,3H);13C NMR(125MHz,CDC13)δ179.42,166.60,160.35(d,JCF=247.4Hz),133.60,131.81,129.27(d,JCF=8.9Hz),129.20(d,JCF=3.7Hz),129.15,127.55,127.39(d,JCF=10.0Hz),123.96(d,JCF=3.3Hz),116.18(d,JCF=23.0Hz),76.90(d,JCF=2.8Hz),76.27,69.26,59.49,58.03,20.04.
N-(((3S,4R,5S)-3-(2,3-二氟苯基)-4-(羥甲基)-5-((三苯甲基氧基)甲基)四氫呋喃-3-基)氨基硫代甲酰基)苯甲酰胺.將手性鹽23(44.21g,0.051mol,1.0當量)在乙酸乙酯(133mL)中的懸浮液冷卻至1.7℃。加入NaOH水溶液(1.0M,121mL,0.12mol,2.4當量)同時維持溫度低于5℃。將反應攪拌5min然后經5min加入異硫氰酸苯甲基酯(9.43g,0.058mol,1.12當量)。1.5h之后,加入乙酸乙酯(133mL)。將有機相分離并連續用飽和的NaHCO3水溶液(130mL)和18%NaCl水溶液(44mL)洗滌。將合并的水相用EtOAc(66mL)和CH2Cl2(30mL)萃取。合并所有有機相,過濾并在減壓下濃縮。在用CH2Cl2(88mL)逐出殘余溶劑后作為泡沫分離標題化合物26(34.2g)。
N-(((3S,4R,5S)-3-(2,3-二氟苯基)-4-(羥甲基)-5-((三苯甲基氧基)甲基)四氫呋喃-3-基)氨基硫代甲酰基)苯甲酰胺:
1H NMR(500MHz,DMSO)δ11.97(br s,1H),11.20(br s,1H),7.94(d,J=7.4Hz,2H),7.63(t,J=7.4Hz,1H),7.51(t,J=7.8Hz,2H),7.38-7.21(m,17H),7.13(dd,J=13.4,7.9Hz,1H),5.13(t,J=4.4Hz,1H),5.07(d,J=9.7Hz,1H),4.47(d,J=9.8Hz,1H),4.24-4.16(m,1H),3.68(t,J=4.6Hz,2H),3.19(dd,J=10.1,2.9Hz,1H),3.01(dd,J=10.1,4.8Hz,1H),2.63-2.52(m,1H);13C NMR(125MHz,DMSO)5180.06,168.14,150.75(dd,JCF=244.6,13.2Hz),147.93(dd,JCF=247.5,13.5Hz),144.15,133.46,132.55.131.90(d,JCF=6.4Hz),129.04,128.86,128.67,128.28,127.43,124.41,124.25,116.12(d,JCF=17.0Hz),86.27,79.75,75.03,68.04,64.63,58.16,53.35.
HRMS計算C39H34F2N2O4S[M+H]+663.2129;實測663.2200.
((2S,3R,4S)-4-氨基-4-(2-氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇(2R,3R)-2,3-雙(苯甲酰基氧基)琥珀酸酯.向反應器裝入鋅粉(<10微米,3.39g 5.0當量)、6a-(2-氟苯基)-4-((三苯甲基氧基)甲基)六氫呋喃并[3,4-c]異噁唑(5.0g,1.0當量)和THF(30.0mL)。向攪拌的懸浮液加入乙酸(2.97mL,5.0當量)。加入之后,將反應溫度維持在20-30℃持續1小時。然后將反應混合物升溫至35-40℃并維持該溫度同時監測原料的消耗。完成后,將反應混合物冷卻至20-25℃并加入28%NH4OH水溶液(12.5mL)同時維持溫度<40℃。將懸浮液冷卻至20-25℃并濾過硅藻土(0.5Wts)。將固體用MTBE(35.0mL)沖洗。將濾液轉移至分液漏斗中。除去水層,將有機層用水(10.0mL)洗滌。在真空下濃縮有機物(浴溫<40℃)以得到泡沫狀消旋的氨基醇(95-105%粗制收率)。向消旋的氨基醇加入(-)-二苯甲酰基-L-酒石酸(3.74g,1.0當量)。向該混合物加入水(0.76mL,4.0當量),然后丙酮(25.3mL)。將混合物加熱至56-60℃并且然后加入甲苯(30.3mL)同時維持溫度高于50℃。完全加入之后,將溫度保持在56-60℃持續至少30分鐘并且然后以10℃/小時冷卻至17-22℃。將懸浮液在17-22℃攪拌至少二小時。將固體過濾并以兩部分用甲苯/丙酮溶液洗滌(6/5V/V,4.0體積)。在減壓下干燥固體得到白色粉末狀((2S,3R,4S)-4-氨基-4-(2-氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇(2R,3R)-2,3-雙(苯甲酰基氧基)琥珀酸酯(3.4g38%收率)。典型ee%96-97%.
1H NMR(400MHz,d6-DMSO)δ7.92(d,J=7.6Hz,4H),7.61-7.54(m,2H),7.46(t,J=7.6Hz,4H),7.33-7.09(m,19H),5.66(s,2H),4.20(s,1H),4.12(d,J=9.6Hz,1H),4.07(d,J=9.6Hz,1H),3.57(dd,J=11.2,6.0,1H),3.48(dd,J=10.8,6.4,1H),3.12(d,J=8.0Hz,1H),3.03(dd,J=10.0,5.2Hz,1H),2.77(app q,J=7.2Hz,1H).
