八甲基八硅倍半氧烷及其制備方法與應用與流程
本發明涉及一種八甲基八硅倍半氧烷(POSS)的制備方法,屬于有機硅材料及精細化學品領域。
背景技術:
八甲基八硅倍半氧烷(POSS)作為一種有機-無機雜化材料,由于其接近無機硅酸鹽的元素組成,又兼具有機硅的性能特點,利用其改性聚合物有很大的優勢。例如,通過化學鍵合將八甲基八硅倍半氧烷作為橋基引入聚合物中可提高產物的玻璃化溫度,提高材料的硬度和模量。改善產物的熱穩定性,并提高聚合物的阻燃性能。也可將POSS作為添加劑混合至高分子材料中,利用八甲基八硅倍半氧烷的耐熱性本質提高材料的耐高溫性能等。
就八甲基八硅倍半氧烷的制備而言,已有技術都是將甲基三氯硅烷在有機溶劑中與水反應,在濃度很稀的條件下通過三官能度氯硅鍵水解、縮合,得到籠型結構的八甲基八硅倍半氧烷。利用不同的烷基三氯硅烷還可以合成得到含不同取代基的POSS。如氫基八甲基八硅倍半氧烷(H-POSS)、乙烯基八甲基八硅倍半氧烷(Vi-POSS)(參見:Xiong Zang,Lu Cai,Yanhua Yuan,Zhanxiong Li.Fluoroalkylation of Silsesquioxanes-Modified Polysiloxane and Its Surface Property.Journal of Inorganic and Organometallic Polymers and Materials,2015,(25)4:975-981)。
利用三氯硅烷水解縮合法可以一步反應直接得到POSS,但是如果反應物濃度過高會生成大量的高交聯度副產物。因此,三氯硅烷水解縮合法制備POSS要求在反應物濃度很低的條件下進行,一般使用丙酮作為稀釋介質。正因如此,三氯硅烷水解縮合法至少存在以下三方面的不足:(1)必須使用大量的反應介質,這嚴重影響了產品的生產率,限制了設備的生產能力;(2)三氯硅烷水解縮合法制備POSS收率低下,常見的制備工藝報道中收率往往在8%~10%左右;(3)利用甲基三氯硅烷水解時,得到的三官能度硅三醇非常活潑,因此并不總是生成POSS籠型結構,由于不同硅醇分子相互之間的縮合聚合反應還生成大量不溶、不熔的大分子量交聯產物,且這些副產物難以從POSS中除去。使得POSS產品不純,影響其性能。以上缺點使得POSS質量不佳、生產成本高,利用有限的設備難以制得大批量產品,因此嚴重限制了POSS作為優良耐熱材料的應用范圍。
技術實現要素:
本發明的目的是提供一種八甲基八硅倍半氧烷的制備方法,利用四甲基四氫基環四硅氧烷為原料,通過催化水解、親核反應制得POSS,反應選擇性好、收率高,成本低,生產率高,可用于大量制備POSS,為拓寬其應用范圍奠定基礎。
為達到上述發明目的,本發明采用的技術方案是:
一種八甲基八硅倍半氧烷的制備方法,包括如下步驟:
(1)將NaH2PO4·H2O加入NaOH水溶液中制得緩沖液;
(2)在反應器中投入酮類溶劑、Pd/C催化劑、步驟(1)的緩沖液;然后于2~10℃下滴加四甲基四氫基環四硅氧烷;滴加完成后繼續反應30分鐘~6小時;然后調整反應溫度為15~35℃,繼續反應1~24小時;反應結束后抽濾得到濾液;
