一種噻螨酮及其制備方法與流程
本發明涉及殺蟲劑領域,具體涉及一種噻螨酮及其制備方法。
背景技術:
國家對農藥的發展越來越重視,在環保方面,降低農藥對社會和環境的風險成為重中之重。隨著種植結構的變化,農藥三大系列之一的殺蟲劑(殺螨劑)近年來,銷量獲得了快速增長,世界各國也加大了農藥技術開發力度,新技術、新產品不斷涌現。
噻螨酮,化學名稱為:(4RS,5RS)-5-(4-氯苯基)-N-環己基-4-甲基-2-氧代-1,3-噻唑烷-3-羧酰胺。其化學結構式為:
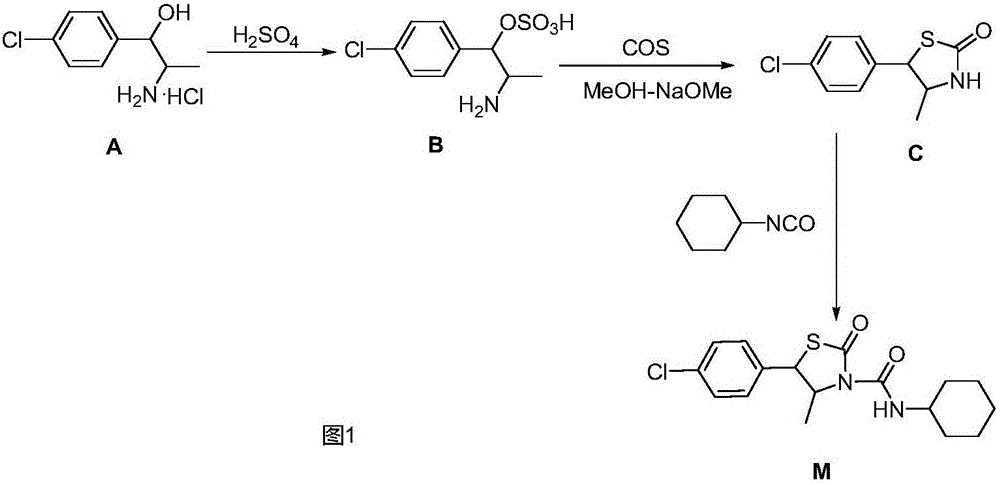
該產品自1985年日本曹達公司開發,其合成路線如圖1所示:以赤-2-氨基-1-(對氯苯基)丙醇鹽酸鹽(A)為起始原料,經硫酸縮合脫水而生成赤-[2-氨基-1-(對氯苯基)]丙基硫酸酯(赤-硫酸酯)(B),然后與氧硫化碳閉環生成取代的反式噻唑烷酮(C),后者再與異氰酸環己酯加成而得到最終產物噻螨酮(M)。該合成工藝所用的起始原料需經多步化學反應合成;其關鍵性中間體反式噻唑烷酮(C)由中間體赤-硫酸酯(B)與氧硫化碳進行關環反應制備,由于氧硫化碳需通過一氧化碳同硫磺蒸汽作用而得到,一氧化碳為劇毒氣體,對工藝設備和操作均有嚴格要求,而且污染環境。因此,該合成工藝存在有工藝流程復雜、合成難度大、操作不安全、污染環境、成本高等缺點。
隨后有人報道對此工藝進行改進,制備方法如圖2所示:采用二硫化碳進行環化,環化后再用雙氧水進行氧化得到反式噻唑烷酮(C),后者再與異氰酸環己酯加成而得到最終產物噻螨酮(M)。但改進后依然需要對原料A預先硫酸酯化,這一步反應往往會造成大量的酸性廢水。
另外有報道,制備方法如圖3所示:以赤-2-氨基-1-(對氯苯基)丙醇鹽酸鹽(A)為原料,在堿存在下與二硫化碳及芐氯反應生成化合物(D),反應產物D在二氯甲烷中與氯化亞砜一起進行化合反應生成(E),然后,將生成物E在鹽酸水溶液中加熱進行回流開環成酮反應生成(F),再經環合得取代的反式噻唑烷酮(C),最后再與異氰酸環己酯加成而得到最終產物噻螨酮(M)。此路線反應步驟長、操作繁瑣、污染較大、收率也低,不適合工業化生產。
因此,需要一種噻螨酮的綠色化生產工藝路線,適合工業化生產,能夠簡化工藝流程,提高反應收率,降低合成難度,減少副產物的形成,降低制備過程中的環境污染問題,并且降低生產成本。
技術實現要素:
本發明針對上述問題,提供一種噻螨酮及其制備方法,適合工業化生產,能夠簡化工藝流程,提高反應收率,降低合成難度,減少副產物的形成,降低制備過程中的環境污染問題,并且降低生產成本。
本發明解決上述問題所采用的技術方案是:一種噻螨酮的制備方法,以對氯苯丙酮為起始原料,和亞硝酸正丁酯以70:47的質量比反應生成酮肟物,然后進行氫化還原為2-氨基-1-對氯苯基丙醇鹽酸鹽。
進一步地,2-氨基-1-對氯苯基丙醇鹽酸鹽直接與二硫化碳反應合成反式-5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮,再經雙氧水氧化生成5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮,5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮再同異氰酸環己酯進行加成反應而得到最終產物噻螨酮。
進一步地,酮肟物的制備過程具體為:向反應釜中投入350份對氯苯丙酮、700份石油醚,降溫到10℃~20℃,再加入235份亞硝酸正丁酯,攪拌5h~10h,再降溫至0℃,保溫攪拌0.5h~1.5h,經放料、過濾、石油醚洗滌,烘干得到固體酮肟物。
