改進的環氧體系和胺聚合物體系及其制備方法與流程
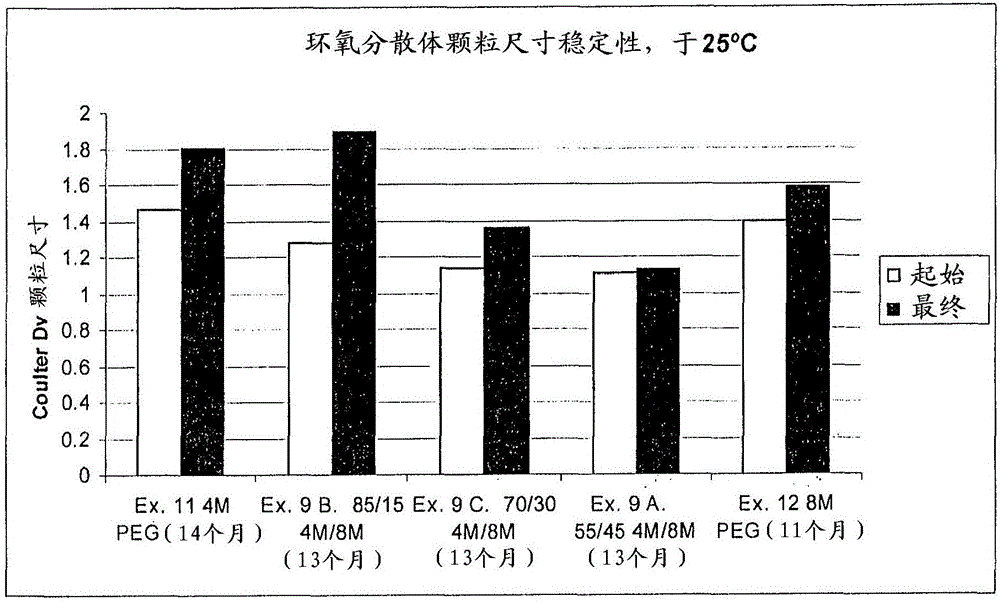
發明領域本發明涉及環氧樹脂的表面活性劑和水性分散體。一方面,本發明涉及改進的胺官能化表面活性劑,并且還涉及通過由聚氧化烯多元醇形成的胺組合物與環氧組合物的反應制備的改進的環氧官能化表面活性劑。發明背景環氧樹脂的水性分散體多年來都是已知的。但是,這些分散體作為涂料成分的性能被認為不如它們的含有溶劑的相似物。已知用于使得環氧組分可乳化的表面活性劑例如壬基酚乙氧基化物、烷基苯酚引發的聚(環氧乙烷)乙醇、烷基苯酚引發的聚(環氧丙烷)聚(環氧乙烷)乙醇和包含聚(環氧丙烷)內嵌段和兩種聚(環氧乙烷)乙醇外嵌段的嵌段共聚物容易遷移到表面界面,據推測,在那里它們對膜性能有不利的影響。此外,由于環氧樹脂水性分散體在工業中被更廣泛地使用,因此需要改進處理性能,例如儲存穩定性。由于表面活性劑分子中胺氮原子的存在,許多含水的環氧分散體的儲存穩定性隨時間內下降。隨著含水的分散體的pH值增加到超過9.8,該儲存穩定性不再以年來計量,而是以月來計量。還期望的是降低固體的顆粒尺寸,或者降低給定固體水平下需要的表面活性劑的量,或者期望使用更少的表面活性劑在給定的顆粒尺寸下分散固體。通常,需要大量的表面活性劑以降低顆粒尺寸并且有效地分散水中的固體,這導致升高了pH值并且降低了儲存穩定性。降低表面活性劑水平以控制pH值導致顆粒尺寸的增加。因此,對形成改進的用于形成環氧樹脂的水性分散體的表面活性劑存在著需要。發明概述本發明的實施方案涉及用于環氧樹脂水性分散體和用于胺聚合物組合物中的表面活性劑。該表面活性劑可以進一步官能化改性用作環氧組合物中的固化劑。一方面,本發明涉及通過由酸封端的聚氧化烯多元醇的共混物形成的酰胺胺組合物與環氧組合物反應制備的改進的環氧官能化表面活性劑。本發明的一方面提供了一種酰胺胺組合物,其包括兩種或更多種酸封端的聚氧化烯多元醇化合物的酸封端的聚氧化烯組合物與二胺化合物的反應產物,其中酸封端的聚氧化烯組合物具有1.1或更高的多分散性并且所述兩種或更多種酸封端的聚氧化烯多元醇化合物具有大約50%到大約100%的來自羥基端基氧化的羧酸端基,且二胺化合物包括伯胺取代基的第一胺取代基和伯胺取代基或仲胺取代基的第二胺取代基,其中反應產物包括具有下式的酰胺胺化合物:其中R1和R2中的每一個包括氫原子或選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的取代基,且R1和R2中的至少一個包括氫原子,R3是二價烴取代基,其選自具有2到18個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組,n為大約18到大約500的平均數,X是氫原子或選自由甲基取代基、乙基取代基、羥甲基取代基以及它們的組合組成的組的取代基,且Y為氫原子或選自甲基取代基、乙基取代基、羥甲基取代基以及它們的組合的組的取代基。本發明的另一方面提供了一種用于形成酰胺胺組合物的方法,其包括提供兩種或更多種酸封端的聚氧化烯多元醇化合物的酸封端的聚氧化烯組合物,其中酸封端的聚氧化烯組合物具有1.1或更高的多分散性并且所述兩種或更多種酸封端的聚氧化烯多元醇化合物具有大約50%到大約100%的來自羥基端基氧化的羧酸端基,提供二胺化合物,其包括伯胺取代基的第一胺取代基和伯胺取代基或仲胺取代基的第二胺取代基,并且使酸封端的聚氧化烯組合物和第一的二胺化合物反應以形成反應產物,其中反應產物包括具有下式的酰胺胺化合物:其中R1和R2中的每一個包括氫原子或選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的取代基,且R1和R2中的至少一個包括氫原子,R3是二價烴取代基,其選自具有2到18個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組,n為大約18到大約500的平均數,X是氫原子或選自由甲基取代基、乙基取代基、羥甲基取代基以及它們的組合組成的組的取代基,且Y為氫原子或選自甲基取代基、乙基取代基、羥甲基取代基以及它們的組合的組的取代基。本發明的另一方面,提供了一種環氧組合物,其包括通過環氧組分與酰胺胺組合物反應制備的反應產物,其中該反應產物包括具有以下結構的環氧官能化表面活性劑:,并且m為1到11,n為1到3,q為0到8,p為大約18到大約500,X為氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合,每一個Y是氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合,且R17可以是烷基基團、芳基基團、酰基基團以及它們的子集和它們的組合。附圖概述當考慮與附圖聯合時,本發明公開內容的優點和其它方面,由于通過參考以下詳細說明而同樣變得更好理解,并對本領域技術人員來說易于領會,其中貫穿附圖的數個圖形,同樣的參考符號標記相同或相似的單元,并且其中:圖1是說明使用本發明描述的材料和方法形成的環氧分散體與現有技術的環氧分散體相比,在25℃下環氧分散體顆粒尺寸穩定性的圖表。發明詳述本發明的實施方案涉及用于環氧樹脂的水性分散體和用于胺聚合物組合物的表面活性劑。該表面活性劑可以進一步官能化改性用作環氧組合物中的固化劑。一方面,本發明涉及通過由酸封端的聚氧化烯多元醇的共混物形成的酰胺胺與組合物環氧組合物反應制備的改進的環氧官能化表面活性劑。本發明的一些實施方案涉及由包含兩種或更多種具有不同分子量的酸封端的聚氧化烯多元醇的化合物(還已知為氧化的聚氧化烯多元醇和羧基化的聚氧化烯)的共混物形成表面活性劑。一種表面活性劑可以是由包含酸封端的聚氧化烯多元醇的化合物共混物制備的聚酰胺胺官能化的聚氧化烯原表面活性劑。該聚酰胺胺官能化的聚氧化烯表面活性劑之后可以進一步反應以形成環氧官能化的表面活性劑,其可以單獨形成或者與環氧樹脂一起原位形成。環氧官能化的表面活性劑可以用于形成水性環氧分散體。環氧官能化的表面活性劑還可以進一步反應以形成胺封端的表面活性劑,該胺封端的表面活性劑可以用于水性胺聚合物分散體,或者作為用于環氧組合物的固化劑。熱固性涂料和纖維尺寸配制品可以由本發明描述的分散體制備。令人驚奇且出乎意料的發現部分氧化的不同分子量的聚氧化烯多元醇的共混物提供了預料不到的超反應性表面活性劑,其用于將環氧官能化和胺官能化的聚合物乳化成“水包樹脂”分散體。觀察到的這種超性能包括降低的顆粒尺寸和改進的保存期限,以及改進(更高)的非揮發性物質含量和改進(更低)粘度的環氧聚合物分散體。本發明的一方面,本發明描述的原表面活性劑的酰胺胺組合物由兩種或更多種具有1.1或更高的聯合多分散性的酸封端的聚氧化烯多元醇化合物的包含酸封端的聚氧化烯多元醇的組合物與二胺化合物的反應產物形成,該聚氧化烯多元醇化合物被氧化為使羥基到羧基的轉化率為大約50%到大約100%,且二胺化合物包括伯胺取代基的第一胺取代基和伯胺取代基或仲胺取代基的第二取代基。酰胺胺化合物和組合物的制造一方面,通過本發明描述的方法形成的酰胺胺化合物,例如聚酰胺胺官能化的聚氧化烯原表面活性劑(本發明中還稱為酰胺胺原表面活性劑)具有下式:R1為氫原子或選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的取代基。R2為氫原子或選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的取代基,且R1和R2中的至少一個包括氫原子,R3是二價烴取代基,其選自具有2到18個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組,m可以是1、2或3,且n可以是大約18到大約500的平均數。對于重復單元,x可以是氫原子或選自由甲基取代基、乙基取代基、羥甲基取代基以及它們的組合組成的組的取代基。Y可以是氫原子或選自甲基取代基、乙基取代基、羥甲基取代基以及它們的組合的組的取代基。氧化烯可以是無規或嵌段聚合的。式(I)的聚酰胺胺官能化的聚氧化烯原表面活性劑可以具有大約8到大約30的胺值,例如大約12到大約24,例如大約14到大約18。該聚酰胺胺官能化的聚氧化烯原表面活性劑可以是親水性的。式(I)的聚酰胺胺官能化的聚氧化烯原表面活性劑可以具有大約200到大約22000的重均分子量(Mw),例如大約2000到大約12000,例如大約4000到大約10000。數均分子量(Mn)可以為大約180到大約20000,例如大約1800到大約11000,例如大約3000到大約8000。數均(標稱)分子量表示聚合物的總重量除以總重量中的分子總數。此外,分子量分布可以進一步表示為與分子量分布分析相關的本領域技術人員通常理解的相關的Z-均分子量(MZ)和Z+1均分子量(MZ+1)。Z-均分子量(MZ)可以為大約300到大約30000,例如大約3000到大約20000,例如大約5000到大約14000。Z+1均分子量(MZ+1)可以為大約400到大約40000,例如大約4000到27000,例如大約6500到大約19000。一方面,多酰胺胺官能化的聚氧化烯原表面活性劑可以由兩種或更多種具有不同分子量的包含酸封端的聚氧化烯多元醇的化合物(還已知為氧化的聚氧化烯多元醇和羧基化的聚氧化烯)與至少一種本發明描述的二胺反應形成。所述至少一種二胺化合物可以包括伯胺取代基的第一胺取代基和伯胺取代基或仲胺取代基的第二胺取代基。該反應可以用過量或不過量的二胺進行,包括大約12:1到大約2:3的胺與酸的當量比。選擇性地,多酰胺胺官能化的聚氧化烯原表面活性劑可以由包含酸封端的聚氧化烯多元醇的化合物與至少一種二胺形成。所述至少一種二胺化合物可以包括伯胺取代基的第一胺取代基和仲胺取代基的第二胺取代基。該反應可以使用過量的二胺進行,包括大約3:1到大約1:2的胺與酸當量比。在一個原表面活性劑的實施方案中,多酰胺胺官能化的聚氧化烯原表面活性劑可以由本發明描述的具有不同分子量的兩種或更多種包含酸封端的聚氧化烯多元醇的化合物的共混物與至少一種二胺反應形成。例如,包含第一酸封端的聚氧化烯多元醇的化合物和具有比包含第一酸封端的聚氧化烯多元醇的化合物分子量更高分子量的包含第二酸封端的聚氧化烯多元醇的化合物的共混物,這些化合物與至少一種二胺反應。該反應可以使用大約12:1到大約1:2的胺與酸的當量比進行。另一方面,多酰胺胺官能化的聚氧化烯原表面活性劑可以通過由兩種或更多種包含酸封端的聚氧化烯多元醇的化合物單獨形成酰胺胺并且之后將反應產物合并為多酰胺胺官能化的聚氧化烯原表面活性劑的共混物而形成。每一個反應都可以與本發明描述的至少一種二胺進行且每一個反應都可以使用相同或不同的二胺。每一個反應都可以使用本發明描述的方法進行且可以使用大約12:1到大約1:2的胺與酸的當量比進行。原表面活性劑組合物的一個實施方胺包括由兩種包含酸封端的聚氧化烯多元醇的化合物一起的共混物形成酰胺胺混合物,第一分子量的酸封端的聚氧化烯多元醇與第二分子量的酸封端的聚氧化烯多元醇的共混比例為大約3:17到大約17:3,例如大約11:9的共混比例。選擇性地,該共混物可以具有大約15wt.%到大約85wt.%,例如大約45%的第一分子量的酸封端的聚氧化烯多元醇,并且具有大約85wt.%到大約15wt.%,例如大約55%的第二分子量的酸封端的聚氧化烯多元醇。第二包含酸封端的聚氧化烯多元醇的化合物具有比第一包含酸封端的聚氧化烯多元醇的化合物的分子量更高的分子量。第一分子量的酸封端的聚氧化烯多元醇可以具有如下所述的大約200到大約5000的重均分子量,例如大約2000到大約5000,且第二分子量的酸封端的聚氧化烯多元醇可以具有如下所述的大于4000到大約16000的重均分子量。包含酸封端的聚氧化烯多元醇的化合物可以具有下式:R3可以是氫原子或具有2到18個碳原子的選自支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的二價烴取代基,且該烴取代基具有羥基端基或羧基端基。對于重復單元,m可以是1-11,且對于每一個相應的包含酸封端的聚氧化烯多元醇的化合物來說n可以是大約18到大約500的平均數。X可以是氫原子、甲基取代基、乙基取代基或羥甲基取代基。Y可以是氫原子、甲基取代基、乙基取代基或羥甲基取代基。包含酸封端的聚氧化烯多元醇的化合物可以是無規或嵌段聚合物。包含聚氧化烯多元醇的化合物可以具有大約50%到大約100%、例如大約70%到大約95%的被氧化形成酸封端的取代基(羧酸端基/羧基端基)的羥基端基。式(IIa)包含酸封端的聚氧化烯多元醇的化合物的重均分子量(Mw)可以是大約200到大約22000,例如大約2000到大約10000,例如大約4000到大約10000。