一種高分散性聚酰亞胺/納米粒子復合薄膜及其制備方法與流程
本發明涉及一種高分散性聚酰亞胺/納米粒子復合薄膜及其制備方法,屬于功能高分子材料領域。
背景技術:
科學技術的發展對材料性能提出了新的要求,納米復合材料作為一種新型材料正受到人們的重視。聚酰亞胺是一種性能優異的高分子材料,它在很寬的溫度范圍內具有優異的耐高溫、耐低溫、耐輻射、優異的機械性能和良好的化學穩定性,因此在航空航天、微電子和電器絕緣等領域具有廣泛的用途。將聚酰亞胺與納米粒子復合可以得到性能更加優良的復合材料。
制備聚酰亞胺/納米粒子復合材料最關鍵問題是納米粒子在聚酰亞胺中的均勻分散問題,目前制備聚酰亞胺/納米粒子復合材料最主要的方法是原位分散聚合法和溶膠-凝膠法。原位分散聚合法是采用超聲分散、機械共混等手段,使納米粒子在聚合物中均勻分散,然后在一定條件下就地聚合,從而得到聚酰亞胺/納米粒子復合材料。這種方法制備的復合材料,納米粒子在聚酰亞胺中的分散很不均勻,通常要對納米粒子的表面進行改性。溶膠-凝膠法是利用無機納米粒子的前驅體在聚酰亞胺或單體的共溶劑中水解和縮合,形成的凝膠不與聚合物發生相分離,從而得到聚酰亞胺/納米粒子復合材料。這種方法的困難是選擇具有良好溶解性能的共溶劑,并且其應用的領域也受到一定的限制。
技術實現要素:
本發明的目的就是解決以上的不足,提供一種高分散性聚酰亞胺/納米粒子復合薄膜及其制備方法,這種方法具有簡單、方便、應用面廣等優點。
本發明采用的技術方案如下:
本發明提供一種聚酰亞胺/納米粒子復合薄膜,該薄膜的組成為聚酰亞胺摻雜納米粒子,其特點是:納米粒子的摻雜方式為:所述納米粒子存在于聚酰胺-胺(PAMAM)樹狀大分子的空腔中,PAMAM通過末端胺基與所述聚酰亞胺中的酸酐基進行化學鍵交聯。
該薄膜的厚度根據實際情況設定,一般為70~100um。
本發明還提供一種聚酰亞胺/納米粒子復合薄膜的制備方法,包括以下步驟:
(1)將設定量的納米粒子與不同代數的PAMAM樹狀大分子混合均勻,得到混合溶液一;
(2)將酸酐封端的聚酰胺酸溶液與所述混合溶液一混合均勻,反應,得到混合溶液二;
(3)將所述混合溶液二進行成膜處理,得到聚酰胺酸凝膠膜;
(4)將步驟(3)的凝膠膜升溫到190~210℃下真空干燥1~3h,然后升溫至230~250℃下再干燥0.5~1.5h,酰胺化后得到聚酰亞胺/納米粒子復合薄膜。
步驟(1)中,所述的PAMAM是目前樹狀大分子化學中研究較為成熟的一類,是三種已經商品化的樹狀大分子之一。樹狀大分子中結構單元每重復一次成為一次繁衍,得到的產物的代數就增加1,據報道,目前聚酰胺-胺(PAMAM)樹狀大分子已合成到10.0代。不同代數的PAMAM合成或獲得已經是本領域技術人員的常規技術手段。本發明中采用的第一、二、和三代PAMAM是通過Tomalia的方法合成的。
本發明中的納米粒子并沒有特殊限定,例如,所述納米粒子為納米銀,納米銅、納米二氧化硅、納米二氧化鈦、納米鈦酸鋇、納米碳酸鈣、納米鈦酸鍶、納米鈦酸鍶鋇等中的一種或幾種。
優選的,所述納米粒子在PAMAM樹狀大分子的質量百分含量為1~15%。
