一種雞溶菌酶和雞β?防御素7融合基因及其制備方法與流程
本發明涉及一種雞溶菌酶和雞β-防御素7融合基因及其制備方法,屬于基因工程技術領域。
背景技術:
目前,由于抗生素濫用,導致耐藥菌株猛增和食品安全危機等公共安全問題層出不窮,尋找高效、無殘留、綠色環保的抗生素替代品已成研究熱點。
溶菌酶主要存在于動植物的組織液和某些微生物體內,如鼻粘液、眼淚、唾液、卵蛋白、枯草桿菌培養物和某些蔬菜中。該酶能水解細菌的細胞壁中N-乙酰氨基葡萄糖和N-乙酰胞壁酸之間的β-1,4-糖苷鍵,故又稱胞壁質酶,即N-乙酰胞壁質糖苷聚糖水解酶。其中對雞蛋清溶菌酶的研究最清楚,它是由129個氨基酸殘基構成的一種堿性蛋白,分子量約為14KD,對熱穩定,對堿不穩定,對革蘭氏陽性細菌有較強的殺菌作用。可藥用,具抗菌、清除局部壞死組織、止血、消腫、消炎等作用。在食品工業上可用作防腐劑,還可添加在牙膏中作為防治齲齒的藥用牙膏。在發酵工業上是一種重要的溶菌劑,用于分解細胞壁,制備無菌體提取液。
防御素是動物體內廣泛存在的一類富含半胱氨酸的抗菌肽,對細菌、真菌、病毒等多種病原微生物均具有抑制作用。防御素抗菌機理獨特,不易產生耐受性,是機體抵御外界病原微生物侵襲的重要武器。由于動物防御素具有抗菌譜廣、不易產生耐藥、安全性高等特點,現已成為抗菌藥物研究的熱點。研究表明,禽類防御素(Av BD)均屬于β-防御素,具備廣譜抗菌活性,有開發成抗菌藥物的潛力。目前關于禽類防御素的研究較少,僅在雞、鴨、鵝等常見家禽和少數幾種野禽中發現了數十種防御素或防御素基因。
以往對溶菌酶、抗菌肽的研究多是針對單一物質。而馮瑄構建了抗菌肽B-人溶菌酶融合蛋白的表達載體;歐陽萍對人溶菌酶-抗菌肽進行了融合表達,并用于小鼠乳房炎的治療試驗,取得了較好的抗菌療效,但將雞溶菌酶-抗菌肽融合表達的研究,還未見報道。
技術實現要素:
本發明旨在為尋找新的抗生素替代品,提供一種雞溶菌酶和雞β-防御素7融合基因及其制備方法。
本發明為達到以上目的,是通過這樣的技術方案來實現的:提供一種雞溶菌酶和雞β-防御素的融合基因,其核苷酸序列是SEQIDNO:1。
該融合基因的核苷酸序列中包含序列SEQIDNO:3、SEQIDNO:4和SEQIDNO:7。基因的5′端為SEQIDNO:3序列,來自于雞溶菌酶基因,全長393bp。基因的3′端為SEQIDNO:4序列,來自于雞β-防御素,全長132bp。SEQIDNO:3和SEQIDNO:4之間為連接肽編碼序列SEQIDNO:7。
本發明還提供了上述融合基因的制備方法,包括以下步驟:
(1)設計擴增雞溶菌酶和雞β-防御素基因片段的PCR引物;
(2)在雞溶菌酶基因下游引物和雞β-防御素基因上游引物中加上一段連接肽序列,然后通過PCR反應擴增雞溶菌酶和雞β-防御素基因;
(3)根據基因重疊延伸技術,以步驟(2)所得的兩個PCR反應產物的混合物為模版,以雞溶菌酶基因上游引物和雞β-防御素7基因下游引物為引物,分三步進行基因重疊延伸,獲得融合基因。
本發明構建融合基因表達載體時所用的表達載體為pET32a。
本發明構建的融合基因工程菌將融合基因表達載體轉入宿主制備而得。
本發明構建的融合基因工程菌為大腸桿菌。