其數均分子量(Mn)可以是大約180到大約20000,例如大約1800到大約11000,例如大約3000到大約8000。該數均(標稱)分子量表示聚合物的總重量除以聚合物中包含的分子的總摩爾數。此外,分子量分布可以進一步表示為與分子量分布分析相關的本領域技術人員通常理解的相關的Z-均分子量(MZ)和Z+1均分子量(MZ+1)。Z-均分子量(MZ)可以為大約300到大約30000,例如大約3000到大約20000,例如大約5000到大約14000。Z+1均分子量(MZ+1)可以為大約400到大約40000,例如大約4000到27000,例如大約6500到大約19000。對于兩種或更多種包含酸封端的聚氧化烯多元醇的化合物的合并組合物,該組合物可以具有大于大約1.1的多分散性。一方面,這種組合物可以具有大約1.15到大約5的多分散性,例如大約1.22到大約1.70。該多分散性通過重均分子量(MW)除以數均分子量(MN)計算。合適的包含酸封端的聚氧化烯多元醇的化合物的實例包括酸封端的聚氧乙二醇,酸封端的聚丙二醇以及它們的組合。例如,兩種或更多種包含酸封端的聚氧乙二醇的化合物中的每一種可以具有以下分子式:對于每一種相應的聚氧化烯二醇化合物來說,N可以是大約18到大約500的平均數。對于重復單元,m可以是1-11且對于每一種相應的包含酸封端的聚氧乙二醇的化合物來說,n可以是大約18到大約500的平均數。X可以是氫原子、甲基取代基、乙基取代基或羥甲基取代基。Y可以是氫原子、甲基取代基、乙基取代基或羥甲基取代基。包含酸封端的聚氧乙二醇的化合物可以是無規或嵌段聚合物。該包含聚氧乙二醇的化合物可以具有大約50%到100%、例如大約70%到大約95%的被氧化形成羧酸端基的羥基端基。式(IIb)的包含酸封端的聚氧乙二醇的化合物的重均分子量可以是大約200到大約22000,例如大約2000到大約10000,例如大約4000到大約10000。所述數均分子量(Mn)可以是大約180到大約20000,例如大約1800到大約11000,例如大約3000到大約8000。該數均(標稱)分子量表示聚合物的總重量除以聚合物中包含的分子的總摩爾數。此外,分子量分布可以進一步表示為與分子量分布分析相關的本領域技術人員通常理解的相關的Z-均分子量(MZ)和Z+1均分子量(MZ+1)。Z-均分子量(MZ)可以為大約300到大約30000,例如大約3000到大約20000,例如大約5000到大約14000。Z+1均分子量(MZ+1)可以為大約400到大約40000,例如大約4000到27000,例如大約6500到大約19000。在一個實施方案中,包含酸封端的聚氧化烯二醇的化合物的組合物可以通過氧化兩種或更多種聚氧化烯多元醇化合物的混合物形成,例如兩種或更多種聚氧化烯二醇化合物。選擇性地,包含酸封端的聚氧化烯二醇的化合物的組合物可以通過單獨氧化兩種或更多種聚氧化烯多元醇化合物中的每一種而形成,例如兩種或更多種聚氧化烯二醇化合物,并且將相應的包含酸封端的聚氧化烯多元醇的化合物合并為一種組合物。對于單獨氧化并且之后合并或者聯合氧化,兩種聚氧化烯多元醇化合物的共混比例可以為大約3:17到大約17:3的第一分子量的聚氧化烯多元醇與第二分子量的聚氧化烯多元醇,例如大約4:1到大約1:4的共混比例,例如大約11:9。選擇性地,該共混比例通過重量百分數表示,可以為大約15wt.%到大約85wt.%、例如大約45%的第一分子量的聚氧化烯多元醇,以及大約85wt.%到大約15wt.%、例如大約55%的第二分子量的聚烯烴多元醇。第二聚氧化烯多元醇具有高于第一聚氧化烯多元醇的分子量的分子量。聚氧化烯多元醇化合物可以通過聚氧化烯多元醇的氧化而氧化或羧基化/酸化以形成包含酸封端的聚氧化烯多元醇的化合物,其包括但不限于U.S.專利號6,235,931中描述的方法,通過參考到其與本發明要求的方面和說明書并不矛盾的程度而將其并入本發明。在一個形成方法的實施方案中,在水中存在自由基(例如2,2,6,6-四甲基-1-哌啶氧)和無機酸(例如硝酸)的情況下將含氧氣體加入到聚氧化烯多元醇中以便將羥基基團氧化為羧酸基團。如果期望的是二酸封端的聚烯烴多元醇,基本上所有的醇基團都被氧化為羧酸基團。包含酸封端的聚烯烴多元醇的化合物還可以通過Williamson醚合成制備,其中在堿的存在下聚氧化烯多元醇與氯乙酸和/或酯反應。在一個反應條件的實施方案中,溫度可以是大約20℃到大約70℃,且壓力在大約大氣壓到最高大約100psig的范圍內。反應后,從反應器中蒸餾出剩余的無機酸和水。在一個實施方案中,兩種包含酸封端的聚氧化烯多元醇的化合物可以通過氧化第一分子量和第二分子量的聚氧化烯多元醇化合物的混合物形成,例如具有不同分子量的聚氧乙二醇,共混比例為大約3:17到大約17:3的第一分子量的聚氧化烯多元醇與第二分子量的聚氧化烯多元醇,例如大約11:9的共混比例。第一分子量的聚氧化烯多元醇可以具有大約200到大約5000的分子量,且第二分子量聚氧化烯多元醇可以具有大于4000到大約16000的分子量。第二分子量聚氧化烯多元醇具有比第一分子量的聚氧化烯多元醇更高的分子量。本發明描述的聚氧化烯多元醇包括且不限于通過堿金屬和雙金屬氰化物催化劑聚合的聚氧化烯多元醇。該聚氧化烯多元醇可以用水自引發,用任何二醇引發,包括但不限于液態二醇或甘油,用雙酚引發,或者用包括伯胺或仲胺的其他活性氫有機化合物引發。合適的聚烯烴多元醇化合物可以具有下式:R3可以是氫原子或具有2到18個碳原子的選自支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的二價烴取代基,且該烴取代基中的每一個具有羥基端基。對于重復單元,m可以是1-11且對于每一種聚氧化烯多元醇化合物來說n可以是大約18到大約500的平均數。X可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、羥基端基以及它們的子集和其組合。Y可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、羥基端基以及它們的子集和其組合。合適的聚氧化烯多元醇包括聚氧乙二醇(聚醚)(PEG),聚丙烯聚醚二醇,1,2聚丁烯聚醚二醇、1,4聚丁烯聚醚二醇以及它們的組合。每一種聚氧化烯二醇都可以包括聚氧乙二醇單烷基醚或環氧乙烷和環氧丙烷或環氧丁烷的嵌段共聚物的單烷基醚(“聚氧化烯二醇”),或環氧乙烷和環氧丙烷或聚環氧丁烷的嵌段共聚物(“聚氧化烯二醇”)。合適的聚氧乙二醇(PEG)化合物的實例可以具有下式:對于重復單元,m可以是1-11且對于較高分子量的化合物來說n可以是大約18到大約500的平均數,例如大約230,且n對于較低分子量的化合物來說可以是5到150,例如大約91。X可以是氫原子,甲基取代基、乙基取代基、羥甲基取代基、羥基端基以及它們的子集和它們的組合。Y可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、羥基端基以及它們的子集和它們的組合。對于重復單元,m可以是1到11,且對于較高分子量的化合物來說n可以是18到500,例如大約230,對于較低分子量的化合物來說n可以是5到150,例如大約91。不論是單獨的或者是組合的形式,聚氧化烯二醇化合物的數均分子量(Mn)可以為大約180到大約20000,例如大約1800到大約11000,例如大約3000到大約8000。數均(標稱)分子量表示聚合物的總重量除以聚合物中包含的分子總數。不論是單獨的或者是組合的形式,聚氧化烯二醇化合物的重均分子量(Mw)可以為大約200到大約22000,例如大約2000到大約10000,例如大約4000到大約10000。此外,分子量分布可以進一步表示為與分子量分布分析相關的本領域技術人員通常理解的相關的Z-均分子量(MZ)和Z+1均分子量(MZ+1)。Z-均分子量(MZ)可以為大約300到大約30000,例如大約3000到大約20000,例如大約5000到大約14000。Z+1均分子量(MZ+1)可以為大約400到大約40000,例如大約4000到27000,例如大約6500到大約19000。對于兩種或更多種聚氧化烯多元醇的組合組合物,該組合物可以具有大約1.1的多分散性,例如1.15到1.70或1.25到大約1.55,例如1.35到大約1.45,其通過重均分子量(MW)除以數均分子量(Mn)計算。在一個實例中,第一分子量聚氧化烯多元醇化合物可以具有5到150范圍內的n值,例如45到115,例如大約91,條件是第一數均分子量為大約2000到大約5000,例如大約4000且第一重均分子量為大約2200到大約5500,例如大約4400。第二分子量聚氧化烯多元醇化合物可以具有大約18到大約500范圍內的n值,例如大約90到大約365,例如大約230,條件是第二數均分子量為大約4000到大約18000,例如大約4000到大約16000,例如大約8000且第二重均分子量為大約4400到大約17600,例如大約8800。在一個實例中,相應的第一重均分子量和第二重均分子量的聚氧化烯多元醇化合物可以以大約45:55的摩爾比混合。共混物中n的平均值可以為大約100到大約205,例如n可以是大約150。聚氧化烯多元醇通常包含具有變化數目環氧乙烷單元和/或其他氧化烯平均單元的化合物分布,且引用的單元數目是最接近統計學平均值和分布峰值的整實數。本發明中使用的整實數指的是正整數或整數分數的數字。包含酸封端的聚氧化烯多元醇的化合物,不論是單獨的或是組合的形式,其之后可以與至少一種多胺化合物反應,例如至少一種二胺化合物,以形成本發明描述的多酰胺胺官能化的聚環氧乙烷原表面活性劑。該反應在反應容器中在縮合的條件下進行。合適的反應條件包括在大氣壓力下將起始原料加熱到大約100℃到大約250℃范圍內的溫度,并且蒸餾縮合副產物,水。與水一起,過量的多胺化合物也可以通過蒸餾除去。為了進一步改進水和任何過量的多胺化合物的去除,可以在引發反應之后可以使用真空或降低的容器壓力,例如大約10到大約200mmHg(托)。在所述二胺化合物的一個實施方案中,二胺化合物的每一個胺基是伯胺,且二胺化合物具有下式:H2N-R3-NH2(IV).R3是二價烴取代基,其選自具有2到18個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組。該二價烴取代基可以任選在其主鏈中包含一個或多個非反應性的氧和/或氮原子。在R3基團中每個結構中可以平均最多存在4個仲氮和/或叔氮原子的氮原子。在R3基團中氧原子可以以平均最多4個原子或更少存在。因此,式(IV)包括二仲胺化合物,其包括HR2N-R3-NHR2的分子式,且R2是選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組。合適的二胺的實例例如包括間亞二甲苯基二胺,1,3-二(氨基甲基)環己烷,2-甲基-1,5-戊烷二胺,1,3-戊烷二胺,乙二胺,二亞乙基三胺,三亞乙基四胺,聚環氧丙烷二胺,2,2(4),4-三甲基1,6-己烷二胺,異佛爾酮二胺,2,4(6)-甲苯二胺,1,6-己烷二胺,1,2-二氨基環己烷,對二氨基二環己基甲烷(PACM)以及它們的組合。合適的含氧胺例如包括1,10-二氨基-4,7-二氧雜癸烷,1,8-二氨基-3,6-二氧雜辛烷,1,13-二氨基-4,7,10-三氧雜十三烷以及它們的組合。根據式(IV)的胺可以按照相對于包含酸封端的聚氧化烯多元醇的化合物摩爾過量的量添加。該過量的二胺化合物可以按照與羧酸官能團的摩爾比為大約6:1到大約2:1添加,例如大約5:1到大約3:1,例如大約4:1到大約3.2:1。當使用過量的二胺化合物時,在反應之后,可以除去未反應的二胺化合物。此外,單環氧化合物可以與二胺同時引入或相繼引入以便進一步形成多酰胺胺官能化的聚氧化烯原表面活性劑。該單環氧化合物可以是本發明描述的單環氧化合物,并且例如包括C10叔羧酸的縮水甘油酯。該單環氧化合物可以按照一定的量添加,該量足以控制在后續二環氧組分添加中二環氧疏水物的量為大約5%到大約40%疏水物,以便形成環氧封端的表面活性劑和/或環氧分散體。酰胺胺組合物和任選添加的單環氧化合物之間的反應溫度沒有限制。合適的反應溫度在大約60℃到大約150℃的范圍內且在真空或降低的容器壓力下,例如大約1KPa到大約30KPa。合適的單環氧化合物包括:其中R4和R6相同或不同并且是具有2到100個碳原子的任選是支化的烯基取代基,或是支化或線型烷基、環烷基、聚氧烷基;且R5是氫原子或具有1-18個碳原子的支化或未支化的烷基。多于一種類型的R5基團連接在芳香環上。