(3)在反應器中投入步驟(2)的濾液;然后加入三(五氟苯基硼);接著滴加四甲基四氫基環四硅氧烷,進行縮合反應得到八甲基八硅倍半氧烷。
上述技術方案中,步驟(1)中,NaOH水溶液的濃度為0.1mol/L;NaH2PO4·H2O、NaOH水溶液的質量比為(2~5)∶100。
上述技術方案中,步驟(2)中,酮類溶劑、Pd/C催化劑、步驟(1)的緩沖液、四甲基四氫基環四硅氧烷的質量比為(10~100)∶(0.1~1)∶(5~20)∶(10~50)。
上述技術方案中,步驟(2)中,Pd/C催化劑為5%Pd/C催化劑;酮類溶劑為丙酮、丁酮或環己酮;滴加四甲基四氫基環四硅氧烷的時間為30分鐘~2小時;步驟(3)中,滴加四甲基四氫基環四硅氧烷的時間為30分鐘~2小時。
上述技術方案中,步驟(3)中,步驟(2)的濾液、三(五氟苯基硼)、四甲基四氫基環四硅氧烷的質量比為120∶(1~5)∶(10~50)。
上述技術方案中,步驟(3)中,縮合反應溫度為2~80℃,時間為1~120小時。優選的,縮合反應溫度為10~30℃,時間為1~6小時。
上述技術方案中,步驟(3)中,縮合反應結束后,將反應液放置至室溫,抽濾得到固體;然后固體經無水醇類溶劑洗滌、干燥,得到八甲基八硅倍半氧烷。
上述技術方案中,所述無水醇類溶劑為無水甲醇、無水乙醇或干燥異丙醇;固體經無水醇類溶劑洗滌3~5次;干燥溫度為50~100℃,時間為1~24小時。每次使用醇溶劑的質量為固體的10~50倍。
本發明還公開了根據上述八甲基八硅倍半氧烷的制備方法制備得到的八甲基八硅倍半氧烷。
本發明進一步公開了上述八甲基八硅倍半氧烷在制備有機硅材料中的應用。
現有技術以甲基三氯硅烷為原料,由三氯硅烷水解縮合獲得八甲基八硅倍半氧烷(POSS)。反應過程中三氯硅烷水解生成的副產物氯化氫會加速原料水解,因此反應可控性差;而且,反應必須在很稀的溶液中才能生成需要的籠型產物POSS,反應液濃度較高時即生成大量高交聯度副產物而影響產品質量。因此,利用三氯硅烷水解制備POSS的方法產率低,生產成本高,反應難控制、生成副產物氯化氫也對設備要求高。這些因素大大限制了POSS的廣泛應用。本發明克服了上述缺點,利用可控反應以高收率、高生產率地制備POSS,通過兩步反應合成POSS。首先以四甲基四氫基環四硅氧烷(D4H)為起始原料,通過催化水解使D4H硅氧烷環上的硅氫鍵(Si-H)水解成硅羥基(Si-OH),然后利用三(五氟苯基)硼催化硅羥基與另一分子D4H中的硅氫鍵脫水縮合生成硅氧烷鍵(Si-O-Si),合籠得到八甲基八硅倍半氧烷(POSS)。反應式如下式:
由于上述技術方案運用,本發明與現有技術相比具有下列優點:
(1)本發明以環硅氧烷作為原料制備POSS,反應條件溫和,無氯化氫氣體釋放,操作環境好。
(2)本發明利用三(五氟苯基)硼催化縮合制備POSS,反應選擇性好,生產成本低,產率高。
(3)本發明公開的反應條件溫和、制備工藝簡便,原材料易得,適合于工業化生產大量制備POSS。
附圖說明