進一步地,2-氨基-1-對氯苯基丙醇鹽酸鹽的制備過程具體為:在反應釜中投入180份制得的酮肟物、480份甲醇,加入催化劑A 1份~2份,溶解0.5h,氮氣置換2次~4次,再氫氣置換2次~4次,升溫至35℃~40℃,通入氫氣,控制氫氣壓力為0.03Mpa~0.08Mpa,直到加不進氫氣為止,通氫氣完畢,趁熱過濾除去催化劑A,得到醇胺物的甲醇溶液,用鹽酸調PH值到6~6.5,減壓脫去溶劑得到醇胺物粗品,冷卻至40℃~50℃,加入500份乙醇,升溫回流1h~3h,冷卻到0℃~5℃后,保溫1h~3h,所得產物經離心、烘干,得2-氨基-1-對氯苯基丙醇鹽酸鹽。
更進一步地,催化劑A包括:鉑、鈀、鎳中的任一種。
更進一步地,噻螨酮的制備方法,包括以下步驟:
步驟S1,向反應釜中投入350份對氯苯丙酮、700份石油醚,降溫到10℃~20℃,再加入235份亞硝酸正丁酯,攪拌5h~10h,再降溫至0℃,保溫攪拌0.5h~1.5h,經放料、過濾、石油醚洗滌,烘干得到固體酮肟物;
步驟S2,在反應釜中投入180份制得的酮肟物、480份甲醇,加入催化劑A1份~2份,溶解0.5h,氮氣置換2次~4次,再氫氣置換2次~4次,升溫至35℃~40℃,通入氫氣,控制氫氣壓力為0.03Mpa~0.08Mpa,直到加不進氫氣為止,通氫氣完畢,趁熱過濾除去催化劑A,得到醇胺物的甲醇溶液,用鹽酸調PH值到6~6.5,減壓脫去溶劑得到醇胺物粗品,冷卻至40℃~50℃,加入500份乙醇,升溫回流1h~3h,冷卻到0℃~5℃后,保溫1h~3h,所得產物經離心、烘干,得2-氨基-1-對氯苯基丙醇鹽酸鹽;
步驟S3,在反應釜中投入2-氨基-1-對氯苯基丙醇鹽酸鹽180份,8%氫氧化鉀溶液700份、催化劑B1份~3份,攪拌均勻后,再加入二硫化碳77份,然后在80℃~100℃攪拌反應12h~14h,保溫結束后降溫到常溫,然后反應釜中抽入500份石油醚,攪拌0.5h~1.5h,靜置0.5h~2h,分層,有機層去脫溶,脫溶結束后,加入甲醇500份升溫回流2h~4h,降溫到0℃~5℃析出晶體后,再保溫1h~3h,離心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步驟S4,在反應釜中抽入甲醇800份,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮190份,催化劑C 2份~4份,開啟攪拌,然后在25℃加入300份的28%甲醇鈉,加畢后,攪拌半小時,然后在25℃以下滴加160份30%雙氧水,滴加完畢,在15℃~25℃保溫4h~8h,保溫結束后,轉入脫溶釜,減壓脫去甲醇,降溫到室溫,抽入甲苯700份,水500份,攪拌半小時,靜置半小時,分去水層,減壓脫去甲苯,降溫到室溫,加入500份甲醇,升溫回流1h~3h,降溫到0℃~5℃保溫2h~4h,抽濾得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步驟S5,在反應釜中,加入700份甲苯、200份5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和10份催化劑D,調節反應釜溫度為35℃~40℃,開始滴加125份異氰酸環已酯,時間控制在1.5h~2.5h,滴完后,保溫反應4h~8h,取樣中控,合格后,加入400份水,控制溫度為35℃~40℃,繼續攪拌1h。然后將反應混合物抽濾,濾液分去水層,油層用400份水洗一次,分去水層,抽入脫溶釜減壓蒸出甲苯,冷卻到50℃~60℃,加入520份乙醇,升溫回流2h~4h,冷卻到5℃~10℃保溫1h~3h,產物經離心、烘干,得到噻螨酮原藥。