R15是具有3到20個碳原子的二價烷基或芳基取代基。該類別包括烯烴的環氧乙烷(oxirane),包括氧化丁烯,氧化環己烯,氧化苯乙烯;單價醇的縮水甘油醚,單價醇例如甲醇、乙醇、丁醇、2-乙基己醇和十二烷醇;具有至少8個碳原子的醇通過相繼將氧化烯添加到相應的烷醇(ROH)中的氧化烯加合物的縮水甘油醚,例如商品名為的那些;單價酚的縮水甘油醚,例如苯酚、甲基苯酚和在鄰位-或對位-用C1-C21支化或未支化的烷基、芳烷基、烷芳基或烷氧基取代的其他酚,例如壬基酚;單羧酸的縮水甘油酯,例如U.S.Pat.No.3,178,500中描述的辛酸的縮水甘油酯,羊蠟酸的縮水甘油酯,月桂酸的縮水甘油酯,硬脂酸的縮水甘油酯,花生酸的縮水甘油酯以及α,α-二烷基單羧酸的縮水甘油酯,該文獻由此通過參考并入本發明;不飽和醇或不飽和羧酸的縮水甘油酯,例如新癸酸的縮水甘油酯;環氧化油酸甲酯,環氧化油酸正丁酯,環氧化棕櫚酸甲酯,環氧化亞油酸乙酯以及類似物質;烯丙基縮水甘油醚和縮水甘油醛的乙縮醛。單縮水甘油基封端劑的具體實例包括烷基鏈上具有1-18個線型碳原子的烷基縮水甘油醚,例如丁基縮水甘油醚或C8-C14烷基的混合縮水甘油醚,甲苯縮水甘油醚,苯基縮水甘油醚,壬基苯基縮水甘油醚,對叔丁基苯基縮水甘油醚,2-乙基己基縮水甘油醚,新癸酸的縮水甘油醚以及它們的組合。其他合適的單環氧化物的實例包括縮水甘油化單酸和由α-烯烴和縮水甘油氧烷基烷氧基硅烷形成的環氧化物。商購的優選的單環氧樹脂的實例例如包括所有由MomentiveSpecialtyChemicalsofColumbusOhio商購可得的Modifiers62、63、64、65和116以及ResinE-10。脂肪族基單環氧化合物通常是以疏水為特征的,其傾向于改進其中單環氧化合物在低溫下使用的環氧固化劑混合物的聚結性質,并且傾向于降低膜或涂層的玻璃化轉變溫度。較低的玻璃化轉變溫度改進了固化膜的沖擊強度。芳香族基單縮水甘油基單環氧化合物可以具有使固化膜更具剛性、耐化學性和高溫下耐應力性的優點。可以使用這些類型的單環氧化合物中的任意一種,并且使用它們的組合的優點在于在最終產品中獲得溶解性、聚結性、機械強度和耐化學性的整體平衡。選擇性地,在二胺的另一個實施方案中,一個胺基是伯胺,且另一個胺基是仲胺,并且其可以具有下式:R1-HN-R3-NH2(V)。R3是二價烴取代基,其選自具有2到18個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組,并且任選在其主鏈中包含一個和多個非反應性氧原子和/或氮原子。R1是具有1到21個碳原子的支化和線型脂肪族、脂環族或芳香族二價基團,且任選在其主鏈中包含一個或多個非反應性氧或氮原子。R1和R3中的每一個可以進一步具有選自烷基基團(即甲基基團)、羥基基團、烷硫基基團以及它們的組合和它們的子集的組的端基取代基。選擇性地,R1和R3可以構成共有環。選擇性地,式(V)可以被改性以便在第二氮原子上具有第二R1基團。因此,式(V)可以包括式HR1N-R3-NHR1的二仲胺化合物,且R1是選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和它們的子集的組的取代基。合適的包含一個伯胺基團和一個仲胺基團的二胺的實例例如包括N-甲基乙二胺,N-丁基-1,6-己烷二胺,N-環己基-1,3-丙烷二胺,N-(2-氨基乙基)哌嗪,氨基乙基乙醇胺,N-甲基-1,4-環己烷二胺,N-油烯基-1,3-丙烷二胺,N-椰油烷基-1,3-丙烷二胺,N-(甲硫基)乙基-1,3-丙烷二胺,N-(線型或支化癸基)氧丙基-1,3-丙烷二胺,N-(線型或支化十三烷基)氧丙基-1,3-丙烷二胺,N-椰油烷基氧丙基-1,3-丙烷二胺,N-(辛基/癸基)氧丙基-1,3-丙烷二胺以及它們的組合。根據式(V)的胺可以按照羧酸官能團的當量與胺的摩爾數之比為大約2:1到大約1:2添加,例如大約3:2到大約2:3,例如大約3:2到大約1:1。如此,在一些實施方案仲,根據式(V)的胺可以按照相對兩種或更多種包含酸封端的聚氧化烯多元醇的化合物并不摩爾過量的量添加。優選地,不同時或之后向與式(V)的胺化合物的反應中添加單環氧化物。在胺反應和/或單環氧化物反應后可以除去任何過量的胺類物質。選擇性地,根據式(V)的胺可以按照羧酸官能團的當量與胺的摩爾數之比為大約2:1到大約1:2,例如大約3:2到大約2:3,例如大約3:2到大約1:1添加到具有一種或多種包含酸封端的聚氧化烯多元醇的化合物的組合物中。在這種替代實施方案中,單環氧化物可以與式(V)的胺化合物同時或在其之后添加到反應中。在胺反應和/或單環氧化物反應后可以除去任何過量的胺類物質。一方面,本發明涉及用于環氧樹脂水性分散體的改進的環氧官能化表面活性劑。一旦制造了酰胺胺組合物,就通過任選部分封端的酰胺胺組合物與至少一種具有每分子多于一個環氧基團的官能度的二環氧樹脂的反應制備該環氧官能化表面活性劑。該環氧官能化表面活性劑可以是當與過量的環氧官能化組合物反應時原位形成的非離子型表面活性劑。環氧組分可以是環氧樹脂,或是環氧樹脂與酚類化合物的混合物。該酰胺胺組合物的多酰胺胺官能化的聚環氧乙烷原表面活性劑與環氧組分在有效的使胺基基團與環氧基團反應的條件下接觸。環氧官能化表面活性劑可以通過在有效使胺基團與環氧基團反應的條件下,使由聚氧化烯多元醇化合物的原始共混物形成的多酰胺胺官能化的聚環氧乙烷原表面活性劑與至少一種環氧組分反應而制備,該環氧組分具有每基團多于一個環氧基團的官能度。該環氧組分可以具有與胺基基團相同的化學計量比或當量比,或者可以具有相對胺基團化學計量過量或當量過量的環氧取代基。胺與環氧化物的當量比可以是至少1:2,例如在大約1:6到大約1:500的范圍內,例如,在大約1:6到大約1:30的范圍內。該反應典型地在從環境溫度到足以使胺基團和環氧基團反應的升高的溫度下進行,例如在大氣壓力下在大約50℃到大約200℃的范圍內進行到有效制備反應產物的時間。反應的進展可以通過測量反應混合物的環氧當量重量而檢測并且其目標在于制備期望的產物。通常,加熱反應混合物直到被消耗的環氧當量等于添加的胺當量,這通常是一小時或更長。多于一種的環氧樹脂可以與多酰胺胺官能化的聚環氧乙烷原表面活性劑反應。用于制備表面活性劑的環氧組分可以是任何每分子平均具有多于一個環氧基團的具有1,2-環氧基、環氧乙烷、等價物的反應性環氧樹脂,且在一些應用中,每分子中具有大約1.5到大約6.5個環氧基團。該環氧樹脂可以是飽和的或不飽和的,線型或支化的,脂肪族、脂環族、芳香族或雜環的,并且可以帶有本質上不妨礙與羧酸的反應的取代基。這些取代基可以包括溴或氟。環氧樹脂可以是單體或聚合性的,液態或固態,但是其優選在室溫下為液態或低熔點固體。通常環氧組分包含具有變化數量的重復單元的化合物分布。合適的環氧樹脂包括通過表氯醇與包含至少1.5個芳香族羥基基團的化合物在堿性反應條件下進行反應制備的縮水甘油醚。適用于本發明的環氧樹脂的實例除了以上提及的環氧樹脂之外,還包括單環氧化物,二羥基化合物的二縮水甘油醚,環氧酚醛清漆樹脂,環狀脂肪族環氧化物,多羧酸的多縮水甘油酯,含甲基丙烯酸縮水甘油酯的丙烯酸系樹脂以及它們的組合。此外,環氧組分可以是可以與多酰胺胺官能化的聚環氧乙烷原表面活性劑反應的環氧樹脂的混合物。在一個這樣的實施方案中,環氧樹脂可以包括單環氧樹脂和二和/或多官能性環氧樹脂,優選環氧樹脂具有大約0.7到大約1.3的官能度,且環氧樹脂具有至少1.5的官能度,優選為至少1.7,更優選為大約1.8到大約2.5。該混合物可以與酰胺胺組合物分步或同時添加或與之反應。例如,酰胺胺組合物的多酰胺胺官能化的聚環氧乙烷原表面活性劑可以首先與單環氧樹脂反應并且之后與二環氧樹脂反應。在另一個實例中,環氧組分可以與酚醛清漆環氧樹脂和二環氧樹脂以任何順序分步或同時反應。如果期望的話,可以從反應混合物中回收或“原位”制備表面活性劑。為了在期望的環氧組分中原位提供表面活性劑,多酰胺胺官能化的聚環氧乙烷原表面活性劑可以反應到期望的環氧組分中。對于原位方法,環氧組分應當以足以提供未反應的環氧組分與表面活性劑加合物的量存在。該原位的方法可以包括提供其中環氧組分殘基(疏水部分)與所分散的本體環氧樹脂相同的環氧官能化的酰胺胺原表面活性劑,該環氧組分與多酰胺胺官能化的聚環氧乙烷原表面活性劑反應。當來自表面活性劑的疏水部分具有與本體環氧樹脂的IR光譜相同的IR光譜時,環氧組分(疏水部分)的殘基與本體環氧樹脂相同。當回收表面活性劑時,胺與環氧的當量比優選在大約1:30到大約1:6的范圍內。此外,為了在經升級的環氧樹脂(advancedepoxyresin)中原位提供表面活性劑,該酰胺胺組合物可以在升級反應期間與二羥基酚一起反應到二環氧樹脂的混合物中,所述二環氧樹脂例如二羥基酚的二縮水甘油醚,或者可以在升級反應之后反應到樹脂中。在升級反應中,通常二環氧樹脂和二羥基酚允許在升級反應催化劑的存在下以大約7.5:1到大約1.1:1的摩爾比反應,制備具有大約225到大約3500的環氧當量的升級環氧樹脂。基于環氧樹脂,或基于環氧樹脂和酚類化合物,典型的使用大約0.1到大約15重量%的酰胺胺組合物。優選在升級反應之后添加酰胺胺組合物,不論是否分離升級產物或其作為升級產物原樣提供。合適的二環氧樹脂可以包括二官能環氧樹脂,二環氧樹脂,例如二羥基酚的二縮水甘油醚,氫化二羥基酚的二縮水甘油醚,支化或線型脂肪族縮水甘油醚,環氧酚醛清漆樹脂或脂環族環氧樹脂。二羥基酚的二縮水甘油醚例如可以通過使表氯醇與二羥基酚在堿的存在下反應而制備。合適的二羥基酚的實例包括:2,2-二(4-羥基苯基)丙烷(雙酚-A);2,2-二(4-羥基-3-叔丁基苯基)丙烷;1,1-二(4-羥基苯基)乙烷;1,1-二(4-羥基苯基)異丁烷;二(2-羥基-1-萘基)甲烷;1,5-二羥基萘;1,1-二(4-羥基-3-烷基苯基)乙烷以及類似物質。合適的二羥基酚還可以由酚與醛例如甲醛(雙酚-F)的反應獲得。二羥基酚的縮水甘油醚包括以上二羥基酚的二縮水甘油醚與二羥基酚(例如雙酚-A)的升級產物,例如通過參考并入本發明的U.S.專利Nos.3,477,990和4,734,468中描述的那些。例如,可以通過氫化二羥基酚,之后與表氯醇在Lewis酸催化劑的存在下進行縮水甘油化反應,并且之后通過與氫氧化鈉反應形成縮水甘油醚而制備氫化二羥基酚的二縮水甘油醚。合適的二羥基酚的實例是以上列出的那些。例如,可以通過使表氯醇與支化或線型脂肪族二醇或芳香族二醇在Lewis酸催化劑的存在下反應,并且之后通過鹵代醇中間體通過與氫氧化鈉反應轉化為縮水甘油醚制備脂肪族縮水甘油醚。優選的脂肪族縮水甘油醚的實例包括對應于下式的那些:其中:p為1到12的整數,優選為1到4,q為4到24的整數,優選為4到12,并且R11可以是具有以下結構和式的二價脂環族基團:或其中R12和R13各自獨立的為具有下式的亞烴基基團,或二價芳基脂肪族基團:其中R14是亞烴基基團。術語脂肪族或脂環族包括在主鏈中或主鏈上具有氧和/或硫原子的化合物。合適的脂肪族縮水甘油醚的實例例如包括1,4-丁二醇的、環己烷二甲醇的、己二醇的、聚丙二醇的以及類似二醇的和乙二醇類的二縮水甘油醚,還包括三羥甲基乙烷和三羥甲基丙烷的三縮水甘油醚。環氧酚醛清漆樹脂可以通過甲醛和酚縮合,之后再通過與表氯醇在堿的存在下反應而縮水甘油化來制備。酚類例如可以是苯酚、甲基苯酚、壬基苯酚和叔丁基苯酚。優選的環氧酚醛清漆樹脂的實例包括對應于下式的那些:其中R7獨立地為氫原子或C1-C10的烷基基團且r為0-6的實數。環氧酚醛清漆樹脂通常包含具有不同數目的縮水甘油化酚氧亞甲基單元r的化合物分布。通常,引用的單元數目是近似于統計學平均值和分布的峰值的數字。脂環族環氧化物可以通過具有多于一個烯鍵的含環烯烴化合物用過氧化羧酸(例如過乙酸)環氧化而制備。優選的脂環族環氧化物的實例包括對應于下式的那些:或其中R10是任選含有醚基或酯基的二價脂肪族基團或者其與R9或R8一起形成任選含有雜原子的螺環,R9和R8獨立的為氫原子,或R9或R8與R10一起形成任選含有雜原子的螺環,例如氧原子。優選R10含有1到20個碳原子。脂環族環氧化物的實例例如包括:3,4-環氧基環己基甲基-(3,4-環氧基)環己烷羧酸酯;通過大約2摩爾的4-環己烯甲醛與季戊四醇縮合,之后再使雙鍵環氧化制備的二環氧基螺二乙縮醛;二(3,4-環氧基環己基甲基)己二酸酯,二(3,4-環氧基環己基)己二酸酯和乙烯基環己烯二氧化[4-(1,2-環氧基乙基)-1,2-環氧基環己烷]。