一種離子液體及利用其合成Thiazovivin的方法與流程
本發明屬于有機合成技術領域,具體涉及一種離子液體及利用其合成Thiazovivin的方法。
背景技術:
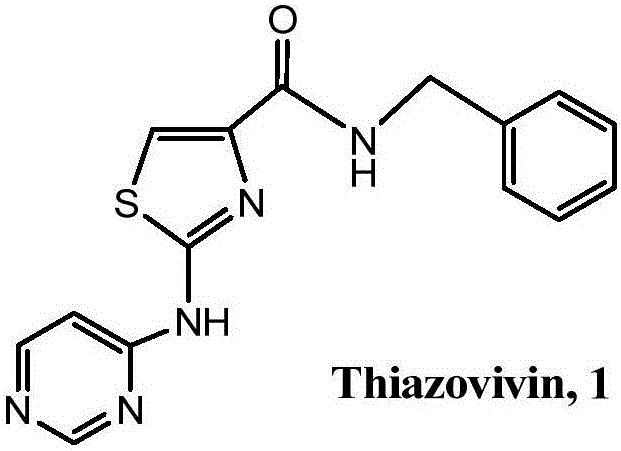
Thiazovivin(1)是一種新型ROCK抑制劑,能顯著增強人胚胎干細胞(hESCs)的存活,使其被酶解后的存活增強30多倍,在無細胞試驗中其IC50為0.5μM。此外,Thiazovivin也能夠有效抑制E-鈣粘蛋白的內吞作用,從而穩定細胞表面的E-鈣粘蛋白,并使細胞-細胞相互作用恢復,來保護hESCs在細胞外基質(ECM)缺乏的懸浮液中免于死亡。Thiazovivin(1)的結構式如圖1所示。
目前報道的Thiazovivin合成方法有固相合成法(WO2010065721)和化學合成法(Synthetic Communications1,43:2876-2882,2013)兩種。固相合成法中的反應屬于異相反應,反應物的活性要比在均相體系中的低,常常需要使用大大過量的反應試劑促使反應完全,且不能使用常規的分析手段如薄層色譜﹑核磁共振﹑質譜等實時監測反應進程;而化學合成法步驟繁瑣,后處理復雜,產物的分離純化需要頻繁的柱層析操作,費時費力。這兩種方法的局限性使得市場上Thiazovivin的價格始終居高不下(>2萬元/克)。因此,本領域亟需開發出新的合成方法來制備Thiazovivin,以提高合成效率、降低生產成本。
離子液體(Ionic liquid,IL)是一種在室溫或接近室溫下呈液態,由有機陽離子和有機或無機陰離子組成的鹽,也稱低溫熔融鹽。離子液體的可設計性強,由于構成它的陰離子和陽離子的可選擇范圍大,因此可以根據需要,簡單改變離子的構成就可以調控它的熔點、粘度、密度、溶解性等物理化學特性。一般情況下,離子液體具有良好的熱穩定性、不揮發、不可燃性、不爆炸及低毒性,因而在有機合成中是環境友好的。
然而,采用離子液枝載技術合成Thiazovivin,本領域尚未見此類報道。
技術實現要素:
針對本領域存在的如上所述的Thiazovivin現有合成方法操作復雜、監控困難以及成本太高等問題,本發明提供一種利用離子液枝載技術合成Thiazovivin的方法。
本發明的技術方案如下:
本發明首先提供一種用于合成Thiazovivin的離子液體,其具有如下通式1所示的結構:
其中R選自
x選自Cl、Br、I、BF4、PF6、ClO4、H2PO4、TS、ClO4、Lac、OAc、DCA、HSO4、NO3、HSO4、TFMS、NTF2、TSA、FSI;
n為0-10中的任意自然數。
上述通式中,碳鏈可以縮短或變長,而苯環部分是必須的,因為這種結構可以在脫除離子液的時候通過TFA(三氟乙酸)選擇性脫掉離子液體而不影響連接在N原子上的另一個芐基;理論上,這部分結構可以為任何形式,在本發明優選的實施例中提供的咪唑型的離子液體,陰離子用的PF6,除此之外,還有很多陰離子,例如:Cl、Br、I、BF4、PF6、ClO4、H2PO4、TS、ClO4、Lac、OAc、DCA、HSO4、NO3、HSO4、TFMS、NTF2、TSA、FSI;都可以跟咪唑形成離子液體。
在本發明最優選的實施例中,所述離子液體為式3所示的化合物3
本發明的一些具體地實施例還提供制備所述的離子液體的方法,其特征在于,包括:
(1)將1,6-二甲氧基-4-羥甲基苯甲醛﹑碳酸鉀和1,3-二溴丙烷在干燥的丙酮溶劑中攪拌回流進行反應直至反應完全,將反應液過濾并減壓蒸干丙酮溶劑,所得混合物經柱層析分離純化,得化合物2