更進一步地,步驟S3中,催化劑B為:甲醇鈉或氫氧化鈉。
更進一步地,步驟S4中,催化劑C包括:過氧化鈉或甲醇鈉。
更進一步地,步驟S5中,催化劑D包括:二氯甲烷、四氫呋喃、三乙胺、二月桂酸二丁基錫中的任一種。
本發明的另一發明目的,在于提供一種噻螨酮,根據上述制備方法制備而成。
本發明的優點是:
1.本發明用綠色化合成工藝開發了噻螨酮農藥,該農藥具有廣譜、低毒、高效、低劑量等優點,有強烈的殺卵、幼、若螨作用,和其他殺螨劑相比,殺卵的效果更為顯著,對植物表皮具有良好的滲透性,每畝單位用量較低僅需有效成分3g~5g,且藥效穩定,持效期長,不受溫度影響,廣泛用于柑橘、蘋果、山楂、棉花、蔬菜等農作物上防止害螨,對人體、環境較為安全,是更新換代的理想產品,具有良好的市場發展前景;
2.本發明在其合成反應的全過程中,所用的原材料絕大部分轉化到最終產物,使用溶劑予以回收循環利用,提高了原子利用率與原子經濟性,減少環境污染;
3.本發明采用一定壓力條件下的二硫化碳成環技術,替代先硫酸酯化再成環的技術和有毒有污染的硫氧化碳成環工藝,大大降低了酸性廢水的排放;
4.本發明采用高效選擇性催化劑,以提高反應收率、減少副產物的形成并降低環境污染;
5.本發明采用石油醚、乙醇等低毒溶劑替代二甲基甲酰胺、二甲亞砜高毒溶劑,以最大限度地降低環境污染。
附圖說明
構成本說明書的一部分、用于進一步理解本發明的附圖示出了本發明的實施方案,并與說明書一起用來說明本發明的制備流程。在附圖中:
圖1是現有技術中日本曹達公司制備噻螨酮的合成技術路線;
圖2是現有技術中一種制備噻螨酮的合成技術路線;
圖3是現有技術中另一種制備噻螨酮的合成技術路線;
圖4是本發明中噻螨酮原藥的制備工藝流程圖;
圖5是本發明中噻螨酮原藥的紅外光譜譜圖;
圖6是本發明中噻螨酮原藥的紫外吸收光譜圖;
圖7是本發明中噻螨酮原藥的核磁共振吸收圖譜;
圖8是本發明中噻螨酮原藥的有效成分質譜圖。
具體實施方式
以下結合附圖對本發明的實施例進行詳細說明,但是本發明可以由權利要求限定和覆蓋的多種不同方式實施。
實施例1
一種噻螨酮的制備方法,包括以下步驟:
步驟S1,酮肟化反應
反應方程式為:
原料中:對氯苯丙酮,純度99%;亞硝酸正丁酯,純度99%;石油醚,溫度90℃~120℃;
操作:向3000升反應釜中投入350Kg對氯苯丙酮、700Kg石油醚,降溫到10℃,再加入235Kg亞硝酸正丁酯,攪拌5h,再降溫至0℃,保溫攪拌0.5h,經放料、過濾、石油醚洗滌,烘干得到固體酮肟物;
步驟S2,醇胺化反應
反應方程式為:
原料中:酮肟物,純度99%;甲醇,純度99.5%;鹽酸,純度30%;乙醇,純度99.5%;
操作:在2000升反應釜中投入180Kg制得的酮肟物、480Kg甲醇,加入催化劑鉑1Kg,溶解0.5h,氮氣置換2次,再氫氣置換2次,升溫至35℃,通入氫氣,控制氫氣壓力為0.03Mpa,直到加不進氫氣為止,通氫氣完畢,趁熱過濾除去催化劑鉑,得到醇胺物的甲醇溶液,用鹽酸調PH值到6,減壓脫去溶劑得到醇胺物粗品,冷卻至40℃,加入500Kg乙醇,升溫回流1h,冷卻到0℃后,保溫1h,所得產物經離心、烘干,得2-氨基-1-對氯苯基丙醇鹽酸鹽;
步驟S3,環合反應
反應方程式為:
原料中:氫氧化鉀,質量百分數8%;