納米粒子與PAMAM樹狀大分子混合均勻采用超聲分散的方法;溫度和時間并沒有特別限定,以分散均勻為目的,超聲分散時間可為30~60min;溫度可為常溫(18~37℃)。
步驟(2)中,所述聚酰胺酸溶液中含有設定量的有機溶劑。所述酸酐封端的聚酰胺酸溶液的制備為本領域技術人員常規知曉的技術手段,其制備方法并沒有特別限定,可以根據現有文獻進行制備,通用方法是利用二胺和二酐反應生成聚酰胺酸,在本發明的某些實施中,優選以下方法:在氮氣保護下,將摩爾比為1.02~1.05:1的二酐(也稱之為二元的酸酐)和二胺(也稱之為二元的胺)加入到有機溶劑中配成固含量10~15%(wt/wt)的溶液,并在冰水浴中反應20~28h(優選24h),得到酸酐封端的聚酰胺酸溶液;
其中,所述二酐為均苯四甲酸二酐,聯苯四甲酸二酐、3,3′,4,4′-二苯甲酮四酸二酐、3,3′,4,4′-二苯醚四酸二酐、2,2′-雙[4-(3,4-二酸苯氧基)苯基]丙烷四酸二酐、2,2′-雙(3,4-二羧苯基)六氟丙烷四酸二酐中任一種。
所述二胺為4,4′-二氨基二苯醚、六氟甲基聯苯二胺、二氨基二苯甲烷、4,4′-雙(4-氨基苯氧基)二苯砜、雙[3,5-二甲基-4-(4-氨基)苯酚]甲烷中的任一種。
所述溶劑為高沸點極性非質子溶劑,包括N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N-甲基-2-吡咯烷酮中的任一種。
所述聚酰胺酸的酸酐基與所述混合溶液一中的PAMAM樹狀大分子中的胺基發生反應。
所述酸酐封端的聚酰胺酸溶液與所述混合溶液一的質量比例為1~10:100。
所述反應的條件為冰水浴;冰水浴時間為10~14h,優選時間為12h。
步驟(3)中,成膜處理的方法并沒有特別的限定,將混合溶液制備成為薄膜為本領域技術人員常規的技術手段。本發明的某些實施例中采用以下方法:將所述混合溶液二澆注至模具上,然后將模具進行干燥,脫除溶劑,得到聚酰胺酸凝膠膜。
為了使最終得到的復合薄膜的性能優異,所述干燥采用真空干燥,真空干燥分為兩個階段,首先在70~90℃真空干燥1~3h,然后在140~160℃真空干燥1~3h。優選的,80℃真空干燥2h,150℃真空干燥2h。
步驟(4)中,優選的,在200℃真空干燥2h,然后在240℃真空干燥1h。
本發明提供一種上述方法制備得到的聚酰亞胺/納米粒子復合薄膜。
上述技術方案中的一個技術方案具有如下有益效果:
本發明利用樹枝狀大分子中存在的大量空腔,將納米粒子分散于PAMAM樹狀大分子的空腔中,再與末端為酸酐基的聚酰胺酸進行反應,最后經去除溶劑,熱酰胺化得到聚酰亞胺/納米粒子復合薄膜。
利用本發明的制備方法,可以使納米粒子在聚酰亞胺基體中均勻分散。另外由于PAMAM末端與聚酰胺酸有化學鍵的連接,所制備的聚酰亞胺/納米粒子復合材料具有較好的耐高溫性、耐低溫、優異的機械性能和良好的化學穩定性。在航空航天、微電子和電器絕緣等領域具有廣泛的用途。
本發明的方法為制備聚酰亞胺/納米粒子復合薄膜提供了一種新的思路。
附圖說明
圖1是納米粒子在PAMAM中的分散示意圖。