對映體過量評估的分析方法:
HPLC參數:
流動相:將120mL 2-丙醇和880mL庚烷(分別用100mL和1000-mL有刻度的量筒測量)和0.5mL三乙胺(用測體積用玻璃移液管測定)加到適合的燒瓶并混合。在使用中線內脫氣。
分析物和雜質的保留時間:
獲得((2S,3R,4S)-4-氨基-4-(2-氟苯基)-2-((三苯甲基氧基)甲基)四氫呋喃-3-基)甲醇(2R,3R)-2,3-雙(苯甲酰基氧基)琥珀酸酯的X射線晶體結構并顯示在圖2中。
E.式IV噻唑基苯甲酰胺的合成
N-((4aS,5S,7aS)-7a-(2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺.將N-((3S,4R,5S)-3-(2-氟苯基)-4-(羥甲基)-5-(三氟甲基)-四氫呋喃-3-基氨基硫代甲酰基)苯甲酰胺24(258.3g,583.8mmol)在CH2Cl2(1.55L)中的溶液冷卻至-19.4℃。加入吡啶(118mL,1.46mol)同時維持溫度在-20℃,并且然后將反應混合物冷卻至-24℃。在另一氮氣吹掃的容器中,加入CH2Cl2(258mL)隨后加入三氟甲磺酸酐(108.0mL,642.2mmol)。經30min將所得溶液加到反應混合物,同時維持溫度低于-19.7℃。完成加入之后,在-20℃至-15℃將反應混合物攪拌30min,并且然后經20min升溫至-11℃。加入NH4Cl飽和水溶液(646mL)和水(390mL)。將混合物升溫至環境溫度并將水層除去。將有機物用預混合的NH4Cl飽和水溶液(646mL)和水(390mL)洗滌。合并水層,并用CH2Cl2(520mL)萃取一次。將有機物合并,并在真空下濃縮以得到淺橙色泡沫狀27(250g,100%)。將殘余物不經純化用于下一階段。
N-((4aS,5S,7aS)-7a-(2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺:
1H NMR(500MHz,CDCl3)δ8.03(d,J=6.7Hz,2H),7.52(t,J=7.0Hz,1H),7.48-7.31(m,4H),7.20(t,J=7.4Hz,1H),7.12(dd,J=12.0,8.4Hz,1H),4.82-4.73(m,1H),4.60(d,J=8.9Hz,1H),4.03(d,J=8.3Hz,1H),3.57(d,J=2.7Hz,1H),3.20(d,J=13.6Hz,1H),2.81(dd,J-13.8,2.5Hz,1H);13C NMR(125MHz,CDCl3)δ171.50,159.57(d,JCF=247.2Hz),134.62,132.49,130.65(d,JCFJ=8.8Hz),129.77,128.51,128.45,125.14(q,JCF=281.8Hz),124.97(d,JCF=3.0Hz),124.66(d,JCF=10.3Hz),117.05(d,JCF=23.5Hz),66.81(d,JCF=5.2Hz),38.90,23.20.
HRMS計算C20H16F4N2O2S[M+H]+425.0947;實測425.0945.
N-((4aS,5R,7aS)-7a-(2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺.在18-22℃下向反應器裝入25(1Wt,1當量)、二氯甲烷(8.0Wt,6.0V)和吡啶(0.509Wt,0.520V,2.50當量)。將混合物冷卻至-20至-15℃。加入三氟甲基磺酸酐(0.807Wt,0.481V,1.10當量)在二氯甲烷(1.32Wt,1.00V)中的溶液同時維持T在-15至-5℃。完成加入之后,在-5至0℃繼續攪拌0.5h并監測25的完全消耗(目標>97%轉化)。加入氯化銨飽和水溶(2.67Wt,2.50V)和水(1.50Wt,1.50V)。將雙相混合物在18-22℃劇烈攪拌并允許分開。保留有機層并將水層用二氯甲烷(4.7Wt,3.5V)萃取。將合并的有機層在減壓下濃縮(T<40℃)以得到28(0.95Wt,假定100%理論)。
N-((4aS,5R,7aS)-7a-(2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺:
1H NMR(500MHz,CDCl3)δ8.11-8.07(m,2H),7.56-7.49(m,1H),7.49-7.33(m,4H),7.26-7.19(m,1H),7.18-7,11(m,1H),4.55(d,J=9.6H2,1H),4.55-4.46(m,1H),4.16(s,2H),4.06(dd,J=9.5,2.8Hz,1H),3.26(dd,J=13.7,3.7Hz,1H),2.93-2.87(m,1H),2.84(dd,J=13.7,3.8Hz,1H),1.42(d,J=6.1Hz,3H);13C NMR(126MHz,cdcl3)δ174.13,160.15(d,JCF=247.1Hz),148.20,135.46,132.50,130.58(d,JCF=8.9Hz),128.97,128.47,128.43(d,JCF=3.5Hz),126.94(d,JCF=10.7Hz),125.00(d,JCF=3.4Hz),117.17(d,JCF=23.1Hz),77.56(d,JCF=5.7Hz),76.60,66.88(d,JCF=4.4Hz),46.16(d,JCF=2.9Hz),23.36,19.67.
N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-((三苯甲基氧基)甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺.在-20℃向N-(((3S,4R,5S)-3-(2,3-二氟苯基)-4-(羥甲基)-5-((三苯甲基氧基)甲基)四氫呋喃-3-基)氨基硫代甲酰基)苯甲酰胺26(34.0g,0.0511mol,1.0當量)在CH2Cl2(204mL)中的溶液加入吡啶(10.3ml,0.128mol,2.50當量)。然后經12min加入三氟甲基磺酸酐(9.46mL,0.0563mol,1.10當量)在CH2Cl2(34mL)中的溶液同時維持溫度低于-17℃。在-20℃將反應攪拌30min然后允許緩慢升溫至5℃。加入NH4Cl飽和水溶液(85mL)和水(32mL)。然后將水層用CH2Cl2(68mL)萃取。將有機相在減壓下濃縮得到橙色泡沫狀標題化合物29(33.6g)。
HRMS計算C39H32F2N2O3S[M+H]+647.2180;實測647.2123.
N-(3-((4aS,5S,7aS)-2-氨基-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-(二氟甲基)吡嗪-2-甲酰胺.將5-(二氟甲基)吡嗪-2-羧酸(30.0g,1.05當量)和(4aS,5S,7aS)-7a-(5-氨基-2-氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺(49.2g,1.00當量)加入反應器中并將EtOAc(443mL)加到混合物以得到懸浮液。在環境溫度下加入(105g,1.10當量,50wt%在EtOAc中)(Archimica,Germany)溶液同時控制內部溫度低于30℃。將反應混合物在40-45℃攪拌>3小時并通過HPLC監測。將反應混合物冷卻至環境溫度并加入水(98mL)。10-15分鐘之后加入28%氫氧化銨(137mL)同時控制溫度低于30℃。加入另外的EtOAc(172mL)并將反應混合物在環境溫度下攪拌30分鐘。分離水相并用EtOAc(246mL)萃回。將有機相合并并用15%NaCl水溶液(98mL)和水(98mL)洗滌。將有機層濾過硅藻土(0.5Wt)并在真空下濃縮以獲得淺褐色固體(定量的粗制收率),其從1-丙醇結晶得到褐色固體狀N-(3-((4aS,5S,7aS)-2-氨基-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-(二氟甲基)吡嗪-2-甲酰胺(54.2g)。
1H NMR(500MHz,DMSO)δ=10.94(s,1H),9.38(d,J=0.9,1H),9.08(s,1H),7.93(dd,J=7.3,2.7,1H),7.90-7.85(m,1H),7.25(t,J=54.0,2H),7.18(dd,J=11.8,8.8,1H),6.04(s,2H),4.68-4.44(m,2H),4.40-4.30(m,1H),4.26(d,J=8.2,1H),3.79(dd,J=8.1,3.1,1H),3.30(s,1H),3.12(dd,J=13.5,3.6,1H),3.00(dd,J=13.5,3.8,1H),2.76(dt,J=7.9,3.7,1H).