脂環族環氧化物包括以下結構和式的化合物:商業的優選環氧樹脂的實例例如包括全部由MomentiveSpecialtyChemicalsofColumbusOhio商購可得的ResinsDPL-862、828、826、825、1001、1002、Resin1510、Modifiers32、62、63、64、65、67、68、71、107、116、ResinDPS155、ResinHPT1050和ResinE-10,還包括由UnionCarbide商購可得的EpoxyResinsERL-4221、-4289、-4299、-4234和-4206。如上所述的酰胺胺組合物與環氧組分的反應形成環氧官能化表面活性劑。對于原位環氧官能化表面活性劑的形成,例如在過量的環氧組分或水性分散體中,環氧官能化表面活性劑占最終組合物的大約1wt.%到大約20wt.%,例如大約1wt.%到大約10wt.%,例如大約1wt.%到大約4wt.%。在環氧官能化表面活性劑的一個實施方案中,該表面活性劑可以具有下式:且m可以是1到11,n可以是1到3,q可以是0到8,例如0到4,且p可以是大約18到大約500。X可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合。每一個Y可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合。R17可以是烷基基團、芳基基團、酰基基團以及它們的子集和它們的組合。R17可以進一步包括1到50個碳原子,并且可以進一步包括氧、氮和硫原子。環氧官能化表面活性劑可以具有下式:對于重復單元,m可以是1到3,n可以是1.2,q可以是1.9到2.3,且p可以是大約81到大約210。X可以是氫原子或甲基取代基。每一個Y可以是氫原子或甲基取代基。R17可以是叔酰基基團。環氧官能化表面活性劑可以轉化為胺官能化化合物。該胺官能化化合物可以用作用于環氧組合物的固化劑或用作表面活性劑用于胺聚合物組合物。如本發明所述的固化劑可以通過使本發明描述的環氧官能化表面活性劑與至少一種胺組分,例如本發明描述的胺組分反應而制備。所述至少一種胺組分可以包括多胺。多胺可以具有至少一個伯胺基團和至少一個仲胺基團。具有第一和第二胺基團的多胺化合物的非限定性實例通過下式表示:H2N-R16-[NH-R16]n-NH2(V),和對于重復單元,n可以是從1到10之間整數的平均數,優選為1到4之間,且R16是具有1到24個碳原子的二價支化或未支化烴基,一種或多種芳基或烷基芳基基團,或者是一種或多種脂環族基團,條件是最初的多胺化合物具有2到18個碳原子。優選地,R16是具有1到10個碳原子的低級亞烴基基團,例如2到6個碳原子。多胺化合物的實例包括亞乙基多胺,亞丁基多胺,亞丙基多胺,亞戊基多胺,亞己基多胺,亞庚基多胺和它們的組合。這些胺的高級同系物和相關的氨基烷基取代的哌嗪也包括在內。多胺的具體實例包括1,2-二氨基乙烷,三(2-氨基乙基)-胺,1,2-和1,3-二氨基丙烷,1,2-和1,4-丁烷二胺,2-甲基-1,5-戊烷二胺,1,6-己烷二胺,1,10-癸烷二胺,1,8-辛烷二胺,1,4,7-三氮雜庚烷,1,4,7,10-四氮雜癸烷,1,9,17-三氮雜十七烷,2,5,8-三甲基-1,4,7,10-四氮雜癸烷,1,4,7,10,13-五氮雜十三烷,1,4,7,10,13,16-六氮雜十六烷,1,5,9-三氮雜壬烷,1,3-和1,4-二(氨基甲基)苯,4,4’-二氨基二苯基甲烷,2,4-二氨基-1-甲基苯,2,6-二氨基-1-甲基苯,多亞甲基多苯基胺,1,2-二氨基環己烷,1-氨基-3-(氨基甲基)-3,5,5-三甲基環己烷,1,3-二(氨基甲基)環己烷,4,4'-二氨基二環己基甲烷以及它們的組合。還可以使用通過兩種或更多種以上列出的亞烴基胺的縮合獲得的高級同系物。本發明描述的固化劑可以通過使環氧官能化組分與至少一種胺組分以活性胺氫原子與環氧基團的比為2:1或更高的比例下反應制備,例如大約5:1到大約30:1,或者例如大約5:1到大約15:1,由此制備胺基封端的產物。本發明描述的固化劑具有至少50的胺氮原子當量重量,優選至少65,例如大約100到大約400。此外,胺官能化合物可以用單環氧化合物,這通過使化合物在有效使剩余的活性胺氫原子與環氧基團在分散前或分散后反應的條件下反應進行。胺封端的產物可以與單環氧化物以剩余的活性胺氫原子與環氧基團的比為3:1或更高的比例下反應以提供封端的產品,例如大約5:1到大約10:1,例如大約8:1。該反應典型的在大約50℃到大約100℃范圍內的溫度下反應有效制備反應產物的時間段。通常,加熱反應混合物直到消耗的胺當量等于環氧當量(基本上所有的環氧基團都消耗掉了)。在胺官能化合物的一個實施方案仲,胺官能化合物可以具有下式并且m可以為1到11,n可以為1到2,q可以為0到8且p可以為大約45到大約500:X可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合。每一個Y可以是氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合。R17可以是烷基基團、芳基基團、酰基基團、以及它們的子集和它們的組合。R17可以進一步包括1到50個碳原子,并且可以進一步包括氧、氮和硫原子。此外,可以將單環氧化合物與多胺同時或相繼引入以便進一步使任何待處理的取代基團反應以形成胺官能化合物。該單環氧化合物可以是本發明描述的單環氧化合物中的任一種。單環氧化物封端劑的具體實例包括在烷基鏈中具有1-18個線型碳原子的烷基縮水甘油醚,例如丁基縮水甘油醚,或C8-C14烷基的混合縮水甘油醚,甲苯縮水甘油醚,苯基縮水甘油醚,壬基苯基縮水甘油醚,對叔丁基苯基縮水甘油醚,2-乙基己基縮水甘油醚,新癸酸的縮水甘油酯以及它們的組合。此外,本發明描述的固化劑可以分散在水溶液中。該分散體可以包含水和本發明描述的固化劑。這種組合物可以通過在封端之前或封端之后在存在或不存在表面活性劑的情況下將水混合到固化劑中而提供。任何用于使固化劑乳化或分散在水溶液中的常規表面活性劑都可以使用。但是,本發明描述的固化劑使自乳化性的并且不需要任何額外的表面活性劑以提供水性固化劑溶液、乳液或分散體。胺官能化合物還指的是胺官能化加合聚合物,其可以分散在水中以獲得透明(white)分散體,該分散體具有大約0.2到大約1的平均微米直徑粒徑Dv,例如大約0.516,且Sa為大約0.2到大約0.7,例如大約0.423。這種分散體中的非揮發性物質含量可以為大約50%到大約55%,例如大約51.6%,且非揮發性胺聚合物的胺值可以為大約225到大約275,例如大約257。這種胺官能化聚合物分散體可以與本發明描述的環氧分散體聯合使用以制備高性能底漆,例如實施例18中描述的。本發明描述的固化劑可以原樣、在有機溶劑中或在水中用于固化液態或固態環氧樹脂。本發明描述的制備本發明描述的固化劑的任何環氧樹脂可以通過本發明描述的固化劑固化。該固化劑可以用于環境涂層應用以及烘烤涂層應用。固化溫度可以取決于應用而變化,其典型的在大約5℃到大約200℃的范圍內。本發明描述的這些固化劑可以有效的用于固化水性環氧樹脂體系。優選的水性環氧樹脂體系是具有350到大約10000的分子量的非離子性分散在水中且具有或不具有二醇醚共溶劑的雙酚-A基環氧樹脂。水性環氧樹脂的商購實例例如包括由MomentiveSpecialtyChemicals,Inc獲得的EPI-REZTMResin3520、3522、33540和5522。本發明描述的固化劑不使用酸性鹽也可與水性分散體相容。這些可固化體系包含水、一種或多種環氧樹脂和一種或多種本發明描述的固化劑。這些水性可固化環氧樹脂體系可以在室溫下或在升高的溫度下固化,或者其可進一步用可商購獲得的叔胺促進劑催化固化,例如2,4,6-三(二甲基氨基甲基苯酚)或其他酚類以便在較低的溫度下固化。這些材料的實例有來自MomentiveSpecialtyChemicals,Inc.的EPI-KURETMCuringAgent3253或來自RohmandHaas的DMP-30。這些低溫典型地在大約5℃到大約20℃的范圍內。對于水性環氧樹脂體系,使用或不使用促進劑的典型的固化溫度都在大約5℃到大約200℃的范圍內。這些固化劑典型的用于配制對涂布的基底具有良好腐蝕保護性的熱固性涂料。水性環氧樹脂分散體水性環氧分散體可以包括水、至少一種本發明描述的環氧樹脂以及多酰胺胺官能化的聚氧化烯原表面活性劑,該原表面活性劑用所述至少一種環氧樹脂在原位環氧官能化,并且可以在與所述分散體的所述至少一種環氧樹脂接觸之前環氧官能化。如上所述本發明形成的水性環氧樹脂分散體表現出多種高于現有技術分散體的改進的性質。一個改進的性質為通過環氧組分改進的穩定性、改進的pH值穩定性以及改進的粘度穩定性證實的改進的貨架穩定性。一方面,與未共混的現有技術中的組合物相比,對于基于本發明用于形成本發明描述的環氧樹脂分散體的表面活性劑的共混物來說,觀察到隨著時間改進的或與之相當的粘度。在一個實例中,觀察到由包含酸封端的聚氧化烯多元醇的組合物共酰胺化形成的水性環氧樹脂分散體在8個月的儲存后具有小于1700cps的粘度,10個月后小于1800cps且15個月后小于1900cps,該組合物為在以下實施例10形成的4000Mw/8000Mw分子量的聚氧乙二醇組分的55:45的共混物。相比之下,基于4000MW的聚氧乙二醇的組合物和基于4600MW的聚氧乙二醇的組合物8個月后各自表現出大于4000cps的粘度。觀察到本發明形成的水性分散體在雙倍粘度或超過6000cps的時間段,具有大約0到大約3的pH單位變化。本發明描述的實施例和表格中顯示了這些改進方案的其他實例。一方面,本發明描述的這里形成的水性環氧樹脂分散體中的平均顆粒尺寸在尺寸上小于1.50μm的數量級,例如大約0.2到大約1.2μm,例如大約0.65到大約0.87μm,并且其可以與現有技術的大約0.74到0.88的尺寸相比擬。期望在盡可能高的環氧聚合物含量的情況下使用盡可能小的顆粒尺寸以獲得改進的經濟性和改進的聚結性,由此獲得最佳膜機械性質和化學性質。在根據本發明描述的用于涂料應用的典型水性分散體中,基于總的分散體,包括環氧官能化表面活性劑的環氧樹脂組分的量(還已知為固體含量或非揮發性物質的含量)可以為大約20到大約75%重量,優選大約55到大約65%重量。通常,基于固體的重量,水和具有每分子多于0.8個環氧官能度的環氧樹脂在以上提及的環氧官能化表面活性劑以大約1wt%到大約6wt%、例如大約2wt%到大約5wt%,例如大約3.5wt%到小于4.5wt%范圍內的量存在的情況下,在有效的提供水包油乳液的條件下混合。如果本發明所述的環氧官能化表面活性劑的效率提高,則分散環氧樹脂所需要的量就降低。從環氧官能化表面活性劑中單獨確認環氧樹脂是方便的,這僅僅是因為環氧官能化表面活性劑可以在環氧樹脂中原位制備。該分散體可以通過將表面活性劑和水添加到環氧樹脂中以便分散或通過如上所述“原位”制備表面活性劑而制備。這些分散體還可以通過將環氧樹脂添加到所述酰胺胺組合物和水中而制備。該表面活性劑可以通過在有效使酰胺胺和環氧樹脂反應的溫度下將多酰胺胺官能化聚氧化烯原表面活性劑添加到環氧樹脂中、或者通過在如上所述升級反應之前或期間將多酰胺胺官能化的聚氧化烯原表面活性劑添加到雙官能化環氧樹脂和二羥基酚中而原位制備。可以使用一種或多種環氧官能化酰胺胺原表面活性劑。任選地,共表面活性劑可以與環氧官能化表面活性劑一起使用。任選地,分散體還包含丙酮。在一個實施方案中,分散體包含丙酮和至少一種非揮發性疏水液態樹脂或樹脂改性劑。丙酮可以以大約0.5wt.%或更高的量存在,更優選以大約1wt.%到大約3wt.%的量存在,例如大約1.5wt.%。有用的涂料組合物可以通過混合胺官能化的環氧樹脂固化劑與以上提及的水性環氧樹脂分散體獲得。以上描述的水性環氧樹脂分散體和固化劑可以用作應用于基底的油漆和涂料的組分,例如,金屬和水泥質結構物。為了驅動涂料組合物固化完成,還可以將可由這些分散體獲得的涂料在升高的溫度下加熱大約30到大約120分鐘,優選在大約50℃到大約120℃的范圍內。為了制備這種油漆和涂料,水性環氧樹脂分散體和/或固化劑主要與增容劑和防腐蝕顏料,以及任選與添加劑,例如表面活性劑、消泡劑、流變改性劑和防污劑(mar)以及潤滑劑(slipreagent)共混。本發明描述的環氧樹脂涂料組合物可以包括其他添加劑,例如彈性體、穩定劑、增容劑、增塑劑、顏料糊、抗氧劑、流平或增稠劑和/或共溶劑、潤濕劑、共表面活性劑、反應性稀釋劑、填料、催化劑以及它們的組合。這些顏料和添加劑的選擇和其用量取決于油漆的有意應用并且通常是本領域技術人員已知的。反應性稀釋劑可以是任何非揮發性、疏水化合物,其在室溫下是液態和可流動的,不論其是原樣或是在疏水性溶劑中,例如二甲苯或丁醇。當某物質滿足根據ASTMD2369-93或ASTMD3960-93的定義時,它就是非揮發性的。