一種廣譜固相萃取填料及其應用的制作方法
本發明涉及一種廣譜固相萃取填料及其應用。
背景技術:
隨著殺蟲劑的使用量不斷增加,其污染問題普遍存在,在各種環境介質中多類殺蟲劑及代謝產物頻繁檢出。環境水體中殺蟲劑及代謝產物在儀器檢測之前,一般需經過富集、凈化等前處理步驟,常用的富集、凈化方式包括液液萃取(liquid-liquid extraction,LLE)、固相萃取(solid phase extraction,SPE)、固相微萃取(solid phase microextraction,SPME)、單液滴微萃取(suspended droplet microextraction,SDME)、攪拌棒吸附萃取(stir bar sorptive extraction,SBSE)等方式。其中,SPE方法最為普遍。然而,由于常用殺蟲劑種類繁多,各化合物之間的性質差異巨大,難以通過使用單一吸附劑涵蓋從極性到非極性的多類化合物,故而,檢測環境中不同類型的殺蟲劑及代謝產物,往往需選擇不同類型的SPE柱填料分批進行,導致實驗強度和成本增加。故而,為有效開展水環境中多農藥殘留同時分析,有必要研發適用于較寬極性化合物的廣譜固相萃取柱填料。
技術實現要素:
本發明的目的是針對現有技術的現狀,提供一種新型固相萃取柱填料的合成方法并裝填固相萃取小柱,可同時測定涵蓋從極性至非極性的水體中多類殺蟲劑殘留。
本發明的目的在于提供一種廣譜固相萃取填料及其應用。
本發明所采取的技術方案是:
一種廣譜固相萃取填料,其合成方法包括以下步驟:將1-乙烯基咪唑、二乙烯基苯、偶氮二異丁腈和乙腈混合,超聲處理后,除去氧氣在密閉條件下于50~70℃反應6~36h;將所得產物粉碎、過篩,篩取粒徑為40~60μm的產物,清洗干凈、干燥,即得廣譜固相萃取填料,所得廣譜固相萃取填料比表面積為300~365m2/g。
進一步的,所述1-乙烯基咪唑和二乙烯基苯的摩爾比為1:5~5:1。
進一步的,所述偶氮二異丁腈的用量為1-乙烯基咪唑和二乙烯基苯二者質量之和的1%~3%。
進一步的,所述1-乙烯基咪唑與乙腈的摩爾體積比為(2~10)mmol:(5~15)mL。
上述廣譜固相萃取填料在檢測和/或富集殺蟲劑及代謝產物中的應用。
進一步的,所述殺蟲劑為新煙堿類殺蟲劑、苯基咪唑類殺蟲劑及其代謝產物、有機磷類殺蟲劑、有機氯類殺蟲劑及其代謝產物、擬除蟲菊酯類殺蟲劑。
進一步的,所述新煙堿類殺蟲劑包括噻蟲嗪、噻蟲胺、吡蟲啉、啶蟲脒、噻蟲啉;所述苯基咪唑類殺蟲劑及其代謝產物包括氟蟲腈、氟蟲腈亞砜、磺基氟蟲腈;所述有機磷類殺蟲劑包括二嗪農、丁基嘧啶磷、甲基對硫磷、馬拉硫磷、毒死蜱、丙溴磷;所述有機氯類殺蟲劑及其代謝產物包括α-六六六、β-六六六、γ-六六六、δ-六六六、七氯、艾氏劑、環氧七氯、α-氯丹、α-硫丹、γ-氯丹、狄氏劑、異狄氏劑、β-硫丹、o,p'-滴滴涕、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑醛、硫丹硫酸鹽、p,p'-滴滴涕、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑酮、甲氧滴滴涕;所述擬除蟲菊酯類殺蟲劑包括七氟菊酯、四氟甲醚菊酯、胺菊酯、聯苯菊酯、甲氰菊酯、三氟氯氰菊酯、芐氯菊酯、氟氯氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯。