13C NMR(126MHz,DMSO)δ160.90(s),156.22(d,J=244.2Hz),149.16(t,J=24.7Hz),148.46(s),146.91(s),143.69(s),140.35(t,J=4.4Hz),134.02(s),130.27(d,J=12.3Hz),122.46(d,J=4.2Hz),121.18(d,J=8.7Hz),116.26(d,J=25.0Hz),112.49(t,J=239.1Hz),83.53(d,J=169.8Hz),78.52(d,J=18.2Hz),77.55(s),65.61(s),35.59(s),22.93(s).
E.式V的氨基噻嗪的合成
(4aS,5S,7aS)-7a-(2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.向N-((4aS,5S,7aS)-7a-(2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺27(250.2g,589.5mmol)在甲醇(1.25L)中的溶液加入K2CO3(81.5g,590.0mmol)。將懸浮液加熱至65℃持續6小時。冷卻至環境溫度之后,在真空下蒸發溶劑。向所得殘余物加入1.0N NaOH水溶液(1.18L)和THF(502mL)。將多相的混合物加熱至45℃持續1小時。將混合物冷卻至環境溫度,加入EtOAc(1.38L)。將水層用EtOAc(0.75L)萃取。將有機物合并,用NaHCO3飽和水溶液(500mL)和NaCl飽和水溶液(500mL)洗滌。將有機物在真空下濃縮以得到棕色油狀標題化合物30(184.1g,91.6%收率,包含殘余溶劑)。
(4aS,5S,7aS)-7a-(2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,DMSO)δ7.49-7.42(m,1H),7.40-7.33(m,1H),7.26-7.15(m,2H),6.26(s,2H),4.77-4.54(m,1H),4.40(d,J=8.0Hz,1H).3.80(dd,J=7.9,2.3Hz,1H),3.24-3.17(m,1H),3.00(dd,J=13.9,3.2Hz,1H),2.85(dd,J=13.9,3.9Hz,1H);13C NMR(125MHz,DMSO)δ159.75(d,JCF=245.1Hz),149.51,131.31(d,JCF=3.9Hz),130.13(d,JCF=8.8Hz),128.08(d,JCF=10.4Hz),128.28(q,JCF=282.1Hz).124.87(d,JCF=3.0Hz),116.80(d,J=23.8Hz).78.77,76.80(q,JCF=30.8Hz),66.31,36.37,23.27.
HRMS計算C13H12F4N2O3S[M+H]+321.0685;實測321.0677.
(4aS,5R,7aS)-7a-(2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.向反應器裝入28(1.0Wt,1.0當量)、碳酸鉀(0.19Wt,0.50當量)和MeOH(4.0Wt,5.0V)。將反應混合物加熱至60-65℃。在60-65℃下繼續攪拌6h并關于28的完全消耗(目標>97%轉化)監測反應。在減壓下將反應混合物濃縮(T<40℃)以除去大部分的甲醇。加入水(1.5Wt,1.5V),隨后加入庚烷(1.1Wt,1.6V)和乙酸乙酯(0.36Wt,0.40V)的混合物。將混合物冷卻至0-5℃并劇烈攪拌1-2h。將混合物過濾并將反應器和濾餅用水(1.0Wt,1.0V)和預冷卻的(0-5℃)的庚烷(0.89Wt,1.3V)和乙酸乙酯(0.29Wt,0.32V)的混合物沖洗。將固體在真空中干燥(T<40℃)以得到31(0.61Wt,85%,淺棕色固體)。
(4aS,5R,7aS)-7a-(2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,CDCl3)δ7.41(td,J=8.2,1.6Hz,1H),7.28-7.20(m,1H),7.15-7.09(m,1H),7.03(ddd,J=12.4,8.1,0.7Hz,1H),4.81-4.42(s,2H),4.61(dd,J=8.7,0.5Hz,1H),4.40-4.27(m,1H),3.80(dd,J=8.7,2.2Hz,1H),3.07(dd,J=13.3,3.9Hz,1H),2.70(dd,J=13.3,3.9Hz,1H),2.56-2.47(m,1H),1.34(d,J=6.1Hz,3II);13C NMR(125MHz,CDCl3)δ160.03(d,JCF=247.0Hz),149.41,130.73(d,JCF=10.0Hz),129.80(d,JCF=4.1Hz),128.83(d,JCF=8.7Hz),124.02(d,JCF=3.4Hz),116.38(d,JCF=23.5Hz),78.54(d,JCF=4.8Hz),76.43,66.97(d,JCF=4.6Hz),44.50(d,JCF=3.7Hz),23.70,19.83.