(2)將所述化合物2溶于干燥的乙腈溶劑中,加入N-甲基咪唑和六氟磷酸鉀;將所得反應液在加熱條件下攪拌回流進行反應直至反應完全,將反應液冷卻至室溫,過濾,減壓蒸干乙腈溶劑,所得混合物用乙醚洗滌,得到所述離子液體。
進一步地,上述步驟(1)中,所述1,6-二甲氧基-4-羥甲基苯甲醛﹑碳酸鉀、1,3-二溴丙烷和丙酮的用量比例為:5mmol∶6mmol∶15mmol∶25ml;碳酸鉀的作用是作為堿來拔除羥甲基中羥基的氫,以便于跟溴丙烷發生反應;丙酮的作用是作為反應溶劑;采用上述比值的好處是:以1,6-二甲氧基-4-羥甲基苯甲醛為標準,一般堿(碳酸鉀)需要多加一點使得反應進行完全,1,3-二溴丙烷過量防止形成兩分子1,6-二甲氧基-4-羥甲基苯甲醛的副產物,丙酮25mL的用量可以上下稍作浮動調整。
所述攪拌回流時間為5小時,這一反應時間是經試驗驗證的最優選的反應時間,5小時可以使該步驟反應完全,小于5小時原料反應不完全,大于5小時則會有副產物生成;
所述柱層析分離純化的流動相為乙酸乙酯∶石油醚=1∶3~1∶2
上述步驟(2)中,所述化合物2、乙腈、N-甲基咪唑和六氟磷酸的用量比例為:1mmol∶5ml∶1.236mmol∶1mmol;N-甲基咪唑和六氟磷酸在這一步中所起的作用是生成上述優選的離子液體中的咪唑結構的原料;
所述加熱條件下攪拌回流指,80℃過夜攪拌回流。這一反應條件和時間是優化得出的最佳反應溫度和反應時間,過夜在化學上一般指反應12小時。
用乙醚洗滌3-4次的作用是:為了除掉化合物2或者咪唑等未反應完的原料,而離子液體是不溶于乙醚的。
基于上述離子液體,本發明另一方面還提供一種合成Thiazovivin的方法,包括如下步驟:
(1)將所述離子液體與芐胺連接得到化合物4;
(2)將上述化合物4與2-溴噻唑-4-羧酸進行酰化反應得到化合物5;
(3)上述化合物5與4-氨基嘧啶進行胺化反應得到化合物6;
(4)將上述化合物6中的離子液體去掉即可得到所述Thiazovivin;
本發明最大的貢獻是:采用所述離子液體作為合成Thiazovivin過程中的載體,并提供了一種全新結構的專用于合成Thiazovivin的離子液體化合物。采用所述離子液體合成Thiazovivin,除了便于跟蹤監測反應是否進行完全,與現有合成方法相比,還具有成本低廉、原材料獲得便捷、操作簡單、產率較高等優勢,對于量產Thiazovivin具有重要意義。
在本發明一些具體的實施例中,所述離子液體為式3所示的化合物3
在一些實施例中,上述步驟(1)中的連接指,將所述離子液體﹑芐胺﹑氰基硼氫化鈉溶于干燥的甲醇溶劑,將所得反應液攪拌至反應完全,去除甲醇溶劑,再溶于二氯甲烷溶劑,依次用1M HCl﹑飽和NaHCO3以及飽和NaCl萃洗,所得有機相用無水Na2SO4干燥,過濾,蒸干二氯甲烷溶劑,得到包含所述化合物4的中間產物。上述步驟中,氰基硼氫化鈉是芐胺和IL發生還原胺化反應所必需的試劑;依次用鹽酸、NaHCO3、NaCl萃洗的目的在于:作為化學反應后處理的標準操作,鹽酸是用來使得反應混合物中的堿性物質如芐胺成鹽,使其溶于水中除掉;飽和NaHCO3是為了洗掉多余的鹽酸,飽和NaCl是為了洗掉多余的無機鹽;
優選地,所述離子液體﹑芐胺﹑氰基硼氫化鈉、甲醇、二氯甲烷的用量比例為1mmol∶2mmol∶4mmol∶10ml∶20ml;這一比值是優化出來的最佳反應原料配比,可以保證產率;上述比值可以稍微變化,但是可能導致產率會有所降低;
所述攪拌指,室溫下攪拌12小時。