一種檢測和/或富集殺蟲劑及代謝產物的方法,包括以下步驟:
1)將上述所述的填料裝入固相萃取柱內,填料上下兩端用篩板固定,得廣譜性殺蟲劑固相萃取柱;
2)依次用甲醇、純水對上述固相萃取柱進行活化,然后將樣品溶液以2~3mL/min的流速進行上樣;
3)洗脫和測定:用乙腈進行洗脫,洗脫液經氮吹濃縮后加入二氯甲烷,混勻后,繼續氮吹濃縮,再加入正己烷,再次氮吹濃縮,最后加入內標并定容,對目標殺蟲劑進行測定。
進一步的,步驟3)中,每0.02~0.2g填料用2~5mL乙腈進行洗脫。
進一步的,步驟3)中,有機磷類殺蟲劑、擬除蟲菊酯類殺蟲劑、苯基咪唑類殺蟲劑及代謝產物濃度用GC-MS(NCI)進行測定;有機氯類殺蟲劑及其代謝產物濃度用GC-MS(EI)進行檢測;新煙堿類殺蟲劑濃度用LC-MS進行測定。
本發明的有益效果是:
1)本發明固相萃取填料的制備方法簡單、快速,比商業化HLB固相萃取填料價格低廉。
2)本發明特定方法制備的固相萃取填料可用于同時涵蓋從極性至非極性多類型殺蟲劑及代謝產物的富集、檢測。
3)本發明固相萃取填料比表面積為300~365m2/g,孔體積與平均孔徑分別為0.092cm3/g和2.22nm。對各種殺蟲劑及代謝產物都具有較好的吸附性能;其中對NNIs(新煙堿類殺蟲劑)的回收率可達76.5%,對PPs(苯基咪唑類殺蟲劑及其代謝產物)的回收率為58.3%~108%,對OPs(有機磷類殺蟲劑)的回收率為35.4%-149%,對OCs(有機氯類殺蟲劑及其代謝產物)的回收率為36.0%-172%(除了硫丹硫酸鹽為214%),對PYRs(擬除蟲菊酯類殺蟲劑)的回收率為26.4%-95.3%。
附圖說明
圖1為實施例1合成的廣譜固相萃取填料;
圖2為實施例1合成的廣譜固相萃取填料的BET測試等溫吸附脫附曲線;
圖3為實施例1合成的廣譜固相萃取填料紅外吸收圖譜;
圖4為1-乙烯基咪唑甲醇溶液、二乙烯基苯甲醇溶液和實施例1合成的廣譜固相萃取填料甲醇清洗液的紫外吸收圖譜。
具體實施方式
一種廣譜固相萃取填料,其合成方法包括以下步驟:將1-乙烯基咪唑、二乙烯基苯、偶氮二異丁腈和乙腈混合,超聲處理后,除去氧氣在密閉條件下于50~70℃反應6~36h;將所得產物粉碎、過篩,篩取粒徑為40~60μm的產物,清洗干凈、干燥,即得廣譜固相萃取填料。
優選的,所述1-乙烯基咪唑和二乙烯基苯的摩爾比為1:5~5:1。
優選的,所述偶氮二異丁腈的用量為1-乙烯基咪唑和二乙烯基苯二者質量之和的1%~3%。
優選的,所述1-乙烯基咪唑與乙腈的摩爾體積比為(2~10)mmol:(5~15)mL。
優選的,所述超聲處理時間為15~30min。
優選的,所述除去氧氣的方法為充入氮氣或惰性氣體。
優選的,所述50~70℃反應6~36h所得產物為塊狀。
優選的,所述清洗的具體操作為:將篩取的產物用甲醇溶液超聲清洗4~6遍,每次超聲清洗20~40min。
優選的,所述干燥的溫度為55~65℃,干燥的時間為20~40h。.