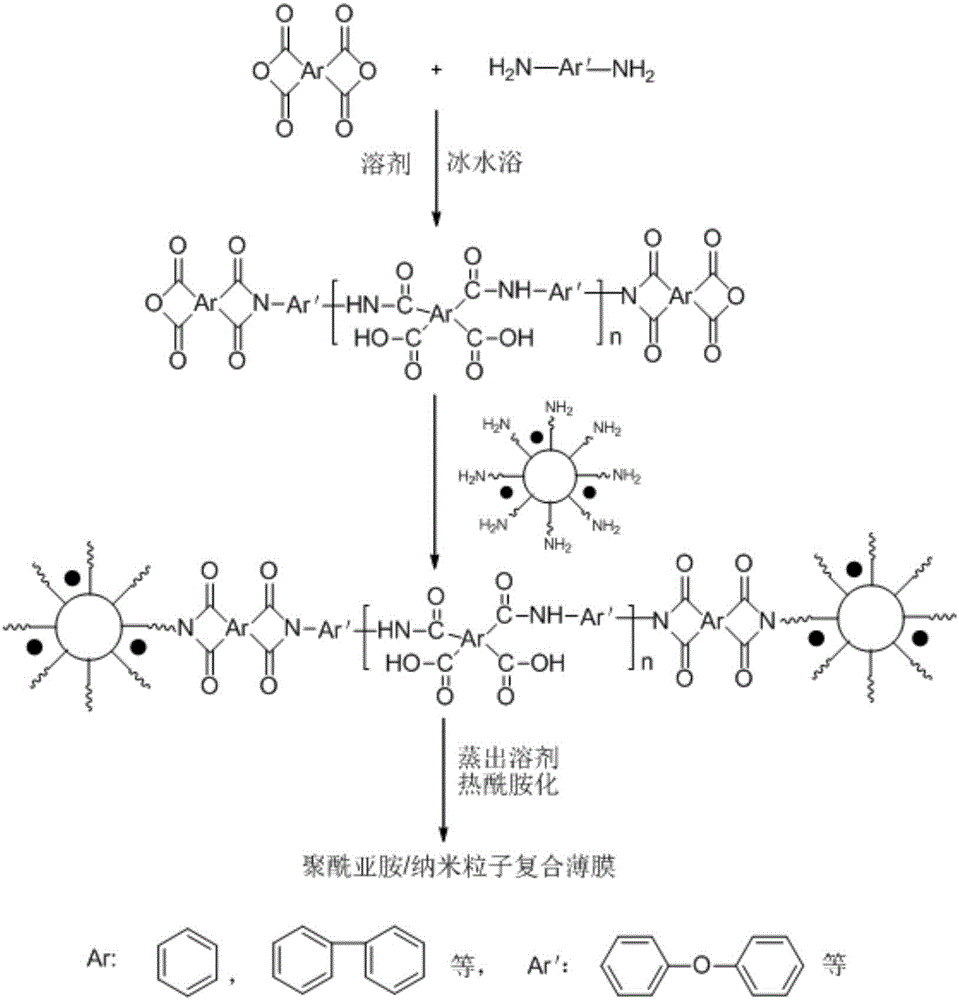
圖2是聚酰亞胺/納米粒子復合膜的制備示意圖,其中,n為100~200,表示二酐,H2N-Ar′-NH2表示二胺。
具體實施方式
本實施例中第一、二和三代PAMAM均是通過Tomalia的方法常規合成的。
實施例1
將0.20g的納米鈦酸鋇加入到5.62g第一代的PAMAM溶液中,超聲分散30min,如圖1所示。在氮氣保護下,將2.2248g的均苯四甲酸二酐和2.0023g的4,4′-二氨基二苯醚加入到28.18g的N-甲基吡咯烷酮中配成固含量15%(wt/wt)的溶液,并在冰水浴中反應24h,得到酐封端的聚酰胺酸溶液。將2.91g分散了納米鈦酸鋇粒子的第一代PAMAM溶液加入到上述聚酰胺酸溶液中,超聲分散數分鐘后,繼續反應12h,得聚酰胺酸粘稠液。取適量聚酰胺酸粘稠液澆注到潔凈的玻璃板模具上。將模具放到真空干燥箱中,80℃真空干燥2h,150℃真空干燥2h,得聚酰胺酸凝膠膜。繼續升溫至200℃下真空干燥1h后,升溫至240℃下再真空干燥1h,得到聚酰亞胺/納米粒子復合薄膜,基本流程如圖2所示。該薄膜的厚度根據實際情況設定,一般為70~100um。經過試驗驗證,納米鈦酸鋇在聚酰亞胺中分散均勻,得到的薄膜具有較好的耐高溫性、耐低溫、優異的機械性能和良好的化學穩定性。
實施例2
將0.3g的納米銀粒子加入到5.45g第二代的PAMAM溶液中,超聲分散40min。在氮氣保護下,將2.2466g的均苯四甲酸二酐和2.0023g的4,4′-二氨基二苯醚加入到42.49g的N,N-二甲基乙酰胺中配成固含量10%(wt/wt)的溶液,并在冰水浴中反應23h,得到酐封端的聚酰胺酸溶液。將2.46g分散了納米銀粒子的第二代PAMAM溶液加入到上述聚酰胺酸溶液中,超聲分散數分鐘后,繼續反應11h,得聚酰胺酸粘稠液。取適量聚酰胺酸粘稠液澆注到潔凈的玻璃板模具上。將模具放到真空干燥箱中,80℃真空干燥2h,150℃真空干燥2h,脫除溶劑,得聚酰胺酸凝膠膜。繼續升溫至200℃下真空干燥2h后,升溫至240℃下再真空干燥1h,得到聚酰亞胺/納米粒子復合薄膜。該薄膜的厚度根據實際情況設定,一般為70~100um。經過試驗驗證,納米銀粒子在聚酰亞胺中分散均勻,得到的薄膜具有較好的耐高溫性、耐低溫、優異的機械性能和良好的化學穩定性。
實施例3
將0.40g的納米二氧化硅粒子加入到3.31g第三代PAMAM溶液中,超聲分散30min。在氮氣保護下,將2.2903g的均苯四甲酸二酐和2.0023g的4,4′-二氨基二苯醚加入到28.62g的N,N-二甲基甲酰胺中配成固含量15%(wt/wt)的溶液,并在冰水浴中反應22h,得到酐封端的聚酰胺酸溶液。將1.95g分散了納米二氧化硅的第三代聚酰胺-胺溶液加入到上述聚酰胺酸溶液中,超聲分散數分鐘后,繼續反應13h,得聚酰胺酸粘稠液。取適量聚酰胺酸粘稠液澆注到潔凈的玻璃板模具上。將模具放到真空干燥箱中,80℃真空干燥2h,150℃真空干燥2h,脫除溶劑,得聚酰胺酸凝膠膜。繼續升溫至200℃下真空干燥2h后,升溫至240℃下再真空干燥1h,得到聚酰亞胺/納米粒子復合薄膜。該薄膜的厚度根據實際情況設定,一般為70~100um。經過試驗驗證,納米二氧化硅粒子在聚酰亞胺中分散均勻,得到的薄膜具有較好的耐高溫性、耐低溫、優異的機械性能和良好的化學穩定性。