在一些實施例中,上述步驟(2)中的酰化反應指,將所述化合物4溶于四氫呋喃溶劑,加入1-羥基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽、N,N-二異丙基乙胺,混勻,然后慢慢加入2-溴噻唑-4-羧酸,攪拌反應直至反應完全,所得反應液用乙酸乙酯和水稀釋,所得有機相依次用1M HCl、飽和NaHCO3溶液洗滌,無水Na2SO4干燥,過濾,蒸干四氫呋喃溶劑,得到包含所述化合物5的中間粗產物。上述反應中,1-羥基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽、N,N-二異丙基乙胺這三種物質是酰胺縮合的必要試劑,加入的順序不分先后;用乙酸乙酯和水稀釋的原因是,乙酸乙酯和四氫呋喃是混溶的,不稀釋的話反應液無法分層;依次用HCl、飽和NaHCO3溶液萃洗的作用是,同樣是作為化學反應后處理的標準操作,鹽酸是用來使得反應混合物中的堿性物質,使其溶于水中除掉;飽和NaHCO3是為了洗掉多余的鹽酸;無水Na2SO4干燥的作用是去除水分,過濾的作用是去除無水Na2SO4;
進一步地,所述四氫呋喃、1-羥基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽、N,N-二異丙基乙胺、2-溴噻唑-4-羧酸、乙酸乙酯、水的用量比例為:5ml∶1mmol∶1mmol∶1mmol∶1mmol∶10ml∶10ml;上述比值是經試驗摸索出的最佳反應配比,能保證產率,可以稍微浮動調整;
所述混勻指,攪拌30分鐘;先攪拌30分鐘的好處是先讓化合物4中的氨基氫與堿反應脫除,然后才能與后面的羧酸反應;
所述攪拌反應指,室溫攪拌10小時,這是優化后的最近反應時間,能使反應完全。
在一些實施例中,上述步驟(3)中的胺化反應指,將所述化合物5溶于乙腈溶劑,加入4-氨基嘧啶,冰浴下分批加入氫化鈉,即每次加少量、緩慢加入,使反應液慢慢反應,防止局部溫度過高使產物分解;10分鐘后,升溫進行反應直至反應完全,冷卻至室溫;10分鐘后升溫是為了先讓氫化鈉與化合物5中的氨基氫拔除,才能與后面的溴代物反應;用乙酸乙酯和水稀釋,使混溶的乙酸乙酯和乙腈分層,所得有機相用無水Na2SO4干燥,過濾,蒸干乙腈溶劑,得到含有所述化合物6的中間粗產物。
具體地,所述乙腈、4-氨基嘧啶、氫化鈉、乙酸乙酯、水的用量比例為:5ml∶1mmol∶2mmol∶10ml∶10ml;這是優化出來的最佳反應條件,可以稍微變化,但是可能產率會有所降低;
所述反應指升溫至100℃反應12小時,優化的反應溫度和反應時間,能保證反應完全;所述氫化鈉指分散在礦物油中的質量分數為60%的氫化鈉。
步驟(1)、(2)、(3)所得的中間粗產物還需經過異相分離方法純化;
所述異相分離方法指,所得的中間粗產物溶解于二氯甲烷溶劑中,加入異丙醚,然后利用旋轉蒸發儀減壓除去二氯甲烷溶劑,待剩余溶劑的總體積約為初始總體積的一半時,停止旋轉蒸發,通過過濾收集沉淀并用乙醚洗滌沉淀得純化產物。
優選地,上述異相分離方法中,所得混合物和二氯甲烷的用量比例為:1g∶10ml;所述異丙醚的用量為二氯甲烷用量的5被體積。
在一些實施例中,上述步驟(4)將化合物6中的離子液體去掉指,將所述化合物6溶于二氯甲烷溶劑,過濾后,加入體積比90%的三氟乙酸溶液進行反應直至反應完全,濃縮,即將反應液蒸干一部分,以便于柱層析上樣;所得的濃縮后的混合物依次經柱層析分離純化和中低壓液相色譜分離純化,得到所述Thiazovivin。
具體地,所述二氯甲烷、三氟乙酸的用量比例為:1ml∶1ml;該比值為優化出來的最佳反應條件,三氟乙酸少了酸度不夠,多了容易導致產物分解;
所述反應指室溫反應6小時;
所述柱層析分離純化的流動相為乙酸乙酯∶石油醚=1∶1→乙酸乙酯∶甲醇=10∶1;采用柱層析梯度洗脫,從極性較小的洗脫液慢慢增加到極性較大的洗脫液。
所述中低壓液相色譜條件為:采用柱長250mm×內徑9.4mm、粒徑5μm的Agilent Eclipse XDB-C18色譜柱,梯度洗脫25分鐘,洗脫劑為體積比80%-90%的甲醇水溶液;