操作:在2000升反應釜中投入2-氨基-1-對氯苯基丙醇鹽酸鹽180Kg,8%氫氧化鉀溶液700Kg、甲醇鈉1Kg,攪拌均勻后,再加入二硫化碳77Kg份,然后在80℃攪拌反應12h,保溫結束后降溫到常溫,然后反應釜中抽入500Kg石油醚,攪拌0.5h,靜置0.5h,分層,有機層去脫溶,脫溶結束后,加入甲醇500Kg升溫回流2h,降溫到0℃析出晶體后,再保溫1h,離心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步驟S4,氧化反應
反應方程式為:
原料中:甲醇鈉的質量分數為28%;雙氧水的質量分數為30%;
操作:在3000升反應釜中抽入甲醇800Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮190Kg,過氧化鈉2Kg,開啟攪拌,然后在25℃加入300Kg的甲醇鈉,加畢后,攪拌半小時,然后在25℃以下滴加160Kg雙氧水,滴加完畢,在15℃保溫4h,保溫結束后,轉入脫溶釜,減壓脫去甲醇,降溫到室溫,抽入甲苯700Kg,水500Kg,攪拌半小時,靜置半小時,分去水層,減壓脫去甲苯,降溫到室溫,加入500Kg甲醇,升溫回流1h,降溫到0℃保溫2h,抽濾得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步驟S5,加成反應
反應方程式為:
原料中:5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮,純度97%;異氰酸環已酯,純度為:99%;
操作:在2000升反應釜中,加入700Kg甲苯、200Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和10Kg二氯甲烷,調節反應釜溫度為35℃,開始滴加125Kg異氰酸環已酯,時間控制在1.5h,滴完后,保溫反應4h,取樣中控,合格后,加入400Kg水,控制溫度為35℃,繼續攪拌1h。然后將反應混合物抽濾,濾液分去水層,油層用400Kg水洗一次,分去水層,抽入脫溶釜減壓蒸出甲苯,冷卻到50℃,加入520Kg乙醇,升溫回流2h,冷卻到5℃保溫1h,產物經離心、烘干,得到噻螨酮原藥。
實施例2
一種噻螨酮的制備方法,包括以下步驟:
步驟S1,向3000升反應釜中投入174Kg對氯苯丙酮、350Kg石油醚,降溫到20℃,再加入117.5Kg亞硝酸正丁酯,攪拌10h,再降溫至0℃,保溫攪拌1.5h,經放料、過濾、石油醚洗滌,烘干得到固體酮肟物;
步驟S2,在2000升反應釜中投入90Kg制得的酮肟物、240Kg甲醇,加入催化劑鈀1Kg,溶解0.5h,氮氣置換4次,再氫氣置換4次,升溫至40℃,通入氫氣,控制氫氣壓力為0.08Mpa,直到加不進氫氣為止,通氫氣完畢,趁熱過濾除去催化劑鈀,得到醇胺物的甲醇溶液,用鹽酸調PH值到6.5,減壓脫去溶劑得到醇胺物粗品,冷卻至50℃,加入250Kg乙醇,升溫回流3h,冷卻到5℃后,保溫3h,所得產物經離心、烘干,得2-氨基-1-對氯苯基丙醇鹽酸鹽;
