一種比率熒光氟離子探針及其制備和應用的制作方法
本發明屬于化學/生物傳感器技術領域,具體涉及一種比率熒光氟離子探針及其制備和在水相氟離子檢測和細胞成像領域中的應用。
背景技術:
生物識別和生物傳感已經成為一個重要的研究領域,尤其是陰離子的檢測變的尤為重要。氟廣泛存在于自然界中,氟化物是人體必須的微量元素之一。但同時含氟化合物是有毒性的,氟化物對人體毒性取決于氟離子的濃度和攝入量。飲用水中含氟的適宜濃度為0.5-1.0mg/L,長期飲用高氟水則會給人體帶來不利影響,嚴重的會引起氟斑牙和氟骨病。氟及氟化物作為重要的化學物質在各個行業有著廣泛的應用。化肥、農藥、化工、石化行業等在生產中使用含氟化學品或生產含氟化工產品,產生了相應的含氟廢水,必須對含氟工業廢水加以處理和監測。因此實現對氟離子簡便、快速的檢測在臨床醫學中表現出越來越重要的作用。
檢測氟離子的機理主要有兩類:一是利用路易斯酸堿相互作用,即硼原子作為路易斯酸與作為路易斯堿的氟離子之間產生強的親和力而實現選擇性識別;二是利用氫鍵作用,即許多氫鍵給體如脲基、吡咯/杯吡咯、酰胺或咪唑能夠與氟離子形成氫鍵從而達到檢測的目的。但是,基于氫鍵相互作用的氟離子探針選擇性較差;基于三芳基硼的氟離子探針選擇性較好,但很難實現水相氟離子檢測。對氟離子的檢測往往很難在水溶液中實現,并且由于檢測過程受到其它多種陰離子的干擾,專一性受到限制。近幾年,利用硅與氟的特殊的相互作用,可以成功實現了純水溶劑體系中氟離子的快速檢測(Angew.Chem.Int.Ed.2010,49,4915–4918)。
現在基于硅與氟相互作用實現氟離子檢測的熒光探針都是基于熒光信號的檢測,熒光信號檢測在實際應用中容易受到背景熒光的干擾,從而降低了檢測的靈敏度和信噪比。熒光比率法是通過兩個波長熒光強度的比值變化檢測目標物,可以通過自我校正消除不定因素的影響,所以比率法是目前檢測離子的最佳選擇。相比有機熒光,具有d6、d8和d10電子結構的磷光重金屬配合物具有優異的光物理性質,如大的斯托克斯位移可以很容易區分激發和發射峰,能使用可見光進行激發,長的發射壽命有利于使用時間分辨技術有效地與背景熒光信號區分開以提高檢測的信噪比和靈敏度等。為了進一步提高檢測的信噪比和靈敏度,并實現水相溶液中的氟離子檢測,有必要設計合成同時基于磷光信號和熒光信號的新型比率熒光氟離子探針。
本發明提供了一種高靈敏度和高選擇性的比率熒光氟離子探針,該探針提高了檢測的準確性,,并可用于細胞標記和成像等領域中。
技術實現要素:
技術問題:本發明的目的在于提供一種比率熒光氟離子探針,所述探針為一類磷光銥配合物,給出該探針的制備方法,并提出這類配合物在檢測氟離子及其在細胞標記、成像中的應用。同時,還利用紫外-可見吸收光譜、熒光發射光譜檢測了該磷光銥配合物對氟離子的響應性能。本發明采用的技術方案如下所述。
技術方案:本發明提供一種比率熒光氟離子探針,所述探針為一類磷光銥配合物,其具有如下結構通式::
其中