在本發明所有的實施例中,上述所有反應步驟均在氮氣保護下進行;
所述反應完全指薄層色譜顯示反應完全;
本發明設計用離子液枝載的方法,即,用離子液體做為載體來合成Thiazovivin,與之前報道的方法相比:離子液枝載合成既有固相合成操作簡便、易于純化的優點,又避免了異相反應效率低、難以實時監測等缺陷,是一種具有規模化生產潛力的Thiazovivin合成方法。
本發明具有如下優點:離子液體是結構明確的小分子可溶性載體,因此可以利用常規分析手段如薄層色譜﹑核磁共振﹑質譜等實時監測反應進程;離子液體載體還具有裝載容量高,易于合成,價格便宜等優勢,因此可以實現Thiazovivin的大量合成。
經查閱,現有合成方法的反應過程中會用到如下幾種物質:
bis(2-oxo-3-oxazolidinyl)phosphinic chloride(BOP-Cl);
Pd2(dba)3(三(二亞芐基丙酮)二鈀)
sodium diethyldithiocarbamate(二乙基二硫代氨基甲酸鈉)
它們不常用、難以獲得,如果通過商購途徑獲取的話,價格非常昂貴,從而導致常規方法制備成本較高、實施便捷程度較低。
而本發明所采用的方法中涉及到的芐胺﹑氰基硼氫化鈉、4-氨基嘧啶、氫化鈉、乙酸乙酯等試劑,均為化學領域常用試劑,易獲取、價格低廉,采用這些原料進行反應或者量產,成本低廉,操作方便。
在產率方面,現有方法合成Thiazovivin的產率約48%,而本發明優化的反應條件下制備的Thiazovivin產率可達到52.1%。
同時,本發明還提供了基于上述合成方法的一鍋法操作,即每一環節生成的中間產物無需提純便直接進入下一步,加入原料進行反應,這種操作方式經驗證是可行的,高效的,且能保證產率達到27.7%,為批量化生產Thiazovivin提供了一種備選方案。
綜上,采用本發明所提供的離子液體及方法合成Thiazovivin,具有操作簡便、成本低廉、得率較高等優點,尤其適用于大規模批量化生產Thiazovivin,以獲得成本交底的Thiazovivin產品。
附圖說明
圖1為Thiazovivin的結構式;
圖2為本發明的一個實施例中所示方法合成Thiazovivin的整體路線圖;
圖3為本發明的一個實施例中所示方法合成離子液體的路線圖;
圖4為本發明的一個優選實施例所示方法合成Thiazovivin的路線圖。
具體實施方式
以下結合具體實施實例進一步闡述本發明,需要說明的是,實例僅用于解釋本發明而不限制本發明的范圍。本發明未特別說明的實驗試劑均為本領域常規試劑,或采用本領域常規方法配制而得,可商購獲得,規格為實驗室純級即可。
實施例1、本發明所述的離子液體及其制備方法
本實施例提供一種用于合成Thiazovivin的離子液體,其特征在于,所述離子液體具有如下通式1所示的結構:
其中R選自
x選自Cl、Br、I、BF4、PF6、ClO4、H2PO4、TS、ClO4、Lac、OAc、DCA、HSO4、NO3、HSO4、TFMS、NTF2、TSA、FSI;
n為0-10中的任意自然數。
上述通式中,碳鏈可以縮短或變長,而苯環部分是必須的,因為這種結構可以在脫除離子液的時候通過TFA選擇性脫掉離子液體,而不影響連接在N原子上的另一個芐基;理論上,這部分結構可以為任何形式,在本發明優選的實施例中提供的咪唑型的離子液體,陰離子用的PF6,除此之外,還有很多陰離子,例如:Cl、Br、I、BF4、ClO4、H2PO4、TS、ClO4、Lac、OAc、DCA、HSO4、NO3、HSO4、TFMS、NTF2、TSA、FSI,都可以跟咪唑形成離子液體。