對于涂料組合物,反應性稀釋劑(還已知為疏水性液體樹脂或樹脂改性劑)必須與涂料組合物中的固化劑相容(例如不能損害耐腐蝕性,或高光澤性等),所述固化劑例如,胺固化劑。基于組分的總量,反應性稀釋劑可以以最多大約25wt.%的量存在,例如大約1到大約10wt.%。優選的反應性稀釋劑例如包括脂肪族單縮水甘油醚,脲醛樹脂或脂肪族單縮水甘油酯。優選的單環氧化物稀釋劑是包含不與水混溶的縮水甘油化C8-20脂肪族醇、C1-18烷基酚縮水甘油醚或縮水甘油化叔羧酸的那些。單環氧化物組分可以包含脂環族和芳香族結構,以及鹵素、硫、磷和其他這樣的雜原子。反應性稀釋劑例如可以是環氧化不飽和烴,例如氧化癸烯和氧化環己烯;單羥基醇的縮水甘油醚,例如2-乙基己醇、十二烷醇和二十烷醇;單羧酸例如己酸的縮水甘油酯;縮水甘油醛的乙縮醛以及類似物質。優選的反應性稀釋劑是單羥基C8-14脂肪醇的縮水甘油醚。反應性稀釋劑可以是由MomentiveSpecialtyChemicals商購獲得的HELOXYTM7Modifier(C8–C10烷基縮水甘油醚),HELOXYTM9Modifier(C10-11烷基縮水甘油醚)和由CytecIndustriesInc商購獲得的CYMELUFTM216-10Resin(烷基化脲醛高固體溶液)。水性分散體還可以包含作為反應性稀釋劑的單環氧化物稀釋劑。主要顏料的實例包括金紅石二氧化鈦,例如由DuPont獲得的2160(Kronos,Inc.)和R-960,黃二氧化鈦,紅氧化鐵,黃氧化鐵和碳黑。增容劑顏料的實例包括偏硅酸鈣,例如10ES(NYCOMinerals,Inc.),硫酸鋇,例如(HarcrosPigments,Inc.)和硅酸鋁,例如170(EnglehardCorp.)。抗腐蝕性顏料的實例包括磷硅酸鈣鍶,例如HALOXSW111(HaloxPigments),鋅離子改性的三磷酸鋁,例如84(TaycaCorp.)以及堿式磷酸鋁鋅水合物,例如ZPA(HeucoTech,Ltd.)。可以在含水環氧油漆和涂料中納入額外的表面活性劑以改進顏料和基底的潤濕。這些表面活性劑典型的是非離子型的,其實例包括X-100和TRITONX-405(DowChemicalCompany)以及104(AirProductsandChemicals)。消泡劑和除泡劑在油漆或涂料制造期間抑制泡沫的產生。有用的消泡劑包括L-475(DrewIndustrialDiv.)、PF-4Concentrate(UltraAdditives)和033(BYK-Chemie)。流變添加劑用于獲得合適的應用性能。有三種類型的添加劑提供含水環氧涂料所要求的的增稠和剪切變稀,即,羥乙基纖維素,有機改性的鋰蒙脫石粘土和締合型增稠劑。250MBR和NATROSOLPlus(Aqualon)是改性的羥乙基纖維素類的實例且LT(RHEOX,Inc.)是鋰蒙脫石粘土的代表性實例。OptifloTM(SouthernClay)是有用的締合型增稠劑。防污劑(Mar)和潤滑劑改進了早期對來自刮擦或輕足印(lightfoottraffic)的磨損的耐受性。聚二甲基硅氧烷和聚環氧乙烷蠟用于這一方面。商購獲得的蠟潤滑劑是MICHEM182(MICHELMAN,INC.)。可固化油漆和涂料組合物可以通過刷涂、噴霧或輥子應用于基底。本發明的水性分散體還可以用作粘合劑和纖維膠料的組分。對于本發明描述的原表面活性劑、表面活性劑、水性分散體和固化劑的其他用途可以包括用于固化混凝土的濕度控制膜以及用于多層金屬層合制品的稀粘合劑。此外,環氧樹脂固化劑可以是任何有效固化(或交聯)分散在水性溶液中的環氧樹脂的固化劑,包括由本發明所述環氧官能化酰胺胺原表面活性劑制備的固化劑。所述固化劑通常是可與水相容的(即可稀釋的和/或可分散的)。其他合適的與分散體一起使用的固化劑或共固化劑包括典型地與環氧樹脂一起使用的那些,例如脂肪族、芳基脂肪族和芳香族胺,多胺,酰胺胺和環氧-胺加合物,蜜胺甲醛樹脂和酚甲醛樹脂。該固化劑表現出不同水平的水相容性,這取決于用于它們的制備的起始原料的特性。優選地,為了在室溫下或更低的溫度下固化,其他合適的固化劑或共固化劑可以具有大約1:0.75到大約1:1.5的環氧當量與胺氫當量的組合比例。這些其他合適的固化劑或共固化劑可用包括聚氧化烯胺固化劑,其為可溶于水或分散在水中并且每分子中包含多于2個活性氫原子的那些,例如二亞乙基三胺,三亞乙基四胺,四乙烯基五胺等。其實例包括1,6-己烷二胺,1,3-戊烷二胺,2,2(4),4-三甲基-1,6-己烷二胺,二(3-氨基丙基)哌嗪,N-氨基乙基哌嗪,N,N’-二(3-氨基丙基)乙二胺,2,4(6)-甲苯二胺,并且還包括脂環族胺,例如1,2-二氨基環己烷,1,4-二氨基-2,5-二乙基環己烷,1,2-二氨基-4-乙基環己烷,1,2-二氨基-4-環己基環己烷,異佛爾酮二胺,降莰烷二胺,4,4'-二氨基二環己基甲烷,1,1-二(4-氨基環己基)乙烷,2,2-二(4-氨基環己基)丙烷,3,3'-二甲基-4,4'-二氨基二環己基甲烷,3-氨基-1-(4-氨基環己基)丙烷,1,3-和1,4-二(氨基甲基)環己烷,以及它們的組合。作為芳脂族胺,特別是使用的其中氨基基團存在于脂肪族基團上的那些胺,例如間-和對二亞甲苯基二胺或它們的氫化產物。所述胺可以單獨使用或作為混合物使用。對于較高溫度的固化應用,氨基塑料樹脂可以用作用于具有高當量重量的環氧樹脂的固化劑,例如高于700。基于環氧樹脂和氨基塑料樹脂的組合重量,通常使用大約5到大約40,例如大約10到大約30,例如大約30重量%的氨基塑料樹脂。合適的氨基塑料樹脂是脲和蜜胺與醛在某些情況中進一步用醇醚化的反應產物。氨基塑料樹脂組分的實例是脲、亞乙基脲、硫脲、蜜胺、苯代三聚氰胺和乙酰三聚氰胺。醛的實例包括甲醛、乙醛和丙醛。氨基塑料樹脂可以烷醇的形式使用,但是優選其以醚的形式使用,其中醚化劑是包含1到8個碳原子的單羥基醇。合適的氨基塑料樹脂的實例是羥甲基脲、二甲氧基羥甲基脲,丁基化的聚合性脲-甲醛樹脂,六(甲氧基甲基)蜜胺,甲基化的聚合性蜜胺-甲醛樹脂和丁基化的聚合性蜜胺-甲醛樹脂。水相容性固化劑的商購實例包括EPI-CURETM8540,8537、8290和6870CuringAgents(由MomentiveSpecialtyChemicals可獲得)、ANQUAMINE401、Casamid360和362固化劑(AirProducts);HardenerHZ350、Hardeners92-113和92-116(Huntsman);BECKOPOXEH659W、EH623W、VEH2133W固化劑(Cytec)和EPOTUF37-680以及37-681固化劑(ReichholdChemicalCo.)。可固化的環氧樹脂組合物可以在大約5℃到大約200℃、例如20℃到大約175℃的范圍內的溫度下固化有效使環氧樹脂固化的時間段。實施例提供以下實施例用于說明如本發明所描述的某些實施方案。這些實施例并不意在限制申請的范圍并且不應當對它們進行那樣的解釋說明。除非另有說明,量均以重量份或體積份、或重量百分數或體積百分數計。測試方法在獲得的乳液或分散體上通過來自BrookfieldEngineeringLaboratories的BrookfieldSynchroLectricViscometer測定粘度。乳液和分散體顆粒尺寸的測定使用來自BrookhavenInstrumentsCorporation和CoulterLS230的BrookhavenBi-DCPParticleSizer完成,除非另有說明。Dn是數均粒徑,Dw是重均粒徑,Dv是體積平均尺寸直徑且Sa是表面積平均粒徑。所有的顆粒尺寸數據均以微米μm計。胺值報告為與一克樣品的堿性氮含量等價的KOH的毫克數,其通過ASTMD2896測試方法的酸-堿滴定測定。酸當量重量/羧基當量重量通過ASTMD1639測試過程測定。胺的氮當量重量理論上通過使用的反應劑當量重量和每批質量平衡成批計算。環氧當量重量通過ASTMD1652和D4142的測試過程測定。組合物和用于形成組合物的方法實例如下所列。實施例1A是現有技術的組合物和氧化具有4600數均分子量和高羧基水平(U.S.專利6,956,086B2實施例1)的聚氧乙二醇(PEG)的方法的對比實施例。向具有大約1150升容量的不銹鋼反應器中加入601.5Kg的PEG4600和45.4Kg的水以形成混合物。將該混合物加熱到60℃并且攪拌以溶解PEG。然后在攪拌下將7.14Kg的67%的硝酸和21.9Kg的水的混合物和5.41Kg的4-羥基-2,2,6,6-四甲基哌啶-1-氧基自由基(4-羥基TEMPO)裝入反應器中。然后封閉反應器出口并且將氧氣加入到反應器中。添加足量的氧使反應器達到并且保持除空氣之外還有25psig(172kPa)的氧氣壓力,所述空氣在加入氧氣之前就存在于反應器中。隨著額外的氧氣從鋼瓶中通過設定在保持添加的氧氣壓力為172kPa的調節器加入,在攪拌下使反應器在57-63℃下保持4小時。此時,鋼瓶壓力降低1975psig(13.62MPa),這表明8.38Kg的氧氣從鋼瓶中轉移到反應器中。然后關閉氧氣流并且在這一溫度下隨著攪拌反應繼續額外的兩個小時以便消耗掉反應器頂部空間中的大部分氧氣。在這一時間段末,加入到反應器頂部空間中的氧氣壓力降低到9.6psig(66kPa)并且氧氣的消耗速率變得非常低。排出剩余的氧氣并且將真空應用于反應器以使絕對壓力降低至13.5kPa。反應器溫度升高到93℃以蒸餾水并且將硝酸保留在頂部集液器中。蒸餾持續1.5小時。中斷真空并且就酸當量重量對產品取樣。該酸當量重量為2382,其對應于93%的醇端基氧化。排空頂部集液器。這種氧化的PEG用于制備實施例4中對比的、現有技術水平的多酰胺胺原表面活性劑。實施例1B是具有4600數均分子量的聚氧乙二醇(PEG)的高羧基水平氧化實施例。向具有大約57升容量的不銹鋼反應器中加入30Kg的PEG4600(具有2300的羥基當量重量)和2.53Kg的水以形成混合物。將該混合物加熱到60℃并且攪拌以溶解PEG。然后在攪拌下將0.18Kg的67%的硝酸和0.45Kg的水的混合物和0.54Kg的4-羥基TEMPO裝入反應器中。然后封閉反應器出口并且從具有調節器的鋼瓶中將氧氣加入到反應器中。添加足量的氧以使反應器達到并且保持在除空氣之外還有25psig(170kPa)的氧氣壓力,所述空氣在加入氧氣之前就存在于反應器中。隨著額外的氧氣從鋼瓶中通過設定在保持添加的氧氣壓力為170kPa的調節器加入,在攪拌下反應器在60℃下保持9.5小時。定時從反應器中取樣并且對干燥的樣品通過滴定測定固體級分(其轉而可以用于計算氧化百分數)的羧基當量重量。9.5小時后取出最終的樣品并且當分析樣品時終止氧氣流入反應器中。干燥樣品的滴定顯示了2440的羧基當量重量,其對應于94.3%的羥基基團向羧基的轉化率。當獲得分析結果時,排空反應器中的氧氣并且用氮氣吹洗反應器。然后,隨著反應器壓力降低到大約1700Pa,將反應溫度升高到大約105℃。將水和其他揮發性物質從反應器中蒸餾大約3小時直到蒸餾物的餾出實質上停止。然后將反應器中殘留的熔融產物全部裝載到多個小桶中。無后干燥時產物具有2410的酸當量重量且在150℃下在空氣烘箱中干燥15分鐘的樣品具有2440的酸當量重量。后者的值對應于94.7%的羥基端基轉化為羧基的氧化程度。實施例2是具有4000數均分子量的聚氧乙二醇(PEG)的高羧基水平氧化實施例。向具有大約57升容量的不銹鋼反應器中加入30Kg的PEG4000(具有2085的羥基當量重量)和2.95Kg的水以形成混合物。將該混合物加熱到60℃并且攪拌以溶解PEG。然后在攪拌下將0.18Kg的67%的硝酸和0.45Kg的水的混合物和0.57Kg的4-羥基TEMPO(溶解于1.06Kg的水中)裝入反應器中。然后封閉反應器出口并且從具有調節器的鋼瓶中將氧氣加入到反應器中。添加足量的氧以使反應器達到并且保持在除空氣之外還有35psig(240kPa)的氧氣壓力,所述空氣在加入氧氣之前就存在于反應器中。隨著額外的氧氣從鋼瓶中通過設定在保持添加的氧氣壓力為240kPa的調節器加入,在攪拌下反應器在56-61℃下保持5.5小時。定時從反應器中取樣并且對干燥的樣品通過滴定測定固體級分(其轉而可以用于計算氧化百分數)的羧基當量重量。5.25小時后固體級分的羧基當量重量為7620,其對應于27.4%的羥基基團向羧基的轉化率。為了提高氧化速率,5.5小時后,將氧氣壓力提高到50psig(比初始空氣的壓力高345kPa)。在反應器排空并且末期終止反應之前,反應在升高的壓力下持續額外的一小時。然后,用氧氣使反應器加壓到高于大氣壓的50psig(345kPa)并且再次加熱到56-61℃,隨著攪拌,混合物在該溫度和氧氣壓力下保持額外的4.5小時。