優選的,所得廣譜固相萃取填料比表面積為300~365m2/g。
上述任一所述的廣譜固相萃取填料在檢測和/或富集殺蟲劑及代謝產物中的應用。
優選的,所述殺蟲劑及代謝產物為新煙堿類殺蟲劑、苯基咪唑類殺蟲劑及其代謝產物、有機磷類殺蟲劑、有機氯類殺蟲劑其代謝產物、擬除蟲菊酯類殺蟲劑。
優選的,所述新煙堿類殺蟲劑包括噻蟲嗪、噻蟲胺、吡蟲啉、啶蟲脒、噻蟲啉;所述苯基咪唑類殺蟲劑及其代謝產物包括氟蟲腈、氟蟲腈亞砜、磺基氟蟲腈;所述有機磷類殺蟲劑包括二嗪農、丁基嘧啶磷、甲基對硫磷、馬拉硫磷、毒死蜱、丙溴磷;所述有機氯類殺蟲劑及其代謝產物包括α-六六六、β-六六六、γ-六六六、δ-六六六、七氯、艾氏劑、環氧七氯、α-氯丹、α-硫丹、γ-氯丹、狄氏劑、異狄氏劑、β-硫丹、o,p'-滴滴涕、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑醛、硫丹硫酸鹽、p,p'-滴滴涕、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑酮、甲氧滴滴涕;所述擬除蟲菊酯類殺蟲劑包括七氟菊酯、四氟甲醚菊酯、胺菊酯、聯苯菊酯、甲氰菊酯、三氟氯氰菊酯、芐氯菊酯、氟氯氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯。
一種檢測和/或富集殺蟲劑及代謝產物的方法,包括以下步驟:
1)將以上任一所述的填料裝入固相萃取柱內,填料上下兩端用篩板固定,得廣譜性殺蟲劑固相萃取柱;
2)依次用甲醇、純水對上述固相萃取柱進行活化,然后將樣品溶液以2-3mL/min的流速進行上樣;
3)洗脫和測定:用乙腈進行洗脫,洗脫液經氮吹濃縮后加入二氯甲烷,混勻后,繼續氮吹濃縮,再加入正己烷,再次氮吹濃縮,最后加入內標并定容,對目標殺蟲劑進行測定。
優選的,步驟1)中,在真空負壓的條件下將填料裝入固相萃取柱內。
優選的,步驟1)中,規格為1~6mL固相萃取柱內裝入0.02~0.2g填料。
優選的,步驟2)中,每0.02~0.2g填料依次用2~5mL甲醇、2~5mL純水進行活化。
優選的,每0.02~0.2g填料用2~5mL乙腈進行洗脫。
優選的,步驟3)中,有機磷類殺蟲劑、擬除蟲菊酯類殺蟲劑、苯基咪唑類殺蟲劑及其代謝產物濃度用GC-MS(NCI)進行測定;有機氯類殺蟲劑及其代謝產物濃度用GC-MS(EI)進行檢測;新煙堿類殺蟲劑濃度用LC-MS進行測定。
優選的,所述新煙堿類殺蟲劑包括噻蟲嗪、噻蟲胺、吡蟲啉、啶蟲脒、噻蟲啉;所述苯基咪唑類殺蟲劑及其代謝產物包括氟蟲腈、氟蟲腈亞砜、磺基氟蟲腈;所述有機磷類殺蟲劑包括二嗪農、丁基嘧啶磷、甲基對硫磷、馬拉硫磷、毒死蜱、丙溴磷;所述有機氯類殺蟲劑及其代謝產物包括α-六六六、β-六六六、γ-六六六、δ-六六六、七氯、艾氏劑、環氧七氯、α-氯丹、α-硫丹、γ-氯丹、狄氏劑、異狄氏劑、β-硫丹、o,p'-滴滴涕、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑醛、硫丹硫酸鹽、p,p'-滴滴涕、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑酮、甲氧滴滴涕;所述擬除蟲菊酯類殺蟲劑包括七氟菊酯、四氟甲醚菊酯、胺菊酯、聯苯菊酯、甲氰菊酯、三氟氯氰菊酯、芐氯菊酯、氟氯氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯。