(4aS,5R,7aS)-7a-(2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺(31)的可選的合成
向反應器裝入硫脲25(1Wt,1當量)、吡啶(2.9Wt,3.0V)和甲苯(2.6Wt,3.0V)。將所得混合物冷卻到-15℃以下。加入三氟甲基磺酸酐(0.800Wt,0.476V,1.10當量)同時維持溫度低于0℃。完成加入之后,在-5–0℃繼續攪拌1.5h并關于完全轉化(≥97.0%)監測反應。加入甲苯(5.2Wt,6.0V)和20wt%氯化銨水溶液(4.2Wt,4.0V)。將所得兩相混合物升溫至15–25℃,攪拌至少15min并且然后允許分開。將較低水層分離,并將上面的有機相除去并保存。將水層用甲苯(2.6Wt,3.0V)萃取。將合并的有機層用水(2.0Wt,2.0V)洗滌并在減壓下濃縮以得到稠的棕色油狀的粗制的28。加入2-丙醇(2.4Wt,3.0V)和3.0M氫氧化鈉水溶液(3.5Wt,3.0V),并將所得反應混合物加熱至80℃持續6h并關于完全轉化(>99.0%)進行監測。然后加入庚烷(1.4Wt,2.0V)和水(10Wt,10V),同時保持內部溫度高于60℃。將所得混合物冷卻至0–5℃并在這一溫度下維持至少2h,然后過濾。將反應器和濾餅用水(1.8Wt,1.8V)和2-丙醇(0.16Wt,0.20V)的混合物沖洗并且然后用庚烷(1.2Wt,1.8V)和乙酸乙酯(0.18Wt,0.20V)的混合物沖洗。將固體干燥以得到異硫脲31(0.57Wt,83%收率)。
N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(羥甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺.將甲酸(116mL)加到N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-((三苯甲基氧基)甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺29(33.30g,0.051mol,1.0當量)中,將混合物在rt下攪拌25min。滴加水(17mL)然后將反應攪拌另外30min。將所得懸浮液過濾,將濾液在真空下通過與甲苯(360mL)共沸蒸餾來干燥。加入甲醇(110mL),隨后加入三乙胺(22mL,0.15mol,3.0當量)并將溶液攪拌2小時。將溶劑在真空下除去并用甲苯(66mL)逐出。將殘余的油溶解在CH2Cl2(250ml)中并連續用HCl水溶液(1M,100mL)、NaHCO3飽和水溶液(67mL)和18%NaCl水溶液(67mL)洗滌。將有機相在減壓下濃縮。將甲苯(66mL)和THF(67mL)加到殘余物然后將溶液濾過硅藻土并在減壓下濃縮以得到標題化合物32(21g)。
1H NMR(500MHz,DMSO)δ8.11(dd,J=16.4,4.2Hz,2H),7.70-7.58(m,1H),7.57-7.44(m,3H),7.43-7.34(m,1H),7.34-7.26(m,1H),4.43(d,J=9.5Hz,1H),4.25-4.16(m,1H),4.10(d,J=9.2Hz,1H),3.59(d,J=4.5Hz,2H),3.30-3.21(m,1H),3.20-3.08(m,2H);13C NMR(125MHz,DMSO)δ169.08,150.95(dd,JCF=245.8,13.1Hz),148.22,148.09(dd,JCF=248.7,13.2Hz),133.46,129.11,128.99,128.62,125.43,124.70,118.16(d,JCF=16.9Hz),81.36,75.29,66.76,62.39,40.77,24.25.
HRMS計算C20H18F2N2O3S[M+H]+405.1084;實測405.1081.
N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺.在氮氣氣氛下將N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(羥甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺32(21.0g,0.519mol,1.0當量)溶解在干燥的THF(105mL)中并將溶液在0℃冷卻。加入N,N-二異丙基乙基胺(40.7mL,0.234mol,4.50當量)、三乙胺三氫氟化物(14.0mL,0.0857mol,1.65當量)和全氟丁烷磺酰基氟化物(22.4mL,0.125mol,2.40當量)同時維持溫度低于5℃。在0℃將反應攪拌3小時然后緩慢升溫至室溫并攪拌11小時。向反應混合物加入飽和NH4Cl(100mL),隨后加入2-甲氧基-2-甲基丙烷(100mL)。將有機相分離并用HCl水溶液(1.0M,100ml)洗滌,濃縮并再溶解在2-甲氧基-2-甲基丙烷(301ml)中。然后將有機相用HCl水溶液(1M,63mL)、飽和NaHCO3水溶液(100mL)洗滌。將最后的水層用2-甲氧基-2-甲基丙烷萃取。將合并的有機相用18%水溶液NaCl(63mL)洗滌并在減壓下濃縮以得到泡沫狀標題化合物33(19.40g)。
N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺:
1H NMR(500MHz,DMSO)δ8.02(s,2H),7.61-7.51(m,1H),7.51-7.38(m,3H),7.39-7.18(m,2H),4.72-4.49(m,2H),4.48-4.37(m,1H),4.41(d,J=9.1Hz,21H),3.04(s,2H);13C NMR(125MHz,DMSO)δ150.92(dd,JCF=245.5,13.3Hz),148.11(dd,JCF=248.2,13.4Hz),132.40,128.99,128.63,125.16,124.94,117.61,83.64(d,JCF=170.2Hz),79.51(d,JCF=18.4Hz),76.17,66.27,23.67.
HRMS計算C20H17F3N2O2S[M+H]+407.1041;實測407.1024.
(4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.在氮氣下,將N-((4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-基)苯甲酰胺33(19.40g,0.048mol,1.0當量)溶解在甲醇(97mL)中。加入1,8-二氮雜雙環[5.4.0]十一-7-烯(8.92mL,0.060mol,1.25當量),并且將溶液加熱至55-60℃。在8h之后,在減壓下濃縮反應混合物,并將殘余物通過硅膠快速柱色譜法純化(5%至100%EtOAc在庚烷中)以得到標題化合物34(9.01g)。
(4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,DMSO)δ7.38-7.28(m,1H),7.26-7.14(m,2H),6.12(s,2H),4.66-4.41(m,2H),4.37-4.27(m,2H),4.29(d,J=8.2Hz,1H),3.74(dd,J=8.2,2.6Hz,1H),3.30(s,1H),3.06-2.90(m,2H),2.79-2.71(m,1H);13C NMR(125MHz,DMSO)δ150.84(dd,J=244.8,14.0Hz),149.71,148.07(dd,JCF=247.8,13.4Hz),133.08(d,JCF=7.7Hz),125.24,124.55(dd,JCF=7.2,4.3Hz),116.58(d,JCF=17.2Hz),83.91·(d,JCF=170.0Hz),79.12(d,JCF=18.2Hz),77.96(d,JCF=4.9Hz),66.22,36.41(d,JCF=3.3Hz),23.40.
HRMS計算C13H13F3N2OS[M+H]+303.0779;實測303.0767.