上述實施例為本發明較佳的實施方式,但本發明的實施方式并不受上述實施例的限制,其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 電泳粒子、電泳粒子的制造方法、電泳分散液、電泳片、電泳裝置和電子設備與流程
- 拋光粒子?分散層復合體及包括此的拋光料漿組合物的制造方法與工藝
- 氧化鋯粒子的有機溶劑分散體及其制備方法與流程
- 拋光粒子?分散層復合體及包括此的拋光料漿組合物的制造方法與工藝
- 一種磁/金納米復合粒子的制備方法與流程
- 金屬納米粒子分散體的制造方法與工藝
- 表面修飾金屬氧化物粒子材料、分散液、聚硅氧烷樹脂組合物、聚硅氧烷樹脂復合體、光半導體發光裝置、照明器具及液晶圖像裝置的制造方法
- 一種高分散負載型貴金屬納米粒子及其制備方法與流程
- 凝膠粒子的水分散物及其制造方法、以及圖像形成方法與流程
- 熱射線遮蔽粒子、熱射線遮蔽粒子分散液、熱射線遮蔽粒子分散體、熱射線遮蔽粒子分散體夾層透明基材、紅外線吸收透明基材、熱射線遮蔽粒子的制造方法與流程
- 拋光粒子?分散層復合體及包括此的拋光料漿組合物的制造方法與工藝
- 表面改性金屬氧化物粒子分散液及其制造方法、表面改性金屬氧化物粒子?硅酮樹脂復合組合物、表面改性金屬氧化物粒子?硅酮樹脂復合物、光學構件以及發光裝置與流程
- 表面修飾金屬氧化物粒子材料、分散液、聚硅氧烷樹脂組合物、聚硅氧烷樹脂復合體、光半導體發光裝置、照明器具及液晶圖像裝置的制造方法
- 一種高分散負載型貴金屬納米粒子及其制備方法與流程
- 凝膠粒子的水分散物及其制造方法、以及圖像形成方法與流程
- 熱射線遮蔽粒子、熱射線遮蔽粒子分散液、熱射線遮蔽粒子分散體、熱射線遮蔽粒子分散體夾層透明基材、紅外線吸收透明基材、熱射線遮蔽粒子的制造方法與流程
- 金屬氧化物粒子分散液、含金屬氧化物粒子組合物、涂膜及顯示裝置的制造方法
- 一種測定不溶物質在溶液中分散性的方法
- 通過液體納米粒子分散體的受控真空冷凍干燥制備纖維狀和層狀多孔微結構和納米結構 ...的制作方法
- 一種冰雹粒子識別方法及裝置與流程
- 電泳粒子、電泳粒子的制造方法、電泳分散液、電泳片、電泳裝置和電子設備與流程
- 電泳粒子、電泳粒子的制造方法、電泳分散液、電泳片、電泳裝置和電子設備與流程
- 拋光粒子?分散層復合體及包括此的拋光料漿組合物的制造方法與工藝
- 氧化鋯粒子的有機溶劑分散體及其制備方法與流程
- 分散裝置以及分散方法與流程
- 拋光粒子?分散層復合體及包括此的拋光料漿組合物的制造方法與工藝
- 表面修飾金屬氧化物粒子材料、分散液、聚硅氧烷樹脂組合物、聚硅氧烷樹脂復合體、光半導體發光裝置、照明器具及液晶圖像裝置的制造方法
- 一種高分散負載型貴金屬納米粒子及其制備方法與流程
- 凝膠粒子的水分散物及其制造方法、以及圖像形成方法與流程