下面結合具體實施例對本發明作進一步的說明,但并不局限于此。
實施例1廣譜固相萃取填料的合成方法
將1-乙烯基咪唑10mmol與二乙烯基苯2mmol溶于5mL的乙腈溶液中,加入偶氮二異丁腈30mg,超聲分散20min后,充氮除氧10min,密封。60℃水域條件下反應24h,得到塊狀產物,再將產物研磨,過篩,篩取粒徑為40-60μm的產物。再將篩取后的聚合物用甲醇溶液超聲清洗5遍,每次30min,洗去多余的反應物,60℃條件下干燥24h,即得廣譜固相萃取填料,備用。
本實施例制備的廣譜固相萃取填料為圖1所示的白色粉末狀顆粒。
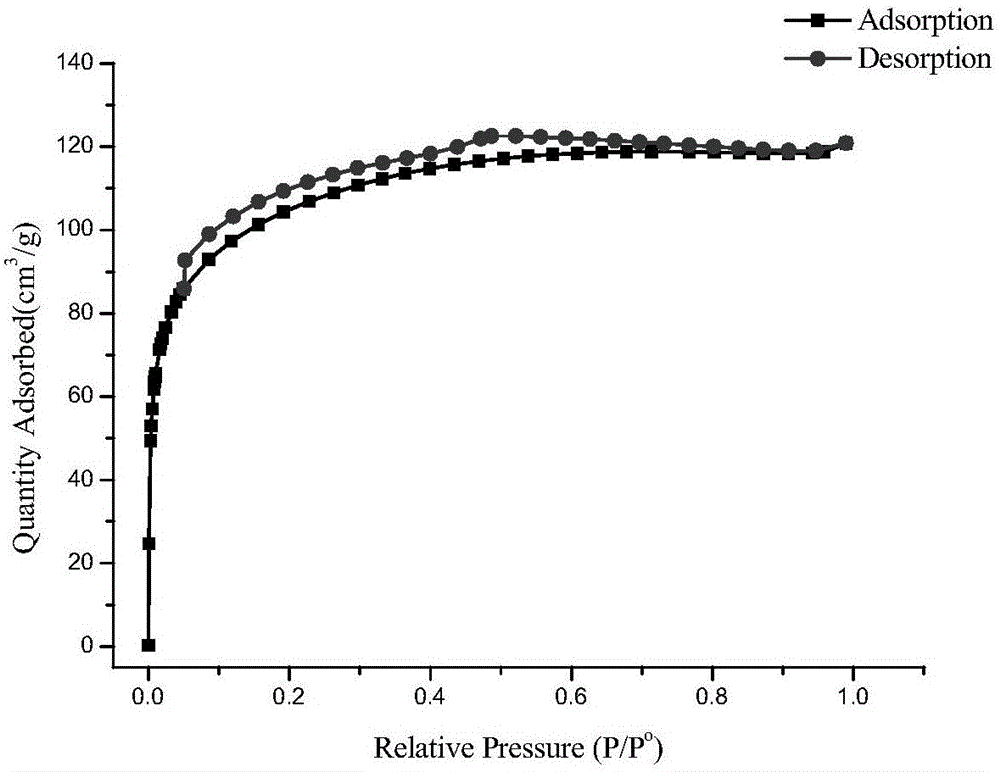
圖2為實施例1合成的廣譜固相萃取填料的BET測試等溫吸附脫附曲線,從圖2的曲線類型可知,合成材料的孔類型為堆積孔,孔的存在有助于對化合物的吸附。BET測試結果表明合成材料的比表面積為341m2/g,孔體積與平均孔徑分別為0.092cm3/g和2.22nm。
圖3為實施例1合成的廣譜固相萃取填料紅外吸收圖譜;從中可以看出,2920cm-1為填料的C-H伸縮振動,1602cm-1、1502cm-1、1447cm-1、1411cm-1為苯環的骨架振動,1288cm-1、1110cm-1、1080cm-1為C-N伸縮振動,915cm-1、827cm-1、800cm-1、710cm-1、665cm-1為二乙烯基苯中不飽和C-H面外變形振動吸收譜帶。
圖4為1-乙烯基咪唑甲醇溶液、二乙烯基苯甲醇溶液和實施例1合成的廣譜固相萃取填料甲醇清洗液的紫外吸收圖譜;從中可以看出,1-乙烯基咪唑的最大吸收峰,位置為230nm,二乙烯基苯的最大吸收峰,位置為238nm;本發明合成的固相萃取填料用甲醇溶液超聲清洗后,甲醇溶液中這兩個位置的紫外吸收都很低(吸光度值在0.1以下),說明1-乙烯基咪唑與二乙烯基苯在甲醇溶液中的含量很低,對本發明制備的填料的使用不會造成干擾。
實施例2廣譜固相萃取填料的合成方法
將1-乙烯基咪唑10mmol與二乙烯基苯2mmol溶于15mL的乙腈溶液中,加入偶氮二異丁腈12mg,超聲分散15min后,充氮除氧10min,密封。50℃水域條件下反應36h,得到塊狀產物,再將產物研磨,過篩,篩取粒徑為40~60μm的產物。再將篩取后的聚合物用甲醇溶液超聲清洗6遍,每次20min,洗去多余的反應物,55℃條件下干燥20h,即得廣譜固相萃取填料,備用。
實施例3廣譜固相萃取填料的合成方法
將1-乙烯基咪唑2mmol與二乙烯基苯10mmol溶于5mL的乙腈溶液中,加入偶氮二異丁腈45mg,超聲分散30min后,充氮除氧10min,密封。70℃水域條件下反應6h,得到塊狀產物,再將產物研磨,過篩,篩取粒徑為40~60μm的產物。再將篩取后的聚合物用甲醇溶液超聲清洗4遍,每次40min,洗去多余的反應物,65℃條件下干燥20h,即得廣譜固相萃取填料,備用。