n為1-10的正整數。
由上述通式可知,該探針由兩部分組成,一部分為熒光的能量給體,一部分為含有異丁基二苯基硅氧基團的離子型銥配合物并作為能量受體,這兩部分通過共價鍵連接在一起。該探針在與氟離子相遇時,銥配合物配體中的硅氧鍵與氟離子發生作用,硅氧鍵會發生斷裂,導致銥配合物激發態和光物理性質的改變從而影響能量給體和受體間的能量轉移效率,外在表現為熒光效應的明顯變化,從而通過對比這種變化,能夠實現對氟離子的比率熒光檢測。
本發明還提供了上述探針分子的制備方法,具體過程為:
(1)在氮氣保護下,向2,7-二溴-芴中加入強堿和1-辛醇,攪拌并加熱至150-220℃,減壓蒸餾出過量的1-辛醇,分離提純得到2,7-二溴-9-辛基芴;
(2)向強堿和2-吡啶基苯并咪唑中加入離子液體,混合攪拌,加入1,6-二溴己烷,在室溫下攪拌,分離提純得到化合物4;
(3)在氮氣保護下,化合物4、2,7-二溴-9-辛基芴、強堿和有機溶劑的混合物加熱到30-110℃,攪拌反應,分離提純得到化合物5;
(4)稱取化合物5、Ar-B(OH)2和鈀催化劑加入到兩頸瓶中,反應體系避光、氮氣保護,將2M的強堿水溶液、有機溶劑經過除氧操作,用注射器注入到反應體系中,升溫至30-110℃,攪拌反應,分離提純得到化合物6;
(5)在氮氣保護下,C^N配體與三水合三氯化銥在乙二醇乙醚/水(3:1,v:v)混合液中50-180℃密閉反應得到相應的銥二氯橋化合物;
(6)再將得到的銥二氯橋化合物與化合物6在有機溶劑混合液中氮氣保護下30-110℃密閉反應,冷卻后得到化合物7;
(7)將上述反應物冷卻至室溫后加入六氟磷酸鉀繼續反應,旋出溶劑,將得的固體用柱層析方法分離,即得到所述磷光氟離子探針;
其中,所述強堿為氫氧化鉀、氫氧化鈉、碳酸鈉等;步驟(2)中的離子液體為1-丁基-3-甲基咪唑鎓六氟磷酸鹽;步驟(3)中的有機溶劑為DMSO;步驟(4)所述的鈀催化劑為四(三苯基磷)鈀等,所述有機溶劑為甲苯及乙醇的混合溶劑,且經過除氧操作;步驟(6)中的有機溶劑為二氯甲烷和甲醇的混合溶劑,且兩者的體積比為2:1。
其中,
n為1-10的正整數。
所述制備方法的制備過程可參照下述方程式:
本發明還提供所述探針的應用,該比率熒光氟離子探針可以用于水相中氟離子的檢測。
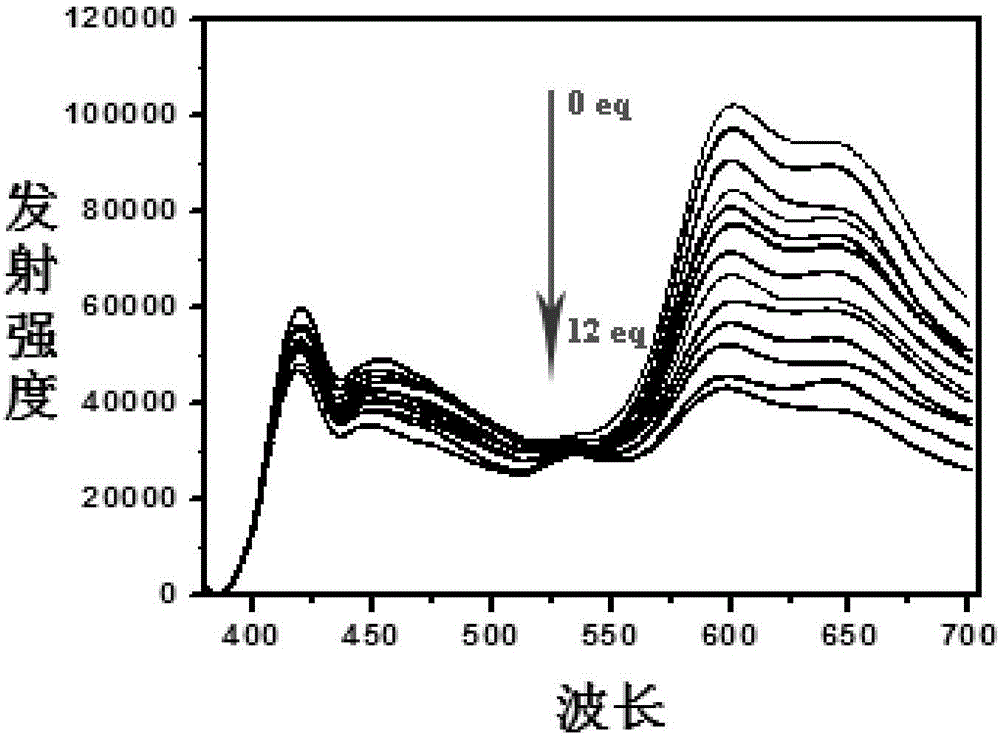
將探針分子溶解在CH3OH/H2O(v/v,2:1,pH 7.2)混合溶液中,逐次加入氟離子的CH3OH/H2O混合溶液中,每次滴加氟離子后,在37℃恒溫水浴中加熱攪拌5min,使氟離子與銥配合物Ir1充分反應,隨后測試其熒光發射光譜,從光譜圖中可以看到,隨著體系中氟離子濃度的增加,化合物在600nm左右處的熒光強度逐漸變弱,而420nm左右處的熒光強度基本不變。
以上結果表明,本發明的化合物對氟離子具有較高的靈敏度,而且可以實現比率法的檢測。