步驟S3,在2000升反應釜中投入2-氨基-1-對氯苯基丙醇鹽酸鹽90Kg,8%氫氧化鉀溶液350Kg、氫氧化鈉1.5Kg,攪拌均勻后,再加入二硫化碳38.5Kg份,然后在100℃攪拌反應14h,保溫結束后降溫到常溫,然后反應釜中抽入250Kg石油醚,攪拌1.5h,靜置2h,分層,有機層去脫溶,脫溶結束后,加入甲醇250Kg升溫回流4h,降溫到5℃析出晶體后,再保溫3h,離心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步驟S4,在3000升反應釜中抽入甲醇400Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮95Kg,甲醇鈉2Kg,開啟攪拌,然后在25℃加入150Kg的甲醇鈉,加畢后,攪拌半小時,然后在25℃以下滴加80Kg雙氧水,滴加完畢,在25℃保溫8h,保溫結束后,轉入脫溶釜,減壓脫去甲醇,降溫到室溫,抽入甲苯350Kg,水250Kg,攪拌半小時,靜置半小時,分去水層,減壓脫去甲苯,降溫到室溫,加入250Kg甲醇,升溫回流3h,降溫到5℃保溫4h,抽濾得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步驟S5,在2000升反應釜中,加入350Kg甲苯、100Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和5Kg四氫呋喃,調節反應釜溫度為40℃,開始滴加62.5Kg異氰酸環已酯,時間控制在2.5h,滴完后,保溫反應8h,取樣中控,合格后,加入200Kg水,控制溫度為40℃,繼續攪拌1h。然后將反應混合物抽濾,濾液分去水層,油層用200Kg水洗一次,分去水層,抽入脫溶釜減壓蒸出甲苯,冷卻到60℃,加入260Kg乙醇,升溫回流4h,冷卻到10℃保溫3h,產物經離心、烘干,得到噻螨酮原藥。
實施例3
一種噻螨酮的制備方法,包括以下步驟:
步驟S1,向3000升反應釜中投入420Kg對氯苯丙酮、840Kg石油醚,降溫到15℃,再加入282Kg亞硝酸正丁酯,攪拌7.5h,再降溫至0℃,保溫攪拌1h,經放料、過濾、石油醚洗滌,烘干得到固體酮肟物;
步驟S2,在2000升反應釜中投入216Kg制得的酮肟物、576Kg甲醇,加入催化劑鎳1.8Kg,溶解0.5h,氮氣置換3次,再氫氣置換3次,升溫至37℃,通入氫氣,控制氫氣壓力為0.05Mpa,直到加不進氫氣為止,通氫氣完畢,趁熱過濾除去催化劑鎳,得到醇胺物的甲醇溶液,用鹽酸調PH值到6.3,減壓脫去溶劑得到醇胺物粗品,冷卻至45℃,加入600Kg乙醇,升溫回流2h,冷卻到2.5℃后,保溫2h,所得產物經離心、烘干,得2-氨基-1-對氯苯基丙醇鹽酸鹽;
步驟S3,在2000升反應釜中投入2-氨基-1-對氯苯基丙醇鹽酸鹽216Kg,8%氫氧化鉀溶液840Kg、甲醇鈉2.4Kg,攪拌均勻后,再加入二硫化碳92.4Kg,然后在90℃攪拌反應13h,保溫結束后降溫到常溫,然后反應釜中抽入600Kg石油醚,攪拌1h,靜置1.2h,分層,有機層去脫溶,脫溶結束后,加入甲醇600Kg升溫回流3h,降溫到2.5℃析出晶體后,再保溫2h,離心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步驟S4,在3000升反應釜中抽入甲醇960Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮228Kg,過氧化鈉3.6Kg,開啟攪拌,然后在25℃加入360Kg的甲醇鈉,加畢后,攪拌半小時,然后在25℃以下滴加192Kg雙氧水,滴加完畢,在20℃保溫6h,保溫結束后,轉入脫溶釜,減壓脫去甲醇,降溫到室溫,抽入甲苯840Kg,水600Kg,攪拌半小時,靜置半小時,分去水層,減壓脫去甲苯,降溫到室溫,加入600Kg甲醇,升溫回流2h,降溫到2.5℃保溫3h,抽濾得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步驟S5,在2000升反應釜中,加入840Kg甲苯、240Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和12Kg三乙胺,調節反應釜溫度為37℃,開始滴加150Kg異氰酸環已酯,時間控制在2h,滴完后,保溫反應6h,取樣中控,合格后,加入480Kg水,控制溫度為37℃,繼續攪拌1h。