圖1是實施例一制備的八甲基八硅倍半氧烷的紅外吸收曲線圖;
圖2是實施例二制備的八甲基八硅倍半氧烷的紅外吸收曲線圖。
具體實施方式
下面結合實施例對本發明作進一步描述:
實施例一
催化水解
在配備回流冷凝管、機械攪拌和加熱裝置的四口燒瓶中,投入丙酮100克、5%Pd/C催化劑0.5克和12克緩沖液(將0.65克的NaH2PO4·H2O加入40克0.1mol/L NaOH水溶液中制得),以冰水浴冷卻至5℃,緩慢滴加24.5克四甲基四氫基環四硅氧烷,30min滴完。加完后反應30min,升溫至25℃繼續反應3h。反應結束后抽濾出Pd/C,抽濾出的Pd/C可循環使用。得到的濾液即為水解產物四甲基四羥基環四硅氧烷,該濾液不必分離,直接進入下一步反應。
縮合
將120克上述濾液轉移至四口燒瓶中,裝配回流冷凝管、機械攪拌和加熱裝置,加入三(五氟苯基硼)2.5克,升溫至30℃后,緩慢滴加27.0克四甲基四氫基環四硅氧烷,45min滴完。加完后保溫反應5h。反應結束后,將反應液冷卻至室溫,抽濾出固體,以無水乙醇洗滌3次,每次使用無水乙醇40克。最后將產物在80℃烘干2h,得到26.1克白色八甲基八硅倍半氧烷產物,收率為48%。
參見附圖1,是本實施例制備的八甲基八硅倍半氧烷的的紅外圖譜,吸收曲線中,在2967.40cm-1處的吸收峰為-CH3中C-H的強對稱伸縮振動引起,1414.59cm-1的吸收峰是由C-H的弱不對稱變形振動引起,1263.79和848.52cm-1處的吸收峰歸屬于Si-CH3中Si-C的伸縮振動及-CH3的彎曲振動,1075.10cm-1處的吸收峰歸屬于Si-O-Si的伸縮振動峰。
實施例二
催化水解
在配備回流冷凝管、機械攪拌和加熱裝置的四口燒瓶中,投入丙酮120克、5%Pd/C催化劑1.0克和12克緩沖液(將0.8克的NaH2PO4·H2O加入40克0.1mol/L NaOH水溶液中制得),以冰水浴冷卻2℃左右,緩慢滴加25.0克四甲基四氫基環四硅氧烷,45min滴完。加完后反應45min,升溫至28℃繼續反應5h。反應結束后抽濾出Pd/C,得到濾液不必分離,直接進入下一步反應。
縮合
將上述水解產物四甲基四羥基環四硅氧烷的濾液121.3克轉移至四口燒瓶中,裝配回流冷凝管、機械攪拌和加熱裝置,加入三(五氟苯基硼)3.0克,升溫至30℃后,緩慢滴加29.0克四甲基四氫基環四硅氧烷,50min滴完。加完后保溫反應6h。反應結束后,將反應液冷卻至室溫,抽濾出固體,以無水甲醇洗滌3次,每次使用無水甲醇35克。最后將產物在90℃烘干1h,得到28.3克白色八甲基八硅倍半氧烷產物,收率為52%。
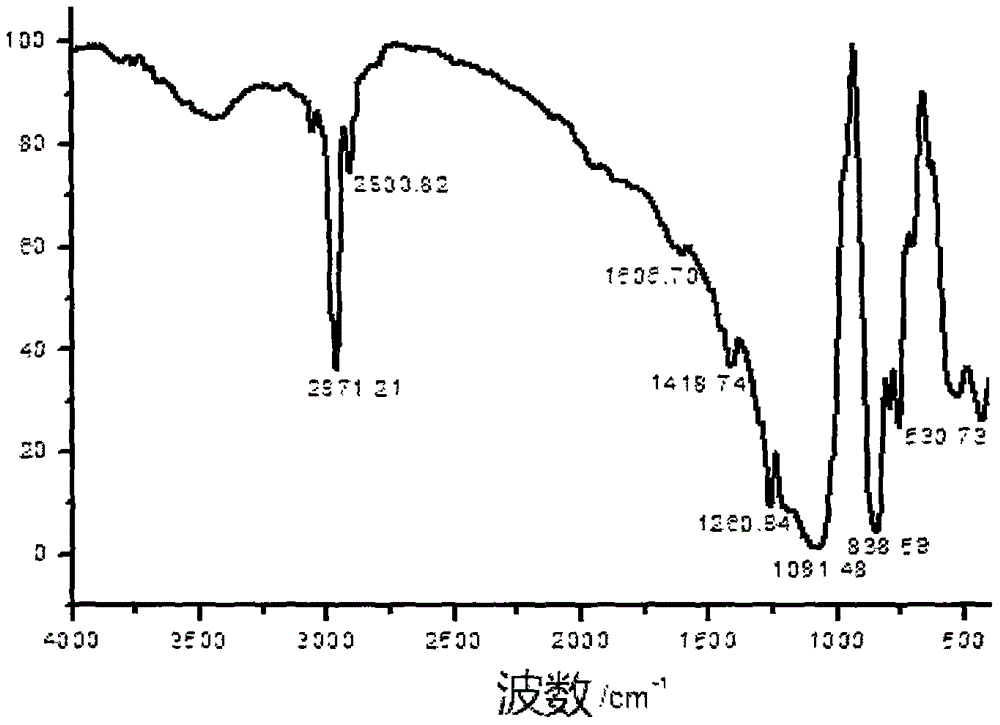
參見附圖2,是本實施例制備的八甲基八硅倍半氧烷的的紅外圖譜,其中,在2971.21cm-1處的吸收峰為-CH3中C-H的強對稱伸縮振動引起,1418.74cm-1的吸收峰是由C-H的弱不對稱變形振動引起,1260.94和838.59cm-1處的吸收峰歸屬于Si-CH3中Si-C的伸縮振動及-CH3的彎曲振動,1081.48cm-1處的吸收峰歸屬于Si-O-Si的伸縮振動峰。