本發明的融合基因表達載體的構建方法:步驟(2)通過設計恰當的引物,通過PCR延伸反應,使雞溶菌酶基因的下游含有完整的連接子序列,而雞β-防御素基因的上游含有部分的連接子序列。步驟(3)根據基因重疊延伸技術,以步驟(2)所得的兩個PCR產物的混合物為模版,不加引物進行互為引物擴增,此時已獲得融合基因。回收擴增產物作為進一步PCR反應的模板,以雞溶菌酶基因的上游引物和雞β-防御素基因的下游引物為引物進行擴增反應,獲得更大量的融合基因產物。然后依次進行了目的片段割膠回收,并插入克隆載體或表達載體、質粒提取等步驟。
本發明還提供了上述融合基因表達載體的構建方法,包括以下步驟:
(1)以PCR擴增融合基因,得到融合基因擴增片段SEQIDNO:1;
(2)采用雙酶切法,雙酶切載體與擴增后的片段,并用T4連接酶將雙酶切后的產物連接到質粒;
(3)將連接產物轉化大腸桿菌獲得陽性克隆子,提取質粒,測序驗證后,得到所述融合基因的表達載體。
本發明同時提供了上述融合基因編碼的融合蛋白,其氨基酸序列是SEQIDNO:2。
該融合蛋白的氨基酸序列SEQIDNO:2同時具有SEQIDNO:5和SEQIDNO:6中的氨基酸序列。
該融合蛋白同時具有雞溶菌酶和雞β-防御素的功能,同時該融合蛋白對金黃色葡萄球菌有明顯的抑菌活性,其最小抑菌濃度低于4mg/ml。
本發明的設計原理:為了防止雞溶菌酶或雞β-防御素在宿主菌中表達成有活性的蛋白形式,而造成的宿主菌的自殺效應,設計連接肽將雞溶菌酶和雞β-防御素連接起來導入宿主細胞,將其融合表達,由于通過這種方式表達的產物蛋白沒有活性,并且融合蛋白的分子量較大,在宿主細胞中大量表達會形成包涵體,有利于之后的蛋白分離純化。
雞溶菌酶和雞β-防御素7,都是存在于動物體內的高效抗菌(抑菌)多肽,對多種微生物發揮抑殺作用,本身無殘留,也不產生賴藥性,作為抗菌藥物、食品添加劑、防腐劑,發揮作用后還能增加食品的蛋白質含量。
具體實施方式
實施例1、PCR引物設計
在保留雞溶菌酶基因和雞β-防御素基因的功能區域基礎上,設計PCR引物;本發明是由連接肽過渡連接的融合蛋白,因此在兩基因的連接區引入連接肽編碼序列(Linker)。
1、雞溶菌酶基因引物:
提取雞輸卵管總RNA,反轉錄成cDNA,以其為模板,根據Genbank公布的雞溶菌酶基因的序列,運用引物設計軟件(primer5.0),設計上下游引物,P1和P2,用于擴增刪除終止密碼子的雞溶菌酶基因。在上游引物中引入BamH I酶切位點序列,下游引物中加入linker序列。
上游引物序列如下,引物名稱為P1:
5′-CGGGATCCGATGAAAGTCTT-3′(劃線部分為BamH I酶切位點);
下游引物序列如下,引物名稱為P2:
5′-linker-CAGCCGGCAGCCTCTG-3′
(linker為5′-tccacctgaaccacctccacctgaaccacctccacc-3′);
2、雞β-防御素7基因引物:
提取雞脊椎總RNA,反轉錄成cDNA,以其為模板,根據Genbank公布的雞β-防御素7基因的序列,運用引物設計軟件(primer5.0),設計上下游引物,P3和P4,用于擴增刪除終止密碼子的雞β-防御素7基因。在下游引物中引入Hind III酶切位點序列,上游引物中加入linker序列。
上游引物序列如下,引物名稱為P3:
5′-linker-AGGCCTATTGATACTTGTAGGCTT-3′
(linker為5′-ggtggaggtggttcaggtggaggtggttcaggtgga-3′);
下游引物序列如下,引物名稱為P4:
5′-AGAAGCTTAGTTATCAGCTCCTCC-3′(劃線部分為Hind III酶切位點)