在該時間段末,取樣并且當分析樣品時終止氧氣流入反應器中。干燥樣品的滴定顯示2597的羧基當量重量,其對應于80.3%的羥基基團向羧基的轉化率。當獲得分析結果時,排空反應器中的氧氣并且用氮氣吹洗反應器。然后用水稀釋產物使固體含量為52%(抑制來自溶液的氧化PEG的結晶)并且從反應器中將其裝載于多個小桶中。最終產品的干燥樣品的滴定顯示了2416的羧基當量重量,其對應于86.3%的羥基基團向羧基的轉化率。這種氧化的PEG用于制備實施例5中描述的和實施例7中共混物中的多酰胺胺原表面活性劑。實施例3是具有8000數均分子量的聚氧乙二醇(PEG)的高羧基水平氧化實施例。向具有大約57升容量的不銹鋼反應器中加入30Kg的PEG8000(具有4057的羥基當量重量)和2.95Kg的水以形成混合物。將該混合物加熱到60℃并且攪拌以溶解PEG。然后如實施例2中那樣,在攪拌下將0.18Kg的67%的硝酸和0.45Kg的水的混合物和0.57Kg的4-羥基TEMPO(溶解于1.06Kg的水中)裝入反應器中。然后封閉反應器出口并且從具有調節器的鋼瓶中將氧氣加入到反應器中。添加足量的氧使反應器達到并且保持在除空氣之外還有50psig(345kPa)的氧氣壓力,所述空氣在加入氧氣之前就存在于反應器中。隨著額外的氧氣從鋼瓶中通過設定在保持添加的氧氣壓力為345kPa的調節器加入,在攪拌下反應器在56-61℃下保持5.8小時。定時從反應器中取樣并且對干燥的樣品通過滴定測定固體級分(其轉而可以用于計算氧化百分數)的羧基當量重量。5小時后固體級分的羧基當量重量為7114,其對應于57.0%的羥基基團向羧基的轉化率。5.8小時后,排空反應器并且在末期終止反應。然后再次將樣品取出并且固體級分的羧基當量重量為6158,其對運關于65.9%的羥基基團向羧基的轉化率。用氧氣使反應器加壓到高于大氣壓的50psig(345kPa)并且再次加熱到56-61℃,隨著攪拌,混合物在該溫度和氧氣壓力下保持額外的2小時。在該時間段末,取出樣品并且當分析樣品時終止向反應器中的氧氣流。干燥樣品的滴定顯示4995的羧基當量重量,其對應于81.2%的羥基基團向羧基的轉化率。當獲得分析結果時,排空反應器中的氧氣并且用氮氣吹洗反應器。然后用水稀釋產物使固體含量為52%(抑制來自溶液的氧化PEG的結晶)并且從反應器中將其裝載于小桶中。最終產品的干燥樣品的滴定顯示了4940的羧基當量重量,其對應于82.1%的羥基基團向羧基的轉化率。這種氧化的PEG用于制備實施例6中描述的和實施例7中共混物中的多酰胺胺原表面活性劑。實施例4A是使用實施例1A的產物作為起始原料(U.S.專利6,956,086B2實施例1)制備現有技術的酰胺胺原表面活性劑組合物的對比實施例。實施例1A中的反應器內容物之后冷卻到68℃并且之后將123.1Kg的2-甲基-1,5-戊烷二胺,DytekA裝入反應器中。在2.5小時的時間段內將反應器溫度升高到202℃,同時允許水和一些二胺的混合物在大氣壓力下蒸餾。當反應器內容物達到該溫度時,排空頂部集液器并且施用真空以除去剩余的二胺。2.75小時后,反應器壓力為3kPa,溫度為209℃并且施用氮氣鼓泡以促進過量二胺的脫除。該脫除過程在真空和氮氣中持續額外的4.5小時。中斷真空并且對反應器內容物進行取樣,其胺氮當量重量為2905。然后將反應器冷卻到104℃并且裝入317.6Kg的水以溶解產物,之后裝入38.1Kg的CARDURAE-10(C10叔羧酸的縮水甘油酯)。該混合物在93℃下攪拌1小時以使CARDURAE-10的環氧基團與氧化的聚氧乙二醇-二胺縮合物的氨基基團反應。測量固體含量并且加入額外的63.5Kg的水以便將固體濃度調節到期望的水平。將該產物裝載到多個圓桶中。最終產物具有63.7%的固體濃度和3281(固體基)的胺氮當量重量。產物(聚氧乙二醇校準物)的凝膠滲透(尺寸排除法)色譜顯示在4590(64.2%的面積)的峰值分子量處的低分子量峰和9268(31.4%的面積,可能對應于偶合物質)的峰值分子量處的高分子量峰。剩余的面積對應于幾百的分子量的物質。在低分子量區域中沒有觀察到特別加寬到對應于鏈斷裂的較低分子量峰。總的混合物具有3758的數均分子量和6260的重均分子量。基于GPC,存在于酰胺胺組合物中的低聚酰胺胺化合物的百分數為31.4%。多分散性,即PD,相對較低且Mz相對較低(表II)。該實施例4A的原表面活性劑在用于表III、IV和V中實施例8顯示的低粘度和小顆粒尺寸的環氧分散體表面活性劑中有效地原位形成。但是,還顯示出來的是粘度、環氧當量重量和顆粒直徑隨著時間顯著的增加,由此按照管理提供了非常有限的少于1年的貨架期。實施例4B是實施例1的產物與烷基化1,3-丙烷二胺縮合物以及它們的水性溶液的制備實施例。1升的4口圓底燒瓶裝備有漿式攪拌器、熱電偶、蒸餾輸出口(distillationtakeoff)和真空輸出口(vacuumtakeoff)/氮氣吹洗口。向該燒瓶中裝入表I所示量的實施例1B的產物(脫除揮發性物質之后)以及以下胺:A,N-油酰基三亞甲基二胺,B,N-(支化的十三烷基氧丙基)三亞甲基二胺和C,N((辛基/癸基)氧丙基)三亞甲基二胺混合物。用氮氣吹洗燒瓶并且通過溫度控制器以最大加熱速率將其加熱到215℃。隨著酰胺形成反應的進行,從反應混合物中蒸餾出水。該反應混合物在大氣壓力下的氮氣中在保持215℃1小時并且之后在這一溫度下在80-160Pa的降低的壓力下保持30分鐘(混合物3和5的情況中為1小時)。然后,反應混合物冷卻到大約90℃并且加入水以制備水溶液。測定水溶液的固體百分數和25℃下的Brookfield粘度。還通過凝膠滲透色譜(GPC)分析該溶液。每種溶液的GPC色譜都顯示了三個峰,其具有MW1000-1200處的低分子量峰、MW4300-4600處的中等分子量峰(與起始原料的主要峰位置接近)和MW9300-9800處的高分子量峰。計算每個峰的面積百分數并且將其顯示于下表I中。表I從表I中可以看到醚二胺B和C制備的縮合物在水溶液中的粘度遠低于長鏈烴二胺A。實施例5是使用實施例2的產物作為起始原料制備酰胺胺原表面活性劑組合物的實施例。向裝備有攪拌器、加熱套、氮氣噴嘴和真空體系的樹脂燒瓶中加入1200.0克來自實施例2的水性羧基化的聚氧環氧乙烷。通過在91℃下真空蒸餾從實施例2的產物中除去水。然后將154.8克2-甲基-1,5-戊烷二胺(InvistaDytekA)加入燒瓶中。給燒瓶加裝冷凝器以允許內容物回流。用氮氣溫和鼓泡的同時,將混合物加熱到181℃并且在該溫度下在回流下混合45分鐘。施用真空和加熱以便蒸餾出過量的DytekA和水以形成酰胺胺化的反應物。在181℃和6.7KPa的絕對壓力下1小時后,羧基化聚氧環氧乙烷和DytekA形成的酰胺胺具有2639的胺氮當量重量。然后在164℃下用濕氮氣的蒸汽鼓泡該批物料以便將胺氮當量重量提高到2865。該批物料冷卻到110℃后,加入去離子水以便將固體降低到70%。在62℃下向該批物料中加入34克CARDURAE-10(C10叔羧酸的縮水甘油酯)。允許該批物料在55-62℃下混合2小時之后,將其稀釋到64.8%的固體并且允許其在室溫下放置過夜。水性封端酰胺胺聚合物的胺氮當量重量測定為3079。這種原表面活性劑之后用于制備如實施例11所示的環氧分散體,觀察到該分散體具有低的粘度。這種原表面活性劑還與來自實施例6的原表面活性劑一起使用以制備實施例9的環氧分散體,與對比的“現有技術水平”的實施例8相比,該環氧分散體表現出更好的貨架穩定性。實施例6是使用實施例3作為起始原料制備酰胺胺原表面活性劑組合物的實施例。向裝備有攪拌器、加熱套、氮氣噴嘴和真空體系的樹脂燒瓶中加入1500.0克來自實施例3的產物的水性羧基化的聚氧環氧乙烷。通過在91℃下真空蒸餾從實施例3的產物中除去水。然后將95.3克2-甲基-1,5-戊烷二胺(InvistaDytekA)加入燒瓶中。給燒瓶加裝冷凝器以允許內容物回流。用氮氣溫和鼓泡的同時,將混合物加熱到179℃并且在該溫度下在回流條件下混合45分鐘。施用真空和加熱以便蒸餾出過量的DytekA和水以形成酰胺胺化的反應試劑。在181℃和5.1KPa的絕對壓力下2.5小時后,羧基化聚氧環氧乙烷和DytekA形成的酰胺胺具有2111的胺氮當量重量。3小時的連續真空蒸餾后胺氮當量重量為4531。然后155℃下用濕氮氣蒸汽鼓泡該批物料以便將胺氮當量重量提高到5013。該批物料冷卻到110℃后,加入去離子水以便將固體降低到70%。在66℃下向該批物料中加入34克CARDURAE-10(C10叔羧酸的縮水甘油酯)。允許該批物料在55-62℃下混合2小時之后,將其稀釋到64.2%的固體并且允許其在室溫下放置過夜。水性封端酰胺胺聚合物的胺氮當量重量測定為5333。這種原表面活性劑之后用于制備如實施例12所示的環氧分散體,該分散體具有低的pH值和出眾的環氧內容物穩定性。這種原表面活性劑還與來自實施例5的分散體共混以形成實施例9的環氧分散體,與對比的“現有技術水平”的實施例8相比,該環氧分散體表現出改進的貨架穩定性。實施例7是使用實施例2和實施例3的產物的共混物作為起始原料的優選本發明酰胺胺原表面活性劑組合物的制備實施例。向裝備有加熱套、攪拌器、氮氣噴嘴以及真空體系的不銹鋼反應器中加入2312.1克來自實施例2的產物水性羧基化聚氧環氧乙烷和1890.0克來自實施例3的產物。通過在最高105℃和6.7KPa的絕對壓力下真空蒸餾從共混物中除去水。然后將418.1克2-甲基-1,5-戊烷二胺(InvistaDytekA)加入該批物料中。給燒瓶加裝冷凝器以允許內容物回流。用氮氣溫和鼓泡的同時,將混合物加熱到184℃并且在該溫度下在回流條件下混合45分鐘。施用真空和加熱以便蒸餾出過量的DytekA和水以形成酰胺胺化的反應物。在171-188℃和13.5到5.1KPa的絕對壓力下4小時后,羧基化聚氧環氧乙烷和DytekA形成的酰胺胺具有3811的胺氮當量重量。該批物料冷卻到110℃后,加入去離子水以便將固體降低到70%。然后在66℃下向該批物料中加入97.3克CARDURAE-10(C10叔羧酸的縮水甘油酯)。允許該批物料在70-83℃下混合2小時之后,將其稀釋到64.3%的固體。封端酰胺胺聚合物的胺氮當量重量測定為3750。正如所預期的PEG4000和PEG8000的共混物的分子量分散性DP,該實施例7氧化的相應PEG酸的共酰胺化共混物還具有1.2895的分子量分散性,這與表II所示實施例5和6的酰胺胺化原表面活性劑的相關共混物的分子量分布相比基本相同但是略低。這種共酰胺化原表面活性劑還用于制備本發明優選的實施例9的環氧分散體,該分散體具有比現有技術水平的實施例更低的粘度、更小的顆粒尺寸和出眾的貨架穩定性。實施例8是對比水性分散體實施例,其包括由根據實施例4中描述的方法制備的原表面活性劑制備的環氧分散體的制備。向裝備有加熱套、攪拌器、氮氣噴嘴、真空體系以及冷凝器的樹脂燒瓶中加入936.9克EPONResin828和285.1克雙酚A。該批物料在攪拌下加熱到115℃。然后向混合物中加入1.0克三苯基膦。該批物料在攪拌下加熱到132℃并且允許其放熱溫升到高至190℃。然后該批物料在177℃-190℃下保持45分鐘。之后將該批物料冷卻到132℃并且加入99.3克丙二醇單甲醚。該批物料回流并且冷卻到105℃。然后在大氣壓力下加入84.1克實施例4A的具有28.7%的高分子量內容物、3164當量重量的氮和64.7%的固體的原表面活性劑。允許該批物料在攪拌下在2.5小時內冷卻到99℃。此時在5分鐘內快速將285.5克去離子水加入到物料中。該批物料在減壓下冷卻到77℃,此時環氧聚合物和原表面活性劑環氧化物形成水包樹脂分散體。分散體在44.0KPa的絕對壓力下混合額外的1.0小時后(將該批物料冷卻到55℃),平均Sa直徑(表面積)顆粒尺寸為0.723微米且平均Dv(體積)顆粒尺寸為1.105微米。此時此時,使該批物料返回到大氣壓力并且向該批物料中加入13.0克丙酮。該批物料在55℃下混合15分鐘并且加入1.26克50%的十二烷基苯磺酸的聚丙二醇溶液以控制pH值接近中性。該批物料混合額外的15分鐘并且之后在10分鐘內加入26.0克HELOXYModifier8。該批物料混合20分鐘并且之后允許其冷卻過夜。第二天早上在攪拌下在1.0小時內將該批物料加熱到52℃。在2小時內用去離子水稀釋該批物料,期間允許溫度冷卻到48℃。該批物料混合額外的20分鐘并且之后通過80目過濾袋過濾。樣品進一步稀釋為55.9%、54.2%和51.8%的非揮發性物質。在稀釋同一天測量該批物料的BrookfieldRVDVI,分別為16,740cP、7,040cP和2,600cP,用錠子5并且在25℃下以20rmp測量。