實施例4廣譜固相萃取填料的合成方法
將1-乙烯基咪唑2mmol與二乙烯基苯10mmol溶于5mL的乙腈溶液中,加入偶氮二異丁腈14.9mg,超聲分散30min后,充氮除氧10min,密封。65℃水域條件下反應28h,得到塊狀產物,再將產物研磨,過篩,篩取粒徑為40~60μm的產物。再將篩取后的聚合物用甲醇溶液超聲清洗4遍,每次40min,洗去多余的反應物,55℃條件下干燥40h,即得廣譜固相萃取填料,備用。
實施例5廣譜固相萃取填料的合成方法
將1-乙烯基咪唑10mmol與二乙烯基苯2mmol溶于15mL的乙腈溶液中,加入偶氮二異丁腈36mg,超聲分散30min后,充氮除氧10min,密封。65℃水域條件下反應28h,得到塊狀產物,再將產物研磨,過篩,篩取粒徑為40~60μm的產物。再將篩取后的聚合物用甲醇溶液超聲清洗4遍,每次40min,洗去多余的反應物,55℃條件下干燥40h,即得廣譜固相萃取填料,備用。
實施例6一種檢測和/或富集殺蟲劑及代謝產物的方法
1)稱取0.02g實施例1制備的廣譜固相萃取填料,在抽真空的條件下,干法裝入規格為3mL聚丙烯材質的固相萃取小柱內,填料上下兩端都用篩板固定,即得廣譜固相萃取小柱。
2)活化:依次用5mL甲醇、5mL純水對固相萃取小柱進行活化。
3)上樣:將樣品水溶液以2~3mL/min的流速往上述活化后的固相萃取小柱進行上樣;樣品水溶液的體積為200mL;
上述樣品水溶液中含有6種有機磷類殺蟲劑(OPs),該6種化合物濃度均為100μg/L;還含有23種有機氯類殺蟲劑及其代謝產物(OCs)、11種擬除蟲菊酯類殺蟲劑(PYRs)、5種新煙堿類殺蟲劑(NNIs)、3種苯基咪唑類殺蟲劑及其代謝產物(PPs),其中由于聯苯菊酯的溶解度較低,其濃度選為100ng/L,其他41種化物濃度均為500ng/L。
上述6種OPs(有機磷類殺蟲劑)為:二嗪農、丁基嘧啶磷、甲基對硫磷、馬拉硫磷、毒死蜱、丙溴磷;
23種OCs(有機氯類殺蟲劑及其代謝產物)為:α-六六六、β-六六六、γ-六六六、δ-六六六、七氯、艾氏劑、環氧七氯、α-氯丹、α-硫丹、γ-氯丹、狄氏劑、異狄氏劑、β-硫丹、o,p'-滴滴涕、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、o,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑醛、硫丹硫酸鹽、p,p'-滴滴涕、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烯、p,p'-2,2-雙(4-氯苯基)-1,1-二氯乙烷、異狄氏劑酮、甲氧滴滴涕;
11種PYRs(擬除蟲菊酯類殺蟲劑)為:七氟菊酯、四氟甲醚菊酯、胺菊酯、聯苯菊酯、甲氰菊酯、三氟氯氰菊酯、芐氯菊酯、氟氯氰菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯;