本實施例還提供一個最為優選的離子液體,即下述式3所示的化合物3
同時,本實施例還提供一種制備上述離子液體的方法,如圖3所示,包括如下步驟:
化合物2的合成:在氮氣保護下,1,6-二甲氧基-4-羥甲基苯甲醛(1.82克,10.0毫摩爾)﹑碳酸鉀(1.66克,12.0毫摩爾)和1,3-二溴丙烷(3.06毫升,30.0毫摩爾)在干燥的丙酮(50毫升)溶劑中攪拌回流5小時。待薄層色譜顯示1,6-二甲氧基-4-羥甲基苯甲醛消耗完后,將反應液過濾并減壓蒸干溶劑,所得混合物經柱層析分離純化(乙酸乙酯:石油醚=1:3~1:2),得化合物2(1.28克,42.32%),為白色固體。
化合物2的表征結果如下:1H NMR(500MHz,CDCl3)δ9.92(s,1H),6.58(s,2H),4.01(t,2H),3.68(s,6H),3.60(t,2H),2.23(m,2H)。其結構式為:
離子液體的合成:在氮氣保護下,化合物2(3.03克,10.0毫摩爾)溶于干燥的乙腈(50毫升)溶劑中,加入N-甲基咪唑(1.00毫升,12.36毫摩爾)和六氟磷酸鉀(KPF6,1.84克,10.0毫摩爾)。反應體系于80℃過夜攪拌回流,待薄層色譜顯示化合物2消耗完后,將反應液冷卻至室溫,過濾,減壓蒸干溶劑,所得混合物用乙醚洗滌3到4次,得純的離子液體(4.14克,92.0%),常溫下為無色油狀液體。
離子液體的表征結果如下:1H NMR(400MHz,CD3OD)δ8.78(s,1H),7.60(s,1H),7.51(s,1H),6.55(s,2H),4.09(t,2H),3.80(s,3H),3.61(s,6H),3.52(t,2H),2.20(m,2H).,2.36(m,2H);HR-ESIMS:m/z 305.1487[M-PF6]+(calcd 305.1496[M-PF6]+for C16H21N2O4).
實施例2、本發明所述的合成Thiazovivin的方法
本實施例提供一種合成Thiazovivin的方法,包括如下步驟:
(1)制備離子液體;
(2)將所述離子液體與芐胺連接得到化合物4;
(3)將上述化合物4與2-溴噻唑-4-羧酸進行酰化反應得到化合物5;
(4)上述化合物5與4-氨基嘧啶進行胺化反應得到化合物6;
(5)將上述化合物6中的離子液體去掉即可得到所述Thiazovivin;
實施例3、采用本發明的方法合成Thiazovivin(1)
本實施例基于實施例2所提供的離子液體,提供采用本發明的方法合成Thiazovivin的具體步驟,如圖2所示,包括如下步驟:
在氮氣保護下,采用實施例2提供的離子液體(4.50克,10.0毫摩爾)﹑芐胺(2.2毫升,20.0毫摩爾)﹑氰基硼氫化鈉(NaCNBH3,2.48克,40.0毫摩爾)溶于干燥的甲醇(100毫升)。反應液于室溫下攪拌12小時,薄層色譜顯示反應完全,減壓蒸干溶劑,再溶于二氯甲烷(200毫升),依次用1M HCl﹑飽和NaHCO3以及飽和NaCl萃洗,有機相用無水Na2SO4干燥,過濾,蒸干溶劑,所得混合物用異相分離方法純化得到化合物4;所述化合物4的結構式如下所示:
將上述化合物4溶于四氫呋喃(THF,100毫升),依次加入1-羥基苯并三唑(HOBT,2.7克,20.0毫摩爾)、1-(3-二甲氨基丙基)-3-乙基碳二亞胺鹽酸鹽(EDCI,3.82克,20.0毫摩爾)、N,N-二異丙基乙胺(DIPEA,3.3毫升,20.0毫摩爾),攪拌30分鐘,然后慢慢加入2-溴噻唑-4-羧酸(4.16克,20.0毫摩爾),室溫攪拌約10小時后,用乙酸乙酯(200毫升)和水(200毫升)稀釋,有機相依次用1M HCl、飽和NaHCO3溶液洗滌,無水Na2SO4干燥,過濾,蒸干溶劑,所得混合物用異相分離方法純化;