G.式VI的硝基苯基噻嗪的合成
(4aS,5S,7aS)-7a-(2-氟-5-硝基苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺鹽酸鹽.向包含(4aS,5S,7aS)-7a-(2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺30(184.1g,574.8mmol)的冷卻的容器中分幾部分加入三氟乙酸(0.954kg)同時將溫度維持在低于20℃。將混合物冷卻至3.5℃并經20min加入硫酸(146mL,2.73mol)同時維持溫度低于5℃。經30min加入發煙硝酸(39.8mL,0.948mol),同時將溫度維持在低于10℃。在0-10℃1.5h后,將反應混合物通過轉移到冷卻至5℃的NaOH(575g,14.4mol)在水(4.6L)中的水溶液來緩慢地猝滅。在21℃將所得懸浮液攪拌1h。然后將懸浮液過濾并將固體用冷水(920mL)沖洗。將固體在真空下干燥直到恒重,然后溶解到乙醇(1.05L)中。將溶液加熱至35℃,并加入濃HCl(55.6mL,0.690mol)同時維持溫度低于40℃。然后將懸浮液冷卻至-5℃,保持1hr并過濾。將固體用冷乙醇(420mL)沖洗并干燥至恒重以獲得標題化合物35(185.0g,87.3%)。
(4aS,5S,7aS)-7a-(2-氟-5-硝基苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺鹽酸鹽:
1H NMR(500MHz,DMSO)δ11.80(s,2H),8.45-8.36(m,1H),8.31(dd,J=6.6,2.5Hz,1H),7.66(dd,J=11.1,9.3Hz,1H),4.96-4.72(m,1H),4.58(d,J=10.0Hz,1H),4.27(d,J=9.9Hz,1H),3.76-3.66(m,1H),3.39(dd,J=14.9,3.6Hz,1H),3.24(dd,J=14.3,4.6Hz,1H);13C NMR(125MHz,DMSO)δ168.34,163.33(d,JCF=257.8Hz),144.58,127.61(d,JCF=11.6Hz),125.84,124.10,119.28(d,JCF=26.5Hz),77.38(q,JCF=31.5Hz),75.99,65.88(d,JCF=4.8Hz),40.36,23.98.
HRMS計算C13H11F4N3O3S[M+H]+366.0536;實測366.0523.
(4aS,5R,7aS)-7a-(2-氟-5-硝基苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.向反應器(反應器1)裝入三氟乙酸(3.7Wt,2.5V)并冷卻至0-5℃。伴隨攪拌分幾部分加入31(1Wt,1當量),同時維持T低于20℃。將儲存罐(the holding vessel)和反應器用三氟乙酸(1.5Wt,1.0V)沖洗。將混合物冷卻到5℃以下,并且然后加入硫酸(1.3Wt,0.72V)同時維持T低于15℃。將所得溶液冷卻至0-5℃并且然后加入發煙硝酸(純度>90%,0.315Wt,0.210V,1.20當量)同時維持T低于25℃。在完全加入之后,繼續攪拌混合物0.5h同時冷卻(浴溫T.0-5℃)并監測反應關于31的完全消耗(目標>98%轉化)。將另一反應器(反應器2)裝入3.0MNaOH(26Wt,23V)并將混合物冷卻至3-7℃。將反應混合物轉移到第二反應器中同時維持T低于30℃。在加入過程中,形成白色沉淀。完成加入之后,在23-27℃下繼續攪拌。將第一反應器用水(2.0Wt,2.0V)沖洗,并將水洗物轉移到反應器2中。若需要,加入另外的3MNaOH使得pH>12。在23-27℃攪拌0.5-1h之后,將混合物過濾并將反應器和濾餅用水(4.0Wt,4.0V)沖洗。將濾餅在減壓下干燥(T<30℃)過夜以得到36(1.12Wt,96.1%收率)。
(4aS,5R,7aS)-7a-(2-氟-5-硝基苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,CDCl3)δ8.39(dd,J=6.9,2.9Hz,1H),8.20-8.14(m,1H),7.21(dd,J=10.8,9.0Hz,1H),4.55(dd,J=9.1,0.9Hz,1H),4.41-4.31(m,1H),3.80(dd,J=9.1,1.8Hz,1H),3.05(dd,J=13.5,3.8Hz,1H),2.74(dd,J=13.5,4.1Hz,1H),2.52-2.42(m,1H),1.37(d,J=6.1Hz,3H);13C NMR(1256MHz,CDCl3)δ163.56(d,JCF=258.9Hz),150.91,144.23,133.73(d,JCF=12.3Hz),126.05(d,JCF=6.2Hz),124.76(d,JCF=10.8Hz),117.53(d,JCF=26.3Hz),78.83(d,JCF=4.0Hz),76.96,66.91(d,JCF=4.8Hz),45.26(d,JCF=3.1Hz),23.88,19.63.
(4aS,5S,7aS)-7a-(2,3-二氟-5-硝基苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.將(4aS,5S,7aS)-7a-(2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺34(9.10g,0.03mol,1.0當量)溶解在三氟乙酸(36.4mL)中,并將溶液冷卻至0℃。加入硫酸(濃硫酸,12.0mL),隨后滴加發煙硝酸(6.9mL)同時維持溫度低于5℃。在0-5℃攪拌4h之后,將反應混合物緩慢加入到劇烈攪拌的NaOH水溶液(43.3g在273mL水中)同時維持溫度低于20℃。將混合物用CH2Cl2(1x 94ml,and 2x 64mL)萃取,并將合并的有機相用NaCl飽和水溶液(46mL)洗滌。將硅藻土(15.0g)加到有機物中,并將混合物過濾,用CH2Cl2(40mL)沖洗。將溶劑蒸發以得到標題化合物37(8.3g),其不經純化直接用于后面的步驟。
(4aS,5S,7aS)-7a-(2,3-二氟-5-硝基苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,DMSO)δ8.41-8.30(m,1H),8.22-8.13(m,1H),6.36(s,2H),4.70-4.46(m,2H),4.43-4.31(m,1H),4.35(d,J=8.7H2,1H),3.69(dd,J=8.5,1.6Hz,1H),3.04-2.91(m,2H),2.86-2.76(m,1H);13C NMR(125MHz,DMSO)δ152.36(dd,JCF=246.8,12.3Hz),151.13,150.32(dd,JCF=238.2,12.2Hz),143.25(dd,JCF=7.9,2.5Hz),134.70(d,JCF=9.5Hz),121.28,113.02(d,JCF=22.3Hz),83.74(d,JCF=170.1Hz),79.44(d,JCF=18.3Hz),78.02(d,J=3.9Hz),36.96,23.28.
HRMS計算C13H12F3N3O3S[M+H]+348.0630;實測348.0614.