5種NNIs(新煙堿類殺蟲劑)為:噻蟲嗪、噻蟲胺、吡蟲啉、啶蟲脒、噻蟲啉;
3種PPs(苯基咪唑類殺蟲劑及其代謝產物)為:氟蟲腈、氟蟲腈亞砜、磺基氟蟲腈。
4)洗脫:用5mL乙腈對上樣后的固相萃取柱進行洗脫,接收乙腈洗脫液。
5)殺蟲劑測定
將上步乙腈洗脫液經氮吹濃縮至約1mL,加入10mL二氯甲烷,充分混勻后,繼續氮吹濃縮至約1mL時,加入10mL正己烷,再次氮吹濃縮,最后用正己烷定容至1mL。
I:OPs(有機磷類殺蟲劑)檢測
取上述定容至1mL溶液中的10μL,加入內標,再次用正己烷定容至0.5mL,即稀釋50倍,OPs類化合物濃度用GC-MS(NCI)進行測定。
II:PYRs、PPs、OCs類殺蟲劑及代謝產物的檢測
另取上述用正己烷定容至1mL的溶液中0.8mL,繼續氮吹濃縮,至大約0.4mL,加入OCs、PYRs及PPs的內標化合物,再用正己烷定容至0.5mL,然后用儀器分別進行以下檢測:
PYRs和PPs類殺蟲劑及其代謝產物用GC-MS(NCI)進行檢測;
OCs類殺蟲劑及其代謝產物用GC-MS(EI)檢測。
III:NNIs類殺蟲劑的檢測
OCs、PYRs及PPs檢測結束后,檢測樣品在柔和的氮氣下吹干,加入NNIs類殺蟲劑的內標,用乙腈定容至1.0mL,經震蕩混勻、過膜,用LC-MS測NNIs類殺蟲劑的濃度。
上述檢測結果顯示,本發明廣譜固相萃取小柱對上述各種殺蟲劑及代謝產物都具有較好的吸附性能;其中對NNIs類殺蟲劑回收率為22.0%~76.5%,對PPs類殺蟲劑及其代謝產物回收率為58.3%~108%,對OPs類殺蟲劑回收率為35.4%~149%,對OCs類殺蟲劑及其代謝產物回收率為36.0%~172%(除了硫丹硫酸鹽為214%),對PYRs類殺蟲劑的回收率為26.4%~95.3%。
實施例7一種檢測和/或富集殺蟲劑及代謝產物的方法
1)稱取0.06g實施例1制備的廣譜固相萃取填料,在抽真空的條件下,干法裝入規格為3mL聚丙烯材質的固相萃取小柱內,填料上下兩端都用篩板固定,即得廣譜固相萃取小柱。
將上述制備的廣譜固相萃取小柱應用于水體多種類型殺蟲劑及代謝產物添加回收率測定,選擇強極性到弱極性的NNIs、PPs、OPs、OCs和PYRs 5類共48種殺蟲劑及代謝產物(見實施例6中所述的48種殺蟲劑及代謝產物),以低(20ng/L)、中(100ng/L)、高(500ng/L)三個濃度進行加標。其中,由于聯苯菊酯在水中溶解度較低,最高濃度水平加標值設為200ng/L,每個濃度梯度4份平行樣,具體步驟如下:
2)活化:依次用5mL甲醇、5mL純水對固相萃取小柱進行活化。
3)上樣:將樣品水溶液以2~3mL/min的流速往上述活化后的固相萃取小柱進行上樣;樣品水溶液的體積為1L。
4)洗脫:固相萃取柱干燥后,用5mL乙腈對上樣后的固相萃取柱進行洗脫,接收乙腈洗脫液。
5)殺蟲劑及代謝物測定
I:OPs、PYRs、PPs、OCs類殺蟲劑及代謝產物的檢測