3、雞溶菌酶和雞β-防御素7融合基因的基因引物:
以所述雞溶菌酶基因和雞β-防御素7基因的PCR擴增產物作為模板,以引物P1和P4為引物,進行PCR擴增,得到雞溶菌酶和雞β-防御素7融合基因。
實施例2、PCR擴增
1、擴增雞溶菌酶基因片段:
此為PCR1,其反應體系為:含Lyz gene質粒0.5μl,上游引物(P1)0.5μl,下游引物(P2)0.5μl,2×HiFi-PCR Master12.5μl,加ddH2O至總體積25μl;擴增條件為:95℃變性5min后進入循環,95℃變性30s,57℃復性30s,72℃延伸30s,30個循環后在72℃延伸10min。
2、擴增雞β-防御素7基因片段:
此為PCR2,其反應體系為:含β-Gal7gene質粒0.5μl,上游引物(P1)0.5μl,下游引物(P2)0.5μl,2×HiFi-PCR Master12.5μl,加ddH2O至總體積25μl;擴增條件為:95℃變性5min后進入循環,95℃變性30s,52℃復性30s,72℃延伸30s,30個循環后在72℃延伸10min。
3、雞溶菌酶基因和雞β-防御素基因的PCR擴增結果
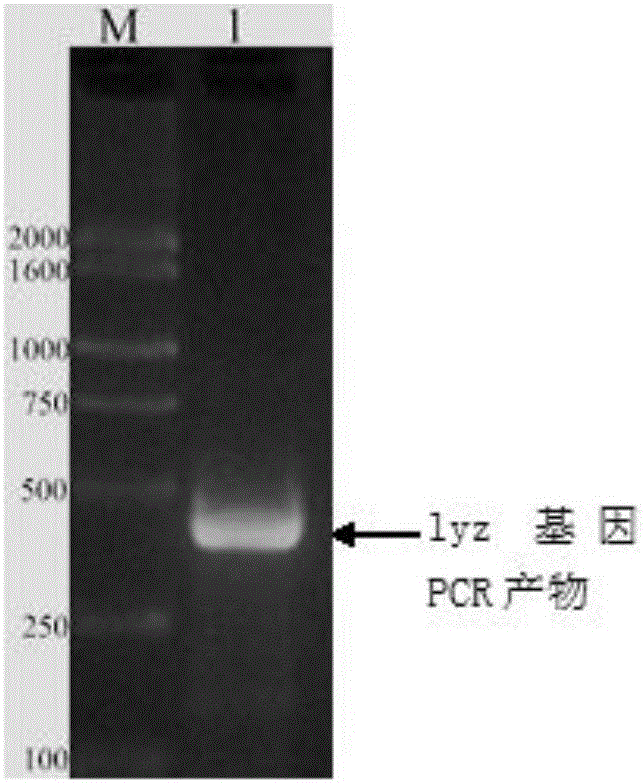
圖1是雞溶菌酶基因Lyz的PCR擴增結果圖,圖2是雞β-防御素7基因β-Gal7的PCR擴增結果圖,從圖中可以看出雞溶菌酶基因和雞β-防御素基因的PCR產物長分別是393bp和132bp,和目的基因的大小一致。
實施例3、融合基因Lyz-β-Gal7的構建(重疊延伸PCR法)
首先,分別進行PCR1反應擴增雞溶菌酶基因Lyz gene和PCR2反應擴增雞抗菌肽基因β-Gal7gene,各取PCR產物25ul分別進行1.5%瓊脂糖凝膠電泳,并用干凈的手術刀切下在100bp至250bp之間、在250bp至500bp之間的目的片段凝膠,并用DNA膠回收試劑盒純化回收凝膠中的DNA片段,取5~10ul回收溶液用分光光度計測OD,計算其中的基因分子濃度,分別取適當體積的Lyz gene回收液和β-Gal7gene回收液使兩基因等量混合,進行PCR3反應構建和擴增Lyz-β-Gal7gene,此處PCR3反應的反應體系和反應條件如表1所示。最后對PCR3反應產物進行1.2%瓊脂糖凝膠電泳和目的片段Lyz-β-Gal7gene膠回收純化。
圖3為融合基因Lyz-β-Gal7的PCR擴增結果圖,從圖中可以看出雞溶菌酶和雞β-防御素融合基因Lyz-β-Gal7的PCR產物為573bp,和目的基因的大小一致。
表1 PCR3構建和擴增Lyz-β-Gal7融合基因的體系和操作
實施例4、融合基因Lyz-β-Gal7的T載體克隆與擴增
Taq DNA聚合酶參與PCR反應延伸DNA鏈時在新生DNA鏈的3′端添加突出的A堿基,該PCR產物與T Vector 3′端突出的T堿基進行AT配對,再受到連接酶的催化,使目的基因與T載體形成環形質粒,可導入E.coli DH5α擴增。
含目的片段Lyz-β-Gal7gene的純化PCR3產物10μl,T Vector2.0μl(約20ng),10×連接酶緩沖液1μl,T4DNA連接酶(濃度U/μl)2μl,50%PEG 4000 2μl,10×Ligation Buffer 2μl,ddH2O 2μl,總體積20μl。16℃連接12h,連接產物可導入E.coli DH5α擴增T-Lyz-β-Gal7或于-20℃冰箱保存。