在相應的%固體下初始粘度(在49℃下1天之后/在25℃下2天之后,以允許排氣和pH值平衡)為15,560cP、5,900cP和1,620cP。這種最終環氧分散體的顆粒尺寸為Sa0.743和Dv0.942微米。樹脂內容物的環氧化物當量重量為504。在49℃下1天之后加上25℃下1天后該批物料的pH值為8.60。如表III所示,這種使用實施例4、標準原表面活性劑制備的環氧分散體具有低的顆粒尺寸、中等初始粘度和相對高的pH值。如表III所示該高的pH值促成了相對快的粘度和環氧當量重量增加的速率。因此粘度和環氧當量重量的增加將現有技術的貨架期限制為小于1年。實施例8的現有技術水平的表面活性劑的結構為以下結構,其中n為1.2。實施例9A是由55:45比例的實施例5和實施例6相應的原表面活性劑的共混物制備的環氧分散體的制備實施例。向裝備有加熱套、攪拌器、氮氣噴嘴、真空體系以及冷凝器的樹脂燒瓶中加入933.3克EPONResin828和289.5克雙酚A。該批物料在攪拌下加熱到105℃。然后向混合物中加入1.0克三苯基膦。該批物料在攪拌下加熱到132℃并且允許其放熱溫升到高至190℃。然后該批物料在177℃-190℃下保持45分鐘。之后將該批物料冷卻到136℃并且加入99.3克丙二醇單甲醚。該批物料回流并且冷卻到105℃。然后在大氣壓力下加入47.0克實施例5和38.4克實施例6(總計85.4g)的物質。允許該批物料在攪拌下在2.0小時內冷卻到99℃。此時,在5分鐘內快速將284.6克去離子水加入到該批物料中。該批物料在減壓下冷卻到77℃,此時環氧聚合物和原表面活性劑環氧化物形成水包樹脂分散體。分散體在44.0KPa的絕對壓力下混合額外的2.5小時后(將該批物料冷卻到55℃),平均Sa直徑(表面積)顆粒尺寸為0.875微米且平均Dv(體積)顆粒尺寸為1.345微米。此時,使該批物料返回到大氣壓力并且向物料中加入13.0克丙酮。該批物料在55℃下混合5分鐘并且加入1.26克50%的十二烷基苯磺酸的聚丙二醇溶液以控制pH值接近中性。該批物料混合額外的10分鐘并且之后在10分鐘內加入26.0克HELOXYModifier8。該批物料混合20分鐘并且之后允許其冷卻過夜。第二天早上在攪拌下在1.0小時內將該批物料加熱到54℃。在2小時內用去離子水稀釋該批物料,期間允許溫度冷卻到50℃。該批物料混合額外的20分鐘并且之后通過80目過濾袋過濾。樣品進一步稀釋為55.7%、54.5%和52.1%的非揮發性物質。25℃下其新鮮的粘度(如實施例8所測量)分別為15,160cP、6,160cP和2,020cP。測量的初始粘度(在49℃下1天之后/在25℃下2天之后)分別為15,960cP、5,100cP和1,480cP。這種最終環氧分散體的顆粒尺寸為Sa0.862和Dv1.428微米。樹脂內容物的環氧化物當量重量為506。在49℃下1天之后該批物料的pH值加上25℃下2天后的pH值為8.18。如表III所示,這種分散體的pH值導致粘度和環氧當量重量增加的速率比標準分散體實施例8更高。如表III所示,這種分散體還具有比實施例11和12二者都更好的貨架穩定性。實施例9B是由85:15比例的實施例5和實施例6各自的原表面活性劑的共混物制備的環氧分散體的制備實施例。這種環氧分散體通過以下與實施例9A相同的過程制備,除了將實施例5和實施例6的活性原表面活性劑的重量比分別調整為85:15。實施例9C是由70:30比例的實施例5和實施例6各自的原表面活性劑的共混物制備的環氧分散體的制備實施例。這種環氧分散體通過以下與實施例9A相同的過程制備,除了將實施例5和實施例6的活性原表面活性劑的重量比分別調整為70:30。實施例10是由根據本發明的實施例7的原表面活性劑產物制備的環氧分散體的制備實施例。向裝備有加熱套、攪拌器、氮氣噴嘴、真空體系以及冷凝器的樹脂燒瓶中加入936.8克EPONResin828、285.6克雙酚A和1.0克三苯基膦。該批物料在攪拌下加熱到105℃。然后向混合物中加入1.0克三苯基膦。用氮氣吹洗燒瓶并且使其壓力降低為20.3KPa的絕對壓力。該批物料在攪拌下加熱到130℃并且允許其放熱溫升到高至194℃。允許在大氣壓力下將該批物料在1.5小時內冷卻到176℃。將該批物料冷卻到151℃并且加入99.3克丙二醇單甲醚。在減壓下使該批物料回流冷卻到99℃。然后在大氣壓力下加入84.6克實施例7的物質。允許該批物料在攪拌下在1.8小時內冷卻到96℃。此時在2分鐘內將285.1克去離子水加入到該批物料中。該批物料在減壓下冷卻到75℃,此時,環氧聚合物和原表面活性劑環氧化物形成水包樹脂分散體。分散體在37.3KPa的絕對壓力下混合額外的1.5小時后(將該批物料冷卻到57℃),平均Sa直徑(表面積)顆粒尺寸為0.869微米且平均Dv(體積)顆粒尺寸為1.274微米。此時,使該批物料返回到大氣壓力并且向物料中加入13.0克丙酮。該批物料在57℃下混合20分鐘并且加入1.26克50%的十二烷基苯磺酸的聚丙二醇溶液以控制pH值接近中性。該批物料混合額外的20分鐘并且之后在10分鐘內加入26.0克HELOXYModifier8。該批物料混合1小時并且之后允許其冷卻到22℃過夜。第二天早上在攪拌下在1.25小時內將該批物料加熱到53℃。然后在2小時內將該批物料稀釋為52.2%的固體,期間允許溫度冷卻到32℃。該批物料混合額外的20分鐘并且之后通過80目過濾袋過濾。這種最終環氧分散體的顆粒尺寸為Sa0.680和Dv1.013微米。25℃下新鮮的物料粘度為1,200cP。樹脂內容物的環氧化物當量重量為513。在49℃下1天之后加上25℃下3天后該批物料的pH值為7.47且根據實施例8的描述在25℃下測量的粘度為1,060cP。如表IV所示,與“現有技術水平”的實施例8以及所有其他實施例相比,這種分散體具有最低的初始粘度分布。這種分散體在顆粒尺寸方面也最低并且在較低的pH下具有出色的環氧當量重量穩定性(參見表III)。據信實施例10的表面活性劑具有以下結構:其中R是C9支化的烷基鏈,n為0、1或2,且通過55份X=81+45份X=192獲得x=131的平均值。實施例11是由根據實施例5描述的方法制備的原表面活性劑制備的環氧分散體的制備對比實施例。向裝備有加熱套、攪拌器、氮氣噴嘴、真空體系以及冷凝器的樹脂燒瓶中加入933.7克EPONResin828和288.2克雙酚A。該批物料在攪拌下加熱到110℃。然后向混合物中加入1.0克三苯基膦。該批物料在攪拌下加熱到132℃并且允許其放熱溫升到高至190℃。然后該批物料在177℃-190℃下保持45分鐘。之后將該批物料冷卻到133℃并且加入99.3克丙二醇單甲醚。該批物料回流并且冷卻到105℃。然后在大氣壓力下加入84.0克通過實施例5的方法制備的物料,但是該物料具有30.8%的高分子量峰含量,2863當量重量的可滴定氮和64.8%的固體。允許該批物料在攪拌下在2.25小時內冷卻到98℃。此時,在10分鐘內快速將285.5克去離子水加入到物料中。該批物料在減壓下冷卻到74℃,此時,環氧聚合物和原表面活性劑環氧化物形成水包樹脂分散體。分散體在44.0KPa的絕對壓力下混合額外的2.0小時后(將該批物料冷卻到55℃),平均Sa直徑(表面積)顆粒尺寸為0.875微米且平均Dv(體積)顆粒尺寸為1.324微米。此時,使該批物料返回到大氣壓力并且向物料中加入13.0克丙酮。該批物料在54℃下混合20分鐘并且加入1.3克50%的十二烷基苯磺酸的聚丙二醇溶液以控制pH值。該批物料混合額外的20分鐘并且之后在10分鐘內加入26.0克HELOXYModifier8。該批物料在54℃下混合35分鐘并且之后允許其在不混合下冷卻到22℃過夜。第二天早上在攪拌下在1小時內非常緩慢地將該批物料加熱到53℃。在1.5小時內用去離子水稀釋該批物料,期間允許溫度冷卻到50℃。該批物料混合額外的20分鐘并且之后通過80目過濾袋過濾。樣品進一步稀釋為55.9%、53.8%和52.0%的非揮發性物質。在稀釋同一天測量該批物料的BrookfieldRVDVI,分別為27,600cP、9,560cP和1,700cP,用錠子5(或者對于高于20,000cP使用錠子6)并且在25℃下以20rmp測量。在相應的%固體下,初始粘度(在49℃下1天之后加上在25℃下2天之后以允許排氣和pH值平衡)為22,200cP、5,980cP和920cP。這種最終環氧分散體的顆粒尺寸為Sa0.835和Dv1.301微米。樹脂內容物的環氧化物當量重量為518。在49℃下1天之后加上25℃下1天后該批物料的pH值為8.79。該分散體具有低的初始粘度,但是粘度和環氧當量重量二者均不穩定并且pH值高。如表IV所示,這種分散體在25℃下儲存期間其粘度增加比實施例8更快。實施例12是由根據實施例6描述的方法制備的原表面活性劑制備的環氧分散體的制備對比實施例。向裝備有加熱套、攪拌器、氮氣噴嘴、真空體系以及冷凝器的樹脂燒瓶中加入933.5克EPONResin828和288.9克雙酚A。該批物料在攪拌下加熱到104℃。然后向混合物中加入1.0克三苯基膦。該批物料在攪拌下加熱到132℃并且允許其放熱溫升到高至190℃。然后該批物料在177℃-190℃下保持55分鐘。之后將該批物料冷卻到141℃并且加入99.3克丙二醇單甲醚。該批物料回流并且冷卻到109℃。然后在大氣壓力下加入84.7克通過實施例6的方法制備的物料,但是該物料具有27.14%的高分子量峰含量,2863當量重量的可滴定氮和64.2%的固體。允許該批物料在攪拌下在2.0小時內冷卻到98℃。此時在10分鐘內快速將285.0克去離子水加入到物料中。該批物料在減壓下冷卻到74℃,此時環氧聚合物和原表面活性劑環氧化物在水分散體中形成樹脂。分散體在33.8KPa的絕對壓力下混合額外的1.0小時后(將該批物料冷卻到54℃),平均Sa直徑(表面積)顆粒尺寸為0.836微米且平均Dv(體積)顆粒尺寸為1.329微米。此時,使該批物料返回到大氣壓力并且向物料中加入13.0克丙酮。該批物料在54℃下混合20分鐘并且加入1.3克50%的十二烷基苯磺酸的聚丙二醇溶液以控制pH值接近中性。該批物料混合額外的20分鐘并且之后在10分鐘內加入26.0克HELOXYModifier8。該批物料混合1小時。在1.5小時內用去離子水稀釋該批物料,期間允許溫度冷卻到50℃。該批物料混合額外的20分鐘并且之后通過80目過濾袋過濾。樣品進一步稀釋為55.8%、53.9%和52.0%的非揮發性物質。在稀釋的同一天測量該批物料的BrookfieldRVDVI,分別為8,060cP、3,660cP和1,680cP,用錠子5并且在25℃下以20rmp測量。在相應的%固體下初始粘度(在49℃下1天之后加上在25℃下2天之后,以允許排氣和pH值平衡)為15,920cP、3,360cP和1,460cP。這種最終環氧分散體的顆粒尺寸為Sa0.873和Dv1.288微米。樹脂內容物的環氧化物當量重量為513。在49℃下1天之后加上25℃下1天后該批物料的pH值為7.29。雖然這種分散體的pH值和環氧當量重量穩定性是優選的,但是其顆粒尺寸較大且初始粘度比優選的實施例10的初始粘度高(如表III和IV所示)。實施例13是55比45比例的分別為4000和8000數均分子量的優選聚氧乙二醇(PEG)的共混物共氧化為羧酸官能化表面活性劑的實施例。55比45比例(20.0磅的PEG4000二醇和16.36磅的PEG8000二醇)的分別為4000和8000數均分子量的優選聚氧乙二醇(PEG)共混物根據與實施例3(還根據U.S.專利6956086B2實施例1概括的方法)相同的方法作為水溶液一起氧化為相應的二羧酸。重復該方法并且共混兩種產物。得到的PEG酸在水中調整為59.4%的濃度且PEG羧酸酯的每酸的平均重量測量為3629。實施例14是使用實施例13作為起始原料的酰胺胺原表面活性劑組合物的制備實施例。實施例13的反應器內容物通過與實施例5相同的方法轉化為酰胺胺原表面活性劑。得到的原表面活性劑具有4065的胺氮當量重量和63.0%的非揮發性物質含量。實施例15是通過實施例8描述的方法使用實施例14的原表面活性劑制備環氧分散體的實施例。由來自實施例14的原表面活性劑得到的環氧分散體具有Dv0.870、Sa0.623、Dn0.633和Dw0.814的典型的平均微米直徑顆粒尺寸分布。分散的聚合物的環氧當量重量為507,52.2%NV下的粘度為1,220cP。這種環氧分散體與實施例16的胺固化劑一起使用以制備實施例17描述的高性能底漆。實施例16是使用來自實施例14的原表面活性劑制備胺固化劑分散體的實施例。以與U.S.