然后將反應混合物抽濾,濾液分去水層,油層用480Kg水洗一次,分去水層,抽入脫溶釜減壓蒸出甲苯,冷卻到55℃,加入624Kg乙醇,升溫回流3h,冷卻到7.5℃保溫2h,產物經離心、烘干,得到噻螨酮原藥。
實施例4
一種噻螨酮的制備方法,包括以下步驟:
步驟S1,向3000升反應釜中投入350Kg對氯苯丙酮、700Kg石油醚,降溫到12℃,再加入235Kg亞硝酸正丁酯,攪拌8h,再降溫至0℃,保溫攪拌1.2h,經放料、過濾、石油醚洗滌,烘干得到固體酮肟物;
步驟S2,在2000升反應釜中投入180Kg制得的酮肟物、480Kg甲醇,加入催化劑鎳1.8Kg,溶解0.5h,氮氣置換2次,再氫氣置換4次,升溫至39℃,通入氫氣,控制氫氣壓力為0.06Mpa,直到加不進氫氣為止,通氫氣完畢,趁熱過濾除去催化劑鎳,得到醇胺物的甲醇溶液,用鹽酸調PH值到6.2,減壓脫去溶劑得到醇胺物粗品,冷卻至42℃,加入500Kg乙醇,升溫回流2.5h,冷卻到4℃后,保溫1.5h,所得產物經離心、烘干,得2-氨基-1-對氯苯基丙醇鹽酸鹽;
步驟S3,在2000升反應釜中投入2-氨基-1-對氯苯基丙醇鹽酸鹽180Kg,8%氫氧化鉀溶液700Kg、氫氧化鈉1.5Kg,攪拌均勻后,再加入二硫化碳77Kg,然后在85℃攪拌反應12.5h,保溫結束后降溫到常溫,然后反應釜中抽入500Kg石油醚,攪拌1.2h,靜置1.5h,分層,有機層去脫溶,脫溶結束后,加入甲醇500Kg升溫回流2.5h,降溫到1℃析出晶體后,再保溫1.5h,離心烘干得到5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮;
步驟S4,在3000升反應釜中抽入甲醇800Kg,投入5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮190Kg,甲醇鈉3.5Kg,開啟攪拌,然后在25℃加入300Kg的甲醇鈉,加畢后,攪拌半小時,然后在25℃以下滴加160Kg雙氧水,滴加完畢,在18℃保溫5h,保溫結束后,轉入脫溶釜,減壓脫去甲醇,降溫到室溫,抽入甲苯700Kg,水500Kg,攪拌半小時,靜置半小時,分去水層,減壓脫去甲苯,降溫到室溫,加入500Kg甲醇,升溫回流1.5h,降溫到1℃保溫2.5h,抽濾得到5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮;
步驟S5,在2000升反應釜中,加入700Kg甲苯、200Kg 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮和10Kg二月桂酸二丁基錫,調節反應釜溫度為38℃,開始滴加125Kg異氰酸環已酯,時間控制在1.8h,滴完后,保溫反應5h,取樣中控,合格后,加入400Kg水,控制溫度為37℃,繼續攪拌1h。然后將反應混合物抽濾,濾液分去水層,油層用400Kg水洗一次,分去水層,抽入脫溶釜減壓蒸出甲苯,冷卻到52℃,加入520Kg乙醇,升溫回流2.5h,冷卻到6℃保溫2.5h,產物經離心、烘干,得到噻螨酮原藥。