圖4為融合基因Lyz-β-Gal7與T Vector連后的質粒圖譜。
實施例5、T載體連接與酶切
連接產物轉化、感受態的制備、陽性克隆篩選、質粒DNA的提取均按文獻所記載的現有技術進行。融合基因片段與和T載體連接并導入宿主菌DH5α后,篩選陽性重組質粒T-Lyz-β-Gal7;然后,提取質粒T-Lyz-β-Gal7,BamH I,Hind III雙酶切,重組質粒Lyz-β-Gal7–T雙酶切,酶切體系如下:10×MBuffer3.0μl,BamH I限制性內切酶(10U/μl)1.0μl,Hind III限制性內切酶(10U/μl)1.0μl,T–Lyz-β-Gal7重組克隆載體20.0μl,加ddH2O至總體積30μl,反應液混勻,37℃酶切過夜。電泳,割膠回收目的基因Lyz-β-Gal7。
圖5為經氨芐青霉素抗性及α-互補篩選得到的陽性重組質粒的PCR擴增結果圖。
實施例6、雞溶菌酶-雞β-防御素7融合基因表達載體的構建:
提取的Lyz-β-Gal7目的片段與BamH I,Hind III雙酶切、純化的質粒pET32a連接,構建表達載體,導入BL21(DE3),篩選陽性克隆。
目的基因和表達載體pET32a片段連接,連接反應體系如下:酶切載體DNA2.0μl,目的基因10.0μl,T4DNA連接酶2.0μl,50%PEG 40002.0μl,10×Ligation Buffer2.0μl,加ddH2O至總體積20μl,于37℃連接過夜。連接產物大腸桿菌BL21感受態細胞,篩選陽性克隆吧。
實施例7、融合表達產物純化及活性檢測
7.1融合表達產物純化
取重組工程菌株pET32a/Lyz-β-Gal7/E.coli BL21單菌落或少量菌液接種于2支裝有3ml含100ug/ml Amp的LB液體培養基的10ml試管中,37℃振蕩培養13~15h活化。按1%取5ml活化菌液接種于500ml含100ug/ml Amp的LB液體培養基(可分裝于多個錐形瓶),37℃180r/min培養3h,加終濃度為0.7mmol/L的IPTG誘導4h。
采用超聲波破碎儀破碎菌懸液,條件設置為:工作時間5s,間隙時間6s,80個循環,冰浴中超聲波破碎菌液20mL左右。對破碎上清液中的目的蛋白用鎳柱進行分離純化,純化步驟如下:
(1)用50ml滅菌離心管收集菌液,用冰冷10ml PBS清洗菌液1次,離心。
(2)重懸于20ml 1×Bind Buffer,冰浴超聲破碎,同實例7,至菌液不在粘稠,分裝至10ml離心管。
(3)4℃12000r/min20min,收集上清,沉淀用5ml 1×Bind Buffer重懸,離心取上清。
(4)1ml Ni-NTA預裝柱,先用10ml滅菌dH2O洗柱,控制流速0.5ml/min。
(5)10ml 1×Bind Buffer進行平衡,流速:0.5ml/min,檢測流出液至A280nm=0為洗干凈。
(6)將準備好的樣品上樣(pET28a-lyz-β-Gal7/BL21表達菌的破碎上清),上清加入柱內(結合時間1h-1.5h)流速控制在0.1-0.5ml/min左右,用一支10ml離心管收集上樣后的流出液。
(7)10ml 1×Wash Buffer洗柱子,分管收集洗脫液10-15管,流速在0.5ml/min左右,至A280nm=0為止。
(8)5ml 1×Elution Buffer洗柱子(收集洗脫液5管)。