實施例三
催化水解
在配備回流冷凝管、機械攪拌和加熱裝置的四口燒瓶中,投入丁酮100克、5%Pd/C催化劑0.9克和12克緩沖液(將0.8克的NaH2PO4·H2O加入40克0.1mol/L NaOH水溶液中制得),以冰水浴冷卻至4℃,緩慢滴加24.4克四甲基四氫基環四硅氧烷,40min滴完。加完后反應50min,升溫至25℃繼續反應3h。反應結束后抽濾出Pd/C,得到濾液,濾液不必分離,直接進入下一步反應。
縮合
將118.6克上述濾液轉移至四口燒瓶中,裝配回流冷凝管、機械攪拌和加熱裝置,加入三(五氟苯基硼)2.1克,升溫至45℃后,緩慢滴加28.2克四甲基四氫基環四硅氧烷,45min滴完。加完后保溫反應10h。反應結束后,將反應液冷卻至室溫,抽濾出固體,以無水乙醇洗滌3次,每次使用無水乙醇30克。最后將產物在85℃烘干2h,得到19.6克白色八甲基八硅倍半氧烷產物,收率為36%。
實施例四
在配備回流冷凝管、機械攪拌和加熱裝置的四口燒瓶中,投入環己酮150克、5%Pd/C催化劑0.85克和10克緩沖液(將0.8克的NaH2PO4·H2O加入40克0.1mol/L NaOH水溶液中制得),以冰水浴冷卻至6℃,緩慢滴加24.5克四甲基四氫基環四硅氧烷,45min滴完。加完后反應45min,升溫至25~26℃繼續反應4h。反應結束后抽濾出Pd/C,得到濾液,濾液不必分離,直接進入下一步反應。
將167.2克上述濾液轉移至四口燒瓶中,裝配回流冷凝管、機械攪拌和加熱裝置,加入三(五氟苯基硼)1.9克,升溫至50℃后,緩慢滴加29.8克四甲基四氫基環四硅氧烷,45min滴完。加完后保溫反應10h。反應結束后,將反應液冷卻至室溫,抽濾出固體,以無水乙醇洗滌3次,每次使用無水乙醇30克。最后將產物在85℃烘干2h,得到21.1克白色八甲基八硅倍半氧烷產物,收率為38%。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種5-[3-(2,5-二乙氧基-4-甲磺酰基-芐基)-脲基]-2-乙氧基-n-異丙基-苯甲酰胺新化 ...的制作方法
- 一種5-[3-(2,5-二乙氧基-4-甲磺酰基-芐基)-脲基]-2-乙氧基-n-丙基-苯甲酰胺新化合 ...的制作方法
- 一種5-[3-(2,5-二乙氧基-4-甲磺酰基-芐基)-脲基]-2-乙氧基-n-乙基-苯甲酰胺新化合 ...的制作方法
- 一種n-(4-乙氧基羰基苯基)-n’-甲基-n’-苯基甲脒的生產系統的制作方法
- 一種n-(4-乙氧基羰基苯基)-n’-甲基-n’-苯基甲脒中間體的生產系統的制作方法
- 一種n-[5-(4-溴苯基)-6-[2-[(5-溴-2-嘧啶基)氧]乙氧基]-4-嘧啶基]-n’-丙基磺酰胺 ...的制作方法
- 一種清除聚二甲基硅烷裂解反應殘留物的方法
- 籠型八聚(乙炔基二甲基硅氧)倍半硅氧烷及其合成方法
- 一種加入γ氨丙基甲基二乙氧基硅烷的功能涂料的制作方法
- 環氧基甲基二乙氧基硅烷瀝青水泥防水橋聯劑及其防水產品和應用