實驗例1
實施例1~4所制的酮肟物的試產數據如表1所示。
表1酮肟物試產數據
實施例1~4所制的2-氨基-1-對氯苯基丙醇鹽酸鹽(β-醇胺物)的試產數據如表2所示。
表2 2-氨基-1-對氯苯基丙醇鹽酸鹽(β-醇胺物)試產數據
實施例1~4所制的5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮的試產數據如表3所示。
表3 5-(4-氯苯基)-4-甲基-2-硫代噻唑烷酮試產數據
實施例1~4所制的5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮的試產數據如表4所示。
表4 5-(4-氯苯基)-4-甲基-2-氧代噻唑烷酮試產數據
實施例1~4所制的噻螨酮原藥的試產數據如表5所示。
表5噻螨酮原藥試產數據
由上述試驗結果可知:酮肟化反應的平均收率為96.8%,所得酮肟物的平均含量≥99%;醇胺化反應的平均收率為95.4%,所得醇胺物的平均含量為≥99%;環合反應的平均收率為93.6%,所得環合物的平均含量≥96%;氧化反應的平均收率為90.8%,所得噻唑烷酮的平均含量≥97%;加成反應平均收率為95.8%,所得最終產物噻螨酮原藥的平均含量為98.8%。
實驗例2
對實施例1~4所制的噻螨酮原藥進行分析及表征。
(1)紅外光譜法:采用溴化鉀壓片法,壓下制成溴化鉀-農藥樣本薄片,以純的溴化鉀薄片為對照,在紅外光譜儀上掃描樣本即得紅外光譜譜圖,掃描范圍為4000cm-400cm。
結果表明,如圖5所示,實施例1~4所制的噻螨酮原藥的有效成份與噻螨酮具有相同的紅外光譜吸收特征。
(2)紫外光譜法:分別取適量待測樣品用乙腈溶解,以乙腈為空白對照,按照儀器操作規程測定待測樣品的紫外吸收。
如圖6所示,實施例1~4所制的噻螨酮原藥的紫外吸收光譜圖之間無明顯差異,皆與噻螨酮標樣的紫外吸收光譜圖十分吻合。從紫外吸收圖譜可見:噻螨酮原藥的紫外光譜與噻螨酮標準品的譜圖相同,在約196nm和223nm附近有較強吸收。
結果表明,實施例1~4所制的噻螨酮原藥的有效成份與噻螨酮具有相同的紫外光譜吸收特征。
(3)核磁共振法:將待測樣品各約10mg,分別放入核磁管中,加入適量d-DMSO,蓋緊管蓋,振蕩使樣本溶解。按照儀器操作規程在核磁共振儀上測定。
如圖7所示,實施例1~4所制的噻螨酮原藥與噻螨酮標樣的核磁共振吸收圖譜之間無明顯差異,各H原子的化學位移和裂分情況也相同。
結果表明,實施例1~4所制的噻螨酮原藥的有效成份與噻螨酮具有相同的核磁共振波譜特征。
(4)質譜法:以液相色譜為分離工具,以MSD為檢測器,在選定的色譜及離子化條件下,對噻螨酮原藥中的主要組分進行分離。通過分析各實施例制備的噻螨酮原藥中有效成分質譜圖,結合原藥生產工藝,對噻螨酮原藥中有效成分進行分離與定性鑒定。
結果顯示,如圖8所示,實施例1~4所制的噻螨酮原藥有效成分和標樣的MS定性分析譜圖之間無明顯差異。根據噻螨酮原藥的合成工藝和質譜數據給出的信息,推斷出實施例1~4所制的噻螨酮原藥有效成分是噻螨酮。
實驗例3
對實施例1~4所制的噻螨酮原藥進行全組分分析鑒定,鑒定結果如表6所示。
表6噻螨酮原藥全組分分析鑒定結果
以上僅為本發明的優選實施例而已,并不用于限制本發明,對于本領域的技術人員來說,本發明可以有各種更改和變化。凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!