專利6,277,928實施例1描述的過程類似的方式,使實施例14的原表面活性劑與過量的EPON1001X75溶液以5.5比94.5的比例反應。這種環氧原表面活性劑加合物后續與相對每當量環氧化物為3摩爾的三亞乙基四胺(TETA)反應。反應完成后,包括2摩爾過量TETA的揮發性物質通過真空蒸餾除去。然后相對每當量未反應的伯胺加入1摩爾單環氧Heloxy62。然后將這種胺官能化的加合聚合物分散在水中以獲得具有Dv0.516和Sa0.423的平均微米尺寸直徑的白色分散體。這種分散體的非揮發性物質的含量為51.6%且非揮發性聚合物的胺值為257。這種胺官能化的聚合物分散體用于與實施例15的環氧分散體組合以制備實施例18中描述的高性能底漆。實施例15的環氧化物與實施例16的胺分散體的MomentiveSF1700油漆性能包括1.25小時的HoursThruDry、7天的H鉛筆SF1700(7DayPencilSF1700ofH)和初始時以及3小時復查的SF1700KU粘度分別為71和68。相比之下,衍生自實施例4的原表面活性劑的商購標準環氧化物和胺分散體包括1.75小時的HoursThruDrySF1700、7天的H鉛筆SF1700和初始時以及3小時復查的SF1700KU粘度分別為54和78。實施例17是使用來自實施例7的原表面活性劑制備胺固化劑分散體的實施例。以與U.S.專利6,277,928實施例1描述的過程類似的方式,使實施例7的原表面活性劑與過量的EPON1001X75溶液以5.5比94.5的比例反應。這種環氧原表面活性劑加合物后續與相對每當量環氧化物為3摩爾的三亞乙基四胺(TETA)反應。反應完成后,包括2摩爾過量TETA的揮發性物質通過真空蒸餾除去。然后相對每當量未反應的伯胺加入1摩爾單環氧Heloxy62。然后將這種胺官能化的加合聚合物分散在水中以獲得具有Dv0.331和Sa0.219的平均微米尺寸直徑的白色分散體。這種分散體的非揮發性物質的含量為51.2%且非揮發性聚合物的胺值為242。這種胺官能化的聚合物分散體用于與實施例15的環氧分散體組合以制備實施例18中描述的高性能底漆。實施例15的環氧化物與實施例17的胺分散體的MomentiveSF1700油漆性能包括1.5小時的HoursThruDry、7天的F+鉛筆SF1700和初始時以及3小時復查的SF1700KU粘度分別為68和73。相比之下,衍生自實施例4的原表面活性劑的商購標準環氧化物和胺分散體包括1.75小時的HoursThruDrySF1700、7天的F+鉛筆SF1700和初始時以及3小時復查的SF1700KU粘度分別為54和78。實施例18是由使用以上通過如下的MomentivepublishedStartingPointFormulation1700定義的酰胺胺原表面活性劑制備的分散體制備底漆的實施例。使300.0lbs(33.33加侖)的本發明實施例15中描述的環氧樹脂分散體與26.0lbs(2.95加侖)的PPH丙二醇苯基醚(來自DowChemical)、3.0lbs(0.35加侖)的2526Defoamer(來自CIBASpecialtyCompany)、100.0lbs(3.1加侖)的R-960顏料(來自DuPont)、100.0lbs(4.12加侖)的10ES(來自NYCOMinerals,Inc.)、67.0lbs(1.83加侖)的SparmiteTMA重晶石(來自ElementisPigmentsInc.)、94.7lbs(3.98加侖)的SW-111(來自HALOXPigments)、7.0lbs(0.3加侖)的濕研磨云母、325目(來自FranklinIndustrialMinerals)混合。對該混合物進行高速分散處理以提供5-6HegmanScale的紋理結構。降低混合速度并且之后加入107.5lbs(11.94加侖)的EPI-REZResin6520-WH-53(來自MomentiveSpecialtyChemicals)、8.6lbs(0.98加侖)的CoatOSilTM1770Silane(來自MomentivePerformanceProducts)和142.9(7.12加侖)的水。此時,混合物包括959.7lbs(80.0加侖)的組合物。向該混合物中加入180lbs(19.78加侖)的如本發明描述的實施例17的固化劑分散體(來自MomentiveSpecialtyChemicals)和2lbs(0.22加侖)的Raybo60閃光防銹劑(來自Raybo)。該組合物調制為1:1的化學計量比(環氧基:胺當量)。實施例19是使用實施例7的原表面活性劑制備用于纖維膠料的環氧分散體的實施例。來自實施例7的原表面活性劑還可以用于制備用于包含減少的或不包含揮發性溶劑的纖維膠料或粘合劑的含水的環氧分散體。向3升的樹脂罐中加入99.7克液態環氧樹脂,EPON828,102.5克水性實施例7的原表面活性劑和200.0克去離子水。向燒瓶加裝攪拌器和冷水冷凝器以便在混合時物料加熱到180°F期間其包含水蒸氣。允許該批物料在這一溫度下反應1小時并且之后加入1.6克十二烷基苯磺酸。然后進一步用200.0克水稀釋該批物料,允許其冷卻到160°F并且之后在良好的攪拌下在30分鐘內加入1141.8克額外的EPON828。允許在攪拌下在2小時內該批物料冷卻到125°F,并且之后加入140.0克水,然后再加入0.7克Rhodaline640消泡劑。該批物料在125-132°F下混合1.5小時并且之后加入具有11.0克OptifloH600VF的額外的340.3克水將其稀釋到54.2%NV。允許該批物料緩慢的冷卻到25℃之后,16小時內不攪拌,通過Brookfield錠子5在20rpm下測量的粘度為1,240cP。顆粒尺寸為Dv0.71,分散的聚合物的環氧當量重量為197.2且其pH值為6.9。實施例20是使用實施例13和異癸基氧丙基-1,3-二氨基丙烷作為起始原料制備酰胺胺原表面活性劑組合物的實施例。向裝備有攪拌器、加熱套、氮氣噴嘴和真空體系的3升4口燒瓶中加入2195.1克來自實施例13的水性、羧基化的聚氧化環氧乙烷。通過在91℃下真空蒸餾從實施例13的產物中除去水。然后將118.2克異癸基氧丙基-1,3-二氨基丙烷加入燒瓶中。用氮氣吹洗燒瓶并且通過溫度控制器設定的最大加熱速率將其加熱到215℃。隨著酰胺形成反應的進行,從反應混合物中蒸餾出水。反應混合物在氮氣中在165℃下保持3小時與二甲苯共沸并且捕獲酰胺化收集的水。在減壓下在強的氮氣鼓泡下除去二甲苯和剩余的水。然后將酰胺胺產物冷卻到90℃并且用水稀釋為65.0%的固體。水性封端的酰胺胺聚合物(固體基)的胺氮當量重量測定為3610。這種原表面活性劑用于制備實施例21的低粘度環氧分散體。實施例21是通過實施例10描述的方法使用實施例20的原表面活性劑制備環氧分散體的實施例。通過與實施例10相同的過程制備分散體,但是使用實施例20的原表面活性劑。使用實施例20的原表面活性劑得到的環氧分散體具有Dv1.39、Dn0.84和Dw1.14典型的平均微米直徑顆粒尺寸分布。該分散的聚合物的環氧當量重量為529,53.75%NV下的粘度為360cps且25℃下24小時后分散體的pH值為8.38。下表II顯示了現有技術的方法和組合物與本發明所描述的組合物和方法之間的對比實施例的酰胺胺原表面活性劑的分子量。表II從表II中看到,來自PEG共混物的酰胺胺化或共酰胺化原表面活性劑的多分散性(PD)比來自單獨的PEG化合物的原表面活性劑的多分散性要高。下表III顯示了現有技術的組合物、分散體1、2和3以及本發明描述的組合物、分散體4、5和6之間的對比實施例。環氧分散體的顆粒尺寸越小,pH值越低,環氧分散體5和6的粘度就越低且貨架期就越長,其最優選用于商業應用。表III最終的pH是在室溫下在該時間點對雙(或者超過6,000cP)粘度的pH值測量。下表IV提供了25℃下粘度穩定性與較長貨架期的對比。如下所示,與現有技術的環氧分散體(實施例8、11和12)相比,由共混物(實施例9和10)制備的環氧分散體提供了等同或改進的粘度結果。以下粘度測量以厘泊(cP或cps)計。表IV*25℃下20rmp的Brookfield粘度,錠子5下表V詳細提供了根據本發明描述的組合物和現有技術的組合物之間在25℃下15個月后環氧當量重量穩定性的對比。本發明最優選的實施例10和優選的實施例9具有比現有技術水平的對照實施例8和11更顯著改進的環氧當量重量穩定性。表V下表VI和圖1所示的圖形詳細的提供了如本發明所描述的組合物和現有技術的組合物之間顆粒尺寸穩定性的對比。表VI說明根據本發明所描述的實施例9和10的顆粒尺寸穩定性高于實施例8中顯示的現有技術發展水平對照。圖1顯示了對比根據本發明實施例9A、9B和9C和對比現有技術的實施例11和12形成的環氧分散體在25℃下環氧分散體的顆粒尺寸穩定性圖形。表VIDv變化對照實施例8共混物實施例9共酰胺化實施例10Δ0.6130.4200.312初始Dv0.9421.2481.073Dv@10個月1.5551.6681.385雖然本發明參考特殊的實施方案進行了描述和說明,但是對那些本領域普通技術人員顯而易見的是本發明還適于變化而無需在這里說明。本發明還包括以下實施方案。1.環氧組合物,包括以下物質反應制備的反應產物:環氧組分;和酰胺胺組合物,其中所述反應產物包括具有以下結構的環氧官能的表面活性劑:其中m為1到11,n為1到3,q為0到8,p為大約18到大約500,X為氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合,每一個Y是氫原子、甲基取代基、乙基取代基、羥甲基取代基、它們的子集或它們的組合,且R17可以是烷基基團、芳基基團、酰基基團以及它們的子集和它們的組合。2.項目1的環氧組合物,其中所述環氧組分包含環氧樹脂或者環氧樹脂與酚類化合物的混合物。3.項目1的環氧組合物,其中環氧組分與多酰胺胺組合物的反應產物包含具有以下結構的環氧官能的表面活性劑:其中m是1到3,n是1.2,q是1.9到2.3,且p是大約81到大約210,X是氫原子或甲基取代基,每一個Y是氫原子或甲基取代基,且R17可以是叔酰基基團。4.項目1的環氧組合物,其中多酰胺胺組合物是由以下反應制備的酰胺胺反應產物:兩種或更多種酸封端的聚氧化烯多元醇化合物的酸封端的聚氧化烯組合物,其中酸封端的聚氧化烯組合物具有1.1或更高的多分散性并且所述兩種或更多種酸封端的聚氧化烯多元醇化合物具有大約50%到大約100%的來自羥基端基氧化的羧酸端基,和,二胺化合物,其包括伯胺取代基的第一胺取代基和伯胺取代基或仲胺取代基的第二胺取代基。5.項目4的環氧組合物,其中二胺化合物按照胺取代基與酸封端的聚氧化烯組合物的酸取代基化學計量過量提供。6.項目4的組合物,還包含將多酰胺胺組合物與單環氧化合物反應。7.項目4的環氧組合物,其中多酰胺胺組合物包含具有下式的多酰胺胺化合物:其中R1和R2中的每一個包括氫原子或選自具有1到21個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組的取代基,且R1和R2中的至少一個包括氫原子,R3是二價烴取代基,其選自具有2到18個碳原子的支化或線型脂肪族、脂環族、芳香族取代基以及它們的組合和其子集的組,n為大約18到大約500的平均數,X是氫原子或選自由甲基取代基、乙基取代基、羥甲基取代基以及它們的組合組成的組的取代基,且Y為氫原子或選自甲基取代基、乙基取代基、羥甲基取代基以及它們的組合的組的取代基。8.項目4的環氧組合物,其中所述兩種或更多種酸封端的聚氧化烯多元醇化合物的每一種單獨地與二胺化合物反應,之后相互混合以形成多酰胺胺組合物。9.項目1的環氧組合物,其中所述表面活性劑為非離子型的。10.項目1的環氧組合物,其中所述環氧組分包含與胺基團相比化學計量過量的環氧取代基并且表面活性劑包含水性環氧樹脂分散體。11.項目10的環氧組合物,其中環氧官能的表面活性劑包含約1wt.%至約20wt.%的水性環氧樹脂分散體。12.項目10的環氧組合物,其中15個月或更長時間所述分散體包含從約0cps至少于10,000cps的粘度變化。13.項目10的環氧組合物,其中所述水性環氧樹脂分散體包括從約0.2至約1.2μm的顆粒尺寸。14.項目10的環氧組合物,其中所述水性環氧樹脂分散體在雙倍粘度或超過6000cps的時間段,pH變化大約0到大約3。15.項目10的環氧組合物,其中所述多酰胺胺組合物包含6wt.%或更低的水性環氧樹脂分散體。16.熱固性涂料,其包含項目1的環氧組合物。17.纖維膠料,其包含項目1的環氧組合物。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1