將上步乙腈洗脫液經氮吹濃縮至約1mL,加入10mL二氯甲烷,充分混勻后,繼續氮吹濃縮至約1mL時,加入10mL正己烷,再次氮吹濃縮,加入內標,用正己烷進行定容,低、中、高加標濃度分別定容至0.2mL、0.5mL和2.0mL。
OPs、PYRs和PPs類殺蟲劑及其代謝產物濃度用GC-MS(NCI)進行測定;
OCs類殺蟲劑及其代謝產物用GC-MS(EI)檢測。
II:NNIs類殺蟲劑的檢測
OPs、OCs、PYRs及PPs檢測結束后,檢測樣品在柔和的氮氣下吹干,加入NNIs類殺蟲劑的內標,用乙腈定容(低、中、高加標濃度分別定容至0.2mL、0.5mL和2.0mL),經震蕩混勻、過膜,用LC-MS測NNIs類殺蟲劑的濃度。
上述5類殺蟲劑及代謝產物極性從強到弱,在不同的加標濃度條件下,各類殺蟲劑及代謝產物的回收率值見表1。
表1各類殺蟲劑及代謝物在不同加標濃度下化合物回收率值
從表1各類化合物的回收率值結果可知,本發明所合成的廣譜固相萃取填料可用于分析寬極性范圍的殺蟲劑及代謝產物。
實施例8一種檢測和/或富集殺蟲劑及代謝產物的方法
1)稱取0.06g實施例1制備的廣譜固相萃取填料,在抽真空的條件下,干法裝入規格為3mL聚丙烯材質的固相萃取小柱內,填料上下兩端都用篩板固定,即得廣譜固相萃取小柱。
2)活化:依次用5mL甲醇、5mL純水對固相萃取小柱進行活化。
3)上樣:將野外城市河流采集的樣品水溶液(采樣點坐標N 23°6′58″,E 113°23′29″)以2~3mL/min的流速往上述活化后的固相萃取小柱進行上樣;樣品水溶液的體積為1000mL,三份平行樣。
4)洗脫:用5mL乙腈對上樣后的固相萃取柱進行洗脫,接收乙腈洗脫液。
5)殺蟲劑及代謝產物測定
I:OPs、PYRs、PPs、OCs類殺蟲劑及代謝產物的檢測
將上步乙腈洗脫液經氮吹濃縮至約1mL,加入10mL二氯甲烷,充分混勻后,繼續氮吹濃縮至約1mL時,加入10mL正己烷,再次氮吹濃縮,加入內標,用正己烷進行定容至0.2mL。
OPs、PYRs和PPs類殺蟲劑及其代謝產物濃度用GC-MS(NCI)進行測定;
OCs類殺蟲劑及其代謝產物用GC-MS(EI)檢測。
II:NNIs類殺蟲劑的檢測
OPs、OCs、PYRs及PPs檢測結束后,檢測樣品在柔和的氮氣下吹干,加入NNIs類殺蟲劑的內標,用乙腈定容至0.2mL,經震蕩混勻、過膜,用LC-MS測NNIs類殺蟲劑的濃度。
結果:上述采集水樣經處理后,所分析的48個目標殺蟲劑及代謝產物(見實施例6所述的6種OPs,23種OCs、11種PYRs、5種NNIs和3種PPs),在水樣中所檢測到的殺蟲劑及代謝產物濃度如下表2所示;固相萃取小柱應用于野外實際水樣測定,在所測定的48個目標化合物中,檢測到了15種,其檢出濃度范圍在0.87ng/L-56.8ng/L之間。
表2實際水樣檢測結果
上述實施例為本發明較佳的實施方式,但本發明的實施方式并不受上述實施例的限制,其他的任何未背離本發明的精神實質與原理下所作的改變、修飾、替代、組合、簡化,均應為等效的置換方式,都包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!