H.式VII的二氨基噻嗪的合成
(4aS,5S,7aS)-7a-(5-氨基-2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.在氮氣氣氛下將乙醇(0.975L)加到鐵粉(62.5g,1.12mol)中。在環境溫度下加入濃HCl(9.03mL)并將懸浮液加熱至65℃持續1.5h。然后將懸浮液冷卻至50℃,并加入NH4Cl飽和水溶液(299g)。允許反應混合物的溫度到達50℃,并分幾部分加入(4aS,5S,7aS)-7a-(2-氟-5-硝基苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺鹽酸鹽35(75.0g,187.0mol)同時維持溫度低于68℃。30min之后,加入乙醇(0.45L),然后將反應混合物經1h冷卻至20-25℃。將懸浮液攪拌2h并濾過硅藻土(75g),用乙醇(0.972L)沖洗。將溶液在真空下濃縮至棕色固體。加入水(0.9L)隨后加入3.0N NaOH(0.187L,560mmol)同時維持溫度低于35℃。將所得懸浮液在20-25℃攪拌1h。將懸浮液過濾,并將固體用冷水(0.38L)沖洗。在40-45℃將固體在真空下干燥經24h以獲得標題化合物38(57.7g,95.5%)。
(4aS,5S,7aS)-7a-(5-氨基-2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,DMSO)δ6.81(dd,J=12.5,8.6Hz,1H),6.62(dd,J=7.0,2.9Hz,1H),6.50-6.42(m,1H),6.16(s,2H).4.96(s,2H),4.72-4.54(m,1H),4.35(d,J=7.8Hz,1H),3.74(dd,J=7.8,2.5Hz,1H),3.18-3.08(m,1H),3.01(dd,J=13.9,3.0Hz,1H).2.84(dd,J=13.8,3.8Hz,1H);13C NMR(125MHz,DMSO)δ156.20(d,JCF=243.0Hz),148.73,145.49.127.86(d,JCF=11.0Hz),116.79(d,.JCF=24.8Hz),116.10(d,JCF=3.3Hz),114.10(d,JCF=8.0Hz),78.89,76.57(q,JCF=31.0Hz),66.35,36.35,23.11.
HRMS計算C13H13F4N3OS[M+H]+336.0794;實測336.0789.
(4aS,5R,7aS)-7a-(5-氨基-2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.在18-22℃向粉末鐵(6當量)在乙醇(8.0V)中的懸浮液中加入濃HCl(37Wt%,0.60當量)。將所得混合物升溫至65-70℃并攪拌2h。冷卻至50-55℃并加入飽和NH4Cl溶液(4.0wt)。在另一反應器中,將36(1.0Wt,1.0當量)溶解在乙醇(5.0V)和濃HCl(1.0當量)的混合物中。將所得溶液加到鐵漿液反應器中同時將T維持在低于65℃,用乙醇(1.0V)沖洗。將反應混合物在55-65℃攪拌直到36的完全消耗。將反應混合物用6.0V乙醇稀釋并冷卻至15-20℃。攪拌1-2h后,將混合物濾過硅藻土墊(1.0Wt),用15V乙醇沖洗。在真空中濃縮得到橙色固體殘余物,將其溶解在水(8V)中。產生了一些橙色顆粒并通過拋光過濾(polishfiltration)除去。將2V的水用于沖洗。在18-25℃經45min加入3MNaOH(3V)。產物作為白色固體沉淀下來。在18-22℃攪拌1-2h后,將混合物過濾并將濾餅用水沖洗三次(2V每次)。在45℃在真空烘箱中干燥過夜得到灰白色固體狀39,分離收率92%。
(4aS,5R,7aS)-7a-(5-氨基-2-氟苯基)-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,DMSO)δ6.76(dd,J=12.3,8.6Hz,1H),6.59(dd,J=7.1,2.8Hz,1H),6.45-6.36(m,1H),5.91(s,2H),4.87(s,2H),4.29(d,J=8.2Hz,1H),4.20-4.11(m,1H),3.61(dd,J=8.0,2.7Hz,1H),2.93(dd,J=13.3,3.7Hz,1H),2.82(dd,J=13.3,3.7Hz,1H),2.37-2.28(m,1H),1.23(d,J=6.1Hz,3H);13C NMR(126MHz,DMSO)δ151.95(d,JCF=232.6Hz),148.19,145.02,131.37(d,JCF=11.3Hz),116.56(d,JCF=24.6Hz),115.42(d,JCF=3.4Hz),113.38(d,JCF=7.9Hz),78.56(d,JCF=5.4Hz),76.00,66.55(d,JCF=4.9Hz),43.73(d,JCF=3.6Hz),22.96,20.32.
(4aS,5S,7aS)-7a-(5-氨基-2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺.向鐵(8.03g,0.144mol,6.0當量)加入乙醇(66.56mL),隨后加入濃HCl(62%,1.2mL,0.014mol,0.60當量)。將混合物加熱至65℃持續2h,并且然后加入飽和NH4Cl(33%,33.3mL)并將反應溫度維持在55℃。將(4aS,5S,7aS)-7a-(2,3-二氟-5-硝基苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺37(8.32g,0.024mol,1.0當量)和濃HCl(1.93mL,0.024mol,1.0當量)在乙醇(41.6mL)中的溶液加到鐵懸浮液。在55℃將反應攪拌30min,然后加入乙醇(50mL)并允許懸浮液冷卻至20℃,濾過硅藻土(8.3g)并用乙醇(125mL)沖洗。將濾液在減壓下濃縮并加入水(66mL),隨后加入3.0M NaOH水溶液(24mL,0.0179mol,3.0當量)。將所得混合物用CH2Cl2(2x 83mL)萃取。將有機相合并并濾過硅藻土并在減壓下濃縮以得到標題化合物40(7.60g)。
(4aS,5S,7aS)-7a-(5-氨基-2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺:
1H NMR(500MHz,DMSO)δ6.42-6.35(m,2H),6.02(brs,2H),4.66-4.41(m,2H),4.37-4.25(m,1H),4.21(d,J=8.0Hz,1H),3.69(dd,J=7.8,2.2Hz,1H),2.97(qd,J=13.5,3.4Hz,3H),2.74-2.64(m,1H);13C NMR(125MHz,DMSO)δ151.08(dd,JCF=240.1,14.8Hz),148.88,145.40(d,JCF=10.7Hz),139,33(dd,JCF=233.5,13.8Hz),132.53(d,JCF=8.1Hz),109.90,100.70(d,JCF=20.0Hz),83.99(d,JCF=169.9Hz),78.79(d,JCF=18.3Hz),77.99(d,JCF=5.5Hz),66.22,36.24,23.14.
HRMS計算C13H14F3N3OS[M+H]+318.0888;實測318.0874.