(9)收集洗脫峰,放入透析袋于4℃,對0.01M pH7.4PBS透析24h,期間更換6次透析液(每次更換一半,最后一次全部更換),透析結束,用PEG8000濃縮,12000rpm 4℃離心10min,收集上清,用于后續試驗或-80℃保存。用10ml 1×Refresh Buffer洗柱子,直至A280nm=0為止,再用10ml1×Bind Buffer進行平衡,可接著作下一次純化。
純化后的目的蛋白含量測定:收集用鎳柱分離純化的Lyz-β-Gal7融合蛋白溶液,取100ul該蛋白液用鎳柱洗脫液進行適當稀釋,以鎳柱洗脫液作參比,測OD260和OD280,粗略計算蛋白濃度C=1.45*OD280—0.74*OD260(mg/ml)。
圖6為鎳柱分離純化前后超聲破碎上清液的SDS-PAGE圖,圖中可見融合蛋白Lyz-β-Gal7條帶。圖7為超聲破碎上清液用鎳柱進行分離純化后的SDS-PAGE圖,圖中可見雜條帶基本消失。
7.2融合表達產物活性檢測
蛋白活性檢測(Lyz-β-Gal7融合蛋白對金黃色葡萄球菌的抑制):參考混菌瓊脂平板打孔法,將固體培養基冷至50℃時,加入終密度為5×106個/ml細菌(乘以混合體積得出要加的菌體數量,菌體數量除以菌體密度得出體積,而菌體密度由比濁法測定),混勻后倒平板,厚度約0.5~1cm,凝固后用打孔器打兩個孔,右邊孔加入100ul 7mg/ml Lyz-β-Gal7融合蛋白(透明圈直徑達到3.9cm);左邊孔加入等體積的透析液PBS作對照。圖8為Lyz-β-Gal7融合蛋白對金黃色葡萄球菌作用結果圖。
探究Lyz-β-Gal7融合蛋白對金黃色葡萄球菌的最小抑制濃度:重復上述步驟,用打孔器打4個孔,孔1、孔2、孔3、孔4,取出圓孔內的瓊脂塊,分別加入100ul濃度分別為1mg/ml、2mg/ml、3mg/ml、4mg/ml的融合蛋白液。把加樣完后的平板正向平放于裝有少量水的盒子內,37℃培養18~24h。圖9為不同濃度Lyz-β-Gal7融合蛋白對金黃色葡萄球菌作用結果圖。
結果表明,本發明的融合蛋白對金黃色葡萄球菌有明顯的抑菌活性,最小抑菌濃度低于4mg/ml,純化后的蛋白電泳得到單一的、大小與理論分子量一致的目的條帶。
最后,還需要注意的是,以上列舉的僅是本發明的若干個具體實施例。顯然,本發明不限于以上實施例,還可以有許多變形。本領域的普通技術人員能從本發明公開的內容直接導出或聯想到的所有變形,均應認為是本發明的保護范圍。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 奶牛金黃色葡萄球菌乳房炎亞單位的疫苗及其制備方法和應用與流程
- 奶牛金黃色葡萄球菌乳房炎亞單位疫苗的組合物及其制備方法和應用與制造工藝
- 含水流動狀組合物及其制造方法、以及溶菌酶加工品和/或其鹽、及其制造方法與制造工藝
- 一種納米材料/群體感應淬滅酶顆粒改性復合膜制備方法與制造工藝
- 一種檢測淚液中乳鐵蛋白的專用膠體金檢測卡的制造方法與工藝
- 一種雞溶菌酶和雞β?防御素7融合基因及其制備方法與制造工藝
- 一種制備治療敏感菌感染性疾病的藥物鹽酸頭孢他美酯組合物的方法與制造工藝
- 含有復合益生元溶菌酶功能性無糖軟糖及其制備方法與制造工藝
- 運用核酸適配體與溶菌酶特異性識別并形成G四鏈體進行熒光顯現潛指紋的方法與制造工藝
- 用于制備結晶DTPMP的方法與制造工藝