上述得到的化合物5溶于乙腈(100毫升),加入4-氨基嘧啶(1.9克,20.0毫摩爾),冰浴下分批加入氫化鈉(1.4克,分散在礦物油中,氫化鈉質量分數為60%,40.0毫摩爾),10分鐘后,升溫至100℃,12小時后反應完全,冷卻至室溫,用乙酸乙酯(200毫升)和水(200毫升)稀釋,有機相用無水Na2SO4干燥,過濾,蒸干溶劑,所得混合物用異相分離方法純化得到化合物6;
上述得到的化合物6溶于二氯甲烷過濾(100毫升),加入體積比90%三氟乙酸水溶液(100毫升),室溫反應6小時,濃縮,所得混合物依次經柱層析分離純化(乙酸乙酯:石油醚=1:1→乙酸乙酯:甲醇=10:1)和中低壓液相色譜分離純化(分離條件為:Agilent Eclipse XDB-C18(5μm,9.4×250mm)色譜柱,梯度洗脫25分鐘,洗脫劑為80%→90%的甲醇水溶液),得純Thiazovivin(0.86克,52.1%),為淡黃色固體。
Thiazovivin(1)的表征結果如下:Rf=0.22(EtOAc);mp=218℃;1H NMR(500MHz,DMSO-d6):δ11.79(br s,1H),8.80(s,1H),8.44(d,J=6.0Hz,1H),8.30(t,J=6.0Hz,1H),7.77(s,1H),7.12-7.49(m,5H),7.01(dd,J=6.0Hz,J=1.2Hz,1H),4.49(d,J=6.2Hz,2H);13C NMR(125MHz,DMSO-d6):δ160.5,158.0,156.9,156.3,155.9,144.2,139.1,128.1,127.0,126.1,117.2,107.1,42.9;HRMS(ESI):m/z calcd.for C15H13N5OS[M+Na]+334.0733;found 334.0722.
本實施例中的上述每一步制備中,除了最后一步脫除IL得到Thiazovivin不需要外,都需要使用異相分離方法對中間產物(含有化合物4、化合物5、化合物6的粗產物)進行純化,所述異相分離方法操作步驟如下:
將中間產物的反應所得混合物溶解于二氯甲烷(10毫升/克)中,加入5倍體積的異丙醚,然后利用旋轉蒸發儀減壓除去溶劑,待剩余溶劑的總體積約為初始總體積的一半時,停止旋轉蒸發,此時體系中的二氯甲烷全部被除去,離子液體支載的化合物全部沉淀下來,通過過濾收集沉淀并用乙醚洗滌沉淀,所得沉淀即為純的中間產物。
在本發明上述實施例的所有反應步驟中,整個合成路線中的每一步中間產物的獲得,都需要通過薄層色譜來監測反應是否完全,正是由于采用了離子液體作為載體,才使得這一監測手段成為可能,這也是本領域最為簡便通用的方法,幾分鐘就得到結果。
實施例4、本發明所述方法一鍋法操作
基于實施例3的操作步驟,本實施例提供本發明Thiazovivin合成方法的一鍋法操作,本實施例中的一鍋法操作在此重新定義為:上述每個步驟或環節不變,以及加入的原料還是依次進行,但無需提純每一步獲得的中間產物而直接進入下一環節的操作方法。
如圖4所示,大致步驟與實施例3無異,但是在分別獲得化合物4、化合物5、和化合物6這些中間環節,到最后即將獲得中間產物時,只進行洗滌步驟,而省去純化操作,得到的分別是含有化合物4的粗產物、含有化合物5的粗產物、含有化合物6的粗產物,而繼續進入下一環節,加入下一步驟反應所需的原料,進行下一步驟反應所需的操作,由于省去了中間產物的純化環節,使得可以從相對簡單的原料出發,不經中間體的分離,直接獲得目的產物,在操作上更為便捷、更有利于產品的批量化生產。這種方式獲得的Thiazovivin的產率為27.7%。
- 還沒有人留言評論。精彩留言會獲得點贊!