I.式IX的化合物的合成
N-(3-((4aS,5S,7aS)-2-氨基-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-甲氧基吡嗪-2-甲酰胺.將5-甲氧基吡嗪-2-羧酸(26.29g,0.17mol)在N,N’-二甲基咪唑啉-2-酮(160mL)中的懸浮液在環境溫度下攪拌15min,然后冷卻至2.2℃。加入亞硫酰氯(14.7mL,0.202mol)同時維持溫度低于5℃。將所得懸浮液在0-10℃攪拌2小時同時其轉變成澄清的溶液。在另一容器中,將(4aS,5S,7aS)-7a-(5-氨基-2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺38(52.0g,0.155mol)溶解到N,N’-二甲基咪唑啉-2-酮(160mL)中。將所得溶液加到酰氯溶液中同時維持溫度低于10℃。將反應混合物攪拌30min。加入水(780mL)同時維持溫度低于30℃。將所得混合物攪拌30min.,并且然后加入EtOAc(780mL)。向這一混合物加入50%NaOH水溶液(84.8g)直到水層pH達到11。將水層用EtOAc(260mL)萃取。將有機層合并,用飽和NaCl水溶液(260mL)和水(260mL)洗滌。將有機物濾過硅藻土墊(26g)并用EtOAc(260mL)沖洗。將有機物在真空下濃縮以得到固體。向該固體加入1-丙醇(728mL),并將懸浮液加熱至75℃直到形成澄清溶液。將該溶液冷卻至-10℃并保持1小時。將固體過濾,用冷的1-丙醇(104mL)沖洗并在真空下干燥(35℃)直到恒重,得到標題化合物41(62.1g,84.9%)。
N-(3-((4aS,5S,7aS)-2-氨基-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-甲氧基吡嗪-2-甲酰胺:
1H NMR(500MHz,DMSO)δ10.56(s,2H),8.88(d,J=1.2Hz,1H),8.39(d,J=1.2Hz,1H),7.95-7.83(m,2H),7.18(dd,J=12.0,8.8Hz,1H),6.25(s,2H),4.76-4.60(m,1H),4.36(d,J=8.1Hz,1H),4.01(s,3H),3.88(dd,J=7.9,2.3Hz,1H),3.23-3.11(m,2H),2.91(dd,J=13.8,3.6Hz,1H);13C NMR(125MHz,DMSO)δ162.11,161.93,156.13(d,JCF=242.9Hz),149.38,142.01,138.35,135.09,133.98,128.53(d,JCF=11.6Hz),126,06(q,JCF=282.0Hz),123.32,121.93(d,JCF=8.6Hz),116.76(d,JCF=25.1Hz),78.86(d,JCF=6.9Hz),76.94(q,JCF=30.5Hz),66.37,54.75,36.44,23.53.
HRMS計算C19H17F4N5O3S[M+H]+472.1066;實測472.1052.
特定的旋光性[α]D+110.5(c 0.584,MeOH)
特定的旋光性參數:
裝置:
試劑:
甲醇:HPLC等級,Baker(目錄號9093-03)或等同物
儀器參數:
N-(3-((4aS,5R,7aS)-2-氨基-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-(二氟甲基)吡嗪-2-甲酰胺.將5-(二氟甲基)吡嗪-2-羧酸42(0.68wt,1.1當量)加入反應器中并且若需要,通過與甲苯共沸蒸餾干燥。加入DMI(2.0V)并將混合物冷卻至0-5℃。加入亞硫酰氯(0.337V,1.30當量)同時保持內部溫度低于10℃。將所得溶液在4-10℃攪拌直到通過HPLC檢測轉化>95.0%。將混合物冷卻到0℃并加入39(1.0Wt,1.0當量)在DMI(2.5V)中的溶液同時保持內部溫度低于5℃。將容器用DMI(0.50V)沖洗并攪拌反應混合物直到轉化>99%。加入水(12V)并將所得混合物在15-20℃攪拌0.5h。加入EtOAc(15V)和然后50%NaOH水溶液(1.5Wt,5.3當量)同時維持T低于30℃。監測pH以保證其高于10。將水相分離并用EtOAc(5.0V)萃取。將有機層合并并用鹽水(5.0V)洗滌并用水洗滌兩次(5.0V每次)。將混合物濾過硅藻土墊(0.50Wt),用EtOAc(3.0V)沖洗。在40-50℃在減壓下將濾液濃縮。加入1-丙醇(15V)并將混合物升溫至90-100℃同時攪拌直到獲得澄清溶液。將混合物經2h冷卻至0-5℃并攪拌1h。將混合物過濾,用冷的1-丙醇沖洗兩次(2.5V每次)。在45℃在真空烘箱中干燥過夜,得到灰白色固體狀標題化合物43,分離收率81%。
5-(二氟甲基)吡嗪-2-羧酸:
1H NMR(500MHz,DMSO)δ4.72(s,1H),4.57(s,1H),4.32(s,1H),2.06(t,J=54.2Hz,1H);13C NMR(125MHz,DMSO)δ164.34,151.67(t,JCF=26.8Hz),145.45,143.29,141.04(t,JCF=3.4Hz),112.59(t,JCF=242.1Hz).
N-(3-((4aS,5R,7aS)-2-氨基-5-甲基-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-(二氟甲基)吡嗪-2-甲酰胺:
1H NMR(500MHz,CD3OD)δ9.38(s,1H),9.00(s,1H),7.89-7.77(m,2H),7.14(dd,J=11.9,8.8Hz,1H),6.94(t,J=54.3Hz,1H),4.80(s,2H),4.58(d,J=8.9Hz,1H),4.37-4.28(m,1H),3.81(dd,J=8.8,2.4Hz,1H),3.14(dd,J=13.5,4.0Hz,1H),2.88(dd,J=13.5,4.1Hz,1H),2.61-2.54(m,1H),1.33(d,J=6.1Hz,3H);13C NMR(125MHz,CD3OD)δ161.11,156.94(d,JCF-244.9Hz),154.91,150.21(t,JCF=25.8Hz),146.53,143.30,140.02(t,JCF=4.0Hz),133.73(d,JCF=2.6Hz),130.70(d,JCF=11.6Hz),122.05(d,JCF=3.9Hz),121.48(d,JCF=8.8Hz),116.39(d,JCF=25.3Hz),112.96(t,JcF=239.8Hz),77.56(d,JCF=5.3Hz),76.79,66.49(d,JCF=4.8Hz),45.16,45.13,22.60,18.58.
N-(3-((4aS,5S,7aS)-2-氨基-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4,5-二氟苯基)-5-甲氧基吡嗪-2-甲酰胺.將5-甲氧基吡嗪-2-羧酸(4.01g,0.026mol,1.10當量)在N,N’-二甲基咪唑啉-2-酮(22.5mL)中的懸浮液在環境溫度下攪拌15min,然后冷卻至0℃。加入亞硫酰氯(2.24mL,0.031mol,1.3當量)同時維持溫度低于10℃。將所得懸浮液在0-10℃攪拌2小時同時其轉變成澄清溶液。在另一容器中,將(4aS,5S,7aS)-7a-(5-氨基-2,3-二氟苯基)-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺40(7.60g,0.024mol,1.0當量)溶解到N,N’-二甲基咪唑啉-2-酮(22.5mL)中。將所得溶液加到酰氯溶液同時維持溫度低于10℃。將反應混合物攪拌30min。加入水(112mL)同時維持溫度低于30℃。將所得混合物攪拌30min.,并且然后加入EtOAc(112mL)。向這一混合物加入50%NaOH水溶液(10.0g)直到水層pH達到11。將水層用EtOAc(75mL)萃取。將有機物合并,用NaCl飽和水溶液(38mL)和水(38mL)洗滌。將有機物濾過硅膠墊(15g)并用EtOAc(37.5mL)沖洗。將有機物在真空下濃縮以得到固體。向該固體加入1-丙醇(112mL),并將懸浮液加熱至100℃。將混合物冷卻至-10℃并保持1小時。將固體過濾,用冷的1-丙醇(15mL)沖洗并在真空下干燥(35℃)直到恒重以得到標題化合物44(8.18g)。
N-(3-((4aS,5S,7aS)-2-氨基-5-(氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4,5-二氟苯基)-5-甲氧基吡嗪-2-甲酰胺:
1H NMR(500MHz,DMSO)δ10.73(s,1H),8.88(d,J=0.8Hz,1H),8.39(d,J=0.9Hz,1H),8.11-7.87(m,1H),7.73(d,J=5.0Hz,1H),6.07(s,2H),4.68-4.44(m,2H),4.41-4.28(m,1H),4.23(d,J=8.3Hz,1H),4.01(s,3H),3.82(dd,J=8.1,2.4Hz,1H),3.18(dd,J=13.5,3.5Hz,1H),3.01(dd,J-13.5,3.8Hz,1H),2.77-2.69(m,1II);13C NMR(125MHz,DMSO)δ162.24,162.20,150.05(dd,JCF=242.1,14.4Hz),149.37,144.42(dd,JCF=245.5,13.7Hz),142.18,138.09,134.73(dd,JCF=10.3,2.7Hz),134.03,133.22(d,JCF=9.1Hz),116.6,108.42(d,JCF=22.4Hz),83.98(d,JCF=169.9Hz),79.21(d,JCF=18.2Hz),77.96(d,JCF=5.2Hz),66.26,54.78,36.23,23.64..
HRMS計算C19H18F3N5O3S[M+H]+454.1161;實測454.1149.
特定的旋光性[α]D+115.3(c 0.584,MeOH)
特有的旋光性參數:
裝置:
試劑:
甲醇:HPLC等級,Baker(目錄號9093-03)或等同物
儀器參數:
N-(3-((4aS,5S,7aS)-2-氨基-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-(氟甲基)吡嗪-2-甲酰胺.將5-(氟甲基)吡嗪-2-羧酸(32.6g,1.05當量)和(4aS,5S,7aS)-7a-(5-氨基-2-氟苯基)-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-2-胺(70.0g,1.0當量)加入到反應器并將EtOAc(630mL)加到混合物以得到懸浮液。在環境溫度下加入(146g,1.10當量,50wt%于EtOAc中)(Archimica,Germany)的溶液同時控制內部溫度低于30℃。將反應混合物在40-45℃攪拌>3小時并通過HPLC監測。將反應混合物冷卻至15-20℃并加入水(140mL)。10-15分鐘之后充入28%氫氧化銨(175mL)同時控制溫度低于30℃。加入EtOAc(245mL)并將反應混合物在環境溫度下攪拌30分鐘。分離水層并用EtOAc(490mL)萃回。將有機相合并并用15%NaCl水溶液(140mL)和水(140mL)洗滌。將有機層濾過硅藻土(1.0Wt)并用EtOAc(140mL)沖洗。將溶液在真空下濃縮得到淺褐色固體(定量的粗制收率),其從1-丙醇再結晶得到N-(3-((4aS,5S,7aS)-2-氨基-5-(三氟甲基)-4a,5,7,7a-四氫-4H-呋喃并[3,4-d][1,3]噻嗪-7a-基)-4-氟苯基)-5-(氟甲基)吡嗪-2-甲酰胺,白色固體狀(70.0g)。
1H NMR(500MHz,DMSO)δ10.89(s,1H),9.30(s,1H),8.89(s,1H),7.95(dd,J=7.3,2.7Hz,1H),7.94-7.89(m,1H),7.21(dd,J=12.0,8.8Hz,1H),6.22(s,2H),5.71(d,J=46.3Hz,2H),4.77-4.61(m,1H),4.37(d,J=8.1Hz,1H),3.87(dd,J=8.0,2.7Hz,1H),3.20(dt,J=7.0,3.5Hz,1H),3.15(dd,J=13.9,3.1Hz,1H),2.91(dd,J=13.8,3.8Hz,1H).
13C NMR(126MHz,DMSO)δ161.32(s),155.82(d,J=243.4Hz),153.71(d,J=18.7Hz),148.77(s),144.71(d,J=1.9Hz),143.30(s),141.01(d,J=5.6Hz),134.36(d,J=2.0Hz),128.20(d,J=12.1Hz),125.57(q,J=283.0Hz),123.12(d,J=3.6Hz),121.64(d,J=8.6Hz),116.35(d,J=25.2Hz),82.55(d,J=165.8Hz),78.37(s),76.44(q,J=30.6Hz),65.89(d,J=5.3Hz),35.89(s),23.01(s).
- 還沒有人留言評論。精彩留言會獲得點贊!
- N-烯基苯并三唑類氮氧衍生物及其制備方法和應用
- 含有噻二唑衍生物、納米銅和石墨烯的添加劑,添加劑制備方法,含該有該添加劑的潤滑油的制作方法
- 一種具有抗菌活性的手性螺1,2,3-噻二唑衍生物及其合成方法和應用
- 含有噻二唑衍生物和納米銅的潤滑油添加劑,添加劑的制備方法,及含有該添加劑的潤滑油的制作方法
- 作為免疫調節劑的1,3,4-*二唑和1,3,4-噻二唑衍生物的制作方法
- 含有噻二唑衍生物和石墨烯的潤滑油添加劑,添加劑的制備方法,及含有該添加劑的潤滑油的制作方法
- 一類1,3,4-噻二唑衍生物、制備方法及應用
- 一類1,3,4-噻二唑衍生物、合成方法及其應用
- 一種雙磷酸酯唑類衍生物的制備方法
- 一種用于rv減速器的鋰基潤滑脂及其制備方法