配制10μM的探針分子溶液(CH3OH/H2O(v/v,2:1,pH 7.2)),移取2.5mL所配化合物溶液于比色皿中,加入過量的(超過200倍當量)的含CO32-,HSO4-,NO2-,NO3-,CH3COO-,HSO3-,HCO3-,Cl-,Br-的水溶液,分別測其發射光譜。從光譜圖中觀察加入各種陰離子前后化合物在600nm處的熒光強度變化,可以看出,化合物對除氟離子之外的其它常見陰離子的熒光基本沒有響應。
同時,本發明所述的比率熒光氟離子探針可用于細胞內比率成像。
將消化后的細胞接種在96孔板中,每孔的接種密度為104個/孔,在37℃,5%CO2的條件下繼續培養24h。吸除不新鮮的培養液后用不同濃度探針分子溶液(1、5、10、25、50μM)的細胞培養液繼續培養細胞24h。每孔加入10μL MTT(5mg/mL)繼續培養4h后終止培養。吸除培養液,每孔加入150μL DMSO,搖床震蕩10min后使用酶標儀測試OD570。
MTT細胞毒性實驗結果顯示,在配合物的濃度為1~50μM時,培養24h后的細胞存活率均大于80%,證明該配合物具有較低的細胞毒性,可用于細胞成像。
本發明采用的細胞均為人宮頸癌HeLa細胞。將消化后的細胞接種在培養皿中,在37℃,5%CO2的條件下繼續培養24h使之貼壁。將培養好的HeLa細胞用PBS緩沖溶液洗三次,然后用上述配好的探針分子溶液培養15min后,加入NaF水溶液繼續培養15min。參照組則只用探針溶液培養15min。明場成像和熒光成像用共聚焦顯微鏡觀測。共聚焦顯微鏡激發波長為405nm,收集波段通道為410-500nm和550-650nm區域。從圖片可以看出,只用化合物溶液培養的HeLa細胞,在兩個波段區域都有很強的熒光信號。而加入氟離子之后,在410-500nm通道有很強的光信號,在550-650nm通道區域的熒光信號很弱。明場觀察可以看到很好的細胞形態,證明在整個實驗過程中細胞活性良好。
有益效果:
1.本發明的化合物作為氟離子磷光傳感器,可用于對水相中氟離子的高靈敏度、高選擇性檢測。
2.本發明的化合物作為氟離子磷光傳感器,可實現比率法和比色法的檢測,消除了背景熒光的干擾,提高了檢測的準確性。
3.本發明的化合物作為氟離子磷光傳感器能夠實現對水相中氟離子的高靈敏度、高選擇性檢測,尤其是能夠實現對氟離子的比率熒光檢測和生物比率成像。
附圖說明
圖1為實施例1中配合物Ir1的熒光滴定圖。
圖2為實施例1中配合物Ir1對常見陰離子的識別響應圖。
圖3為實施例1中配合物Ir1的MTT細胞毒性實驗。。
圖4為實施例1中配合物Ir1在細胞培養后加入氟離子前后的共聚焦成像圖。
具體實施方式
本發明涉及化合物的結構通式如下:
其中
n為1-10的正整數。
為了更好地理解本發明專利的內容,下面通過具體的實例來進一步說明本發明的技術方案。具體包括合成、性質測定、滴定實驗以及細胞成像等。但這些實施例并不限制本發明。
實施例1:探針Ir1,即配體為的探針材料的制備及應用測驗:
探針Ir1的制備方法如下述方程式和步驟所示:
(1)化合物3的制備:在反應瓶中加入2,7-二溴-芴(10.0g,60.2mmol)、細粉狀的氫氧化鉀(1.69g,30.1mmol)和1-辛醇(40g,0.31mol),攪拌并加熱至190℃反應19h。將反應混合物冷卻至室溫,減壓蒸餾出過量的1-辛醇。粗產物采用干的硅膠柱,以正己烷作為淋洗劑,過柱得到2,7-二溴-9-辛基-芴(86%)。1H NMR(400MHz,CDCl3):δ=7.62(d,J=1.7Hz,2H),7.56(d,J=8.1Hz,2H),7.49(dd,J=1.7Hz and 8.1Hz,2H),3.95(t,J=5.8Hz,1H),1.90-2.04(m,2H,CH2),1.06-1.35(m,12H,6×CH2),0.87(t,J=6.7Hz,3H,CH3);13C NMR(100MHz CDCl3,):δ=149.2,139.0,130.1,127.6,121.1,121.0,47.4,32.6,31.7,29.7,29.2,25.4,22.6,14.0;C21H24Br2元素分析理論(實驗)值(%):C 57.8(57.9),H 5.6(5.5)。
(2)化合物4的制備:在反應瓶中加入氫氧化鉀51mmol和2-吡啶基苯并咪唑10.2mmol,并加入1-丁基-3-甲基咪唑鎓六氟磷酸鹽(離子液體)20ml。混合物預先攪拌5min后,加入1,6-二溴己烷8ml,攪拌反應5h。反應完成后,用乙醚萃取三次,合并油相,采用旋轉蒸發儀除去溶劑。得到的粗產物用柱層析進行分離,淋洗液為體積比為石油醚:乙酸乙酯=3∶1的混合溶液,最后得到無色油狀液體2.2g,產率60%。1H NMR(400MHz,CDCl3):δ=8.70(d,J=4.80Hz,1H),8.41(d,J=7.97Hz,1H),7.88-7.33(m,2H),7.46-7.44(m,1H),7.37-7.30(m,3H),4.84(t,J=7.6Hz,2H),3.38(t,J=6.8Hz,2H),1.95-1.88(m,2H),1.86-1.79(m,2H),1.53-1.44(m,2H),1.42-1.35(m,2H);13C NMR(100MHz,CDCl3,298K):δ=150.79,149.94,148.76,142.71,136.94,136.69,124.82,123.88,123.40,122.68,120.23,110.28,45.36,33.88,32.64,29.94,27.81,26.07。
(3)化合物5的制備:在氮氣保護下,化合物4(4.0mmol)、2,7-二溴-9-辛基芴(4.0mmol)、KOH(20mmol)和DMSO(10mL)的混合物加熱到60℃,攪拌反應6h。原料反應完全后,將混合物倒入大量飽和食鹽水中,用乙酸乙酯萃取三次。所得到的有機相用無水硫酸鈉干燥。干燥完全后,將固體過濾,所得濾液用旋轉蒸發儀在減壓下除去部分溶劑,然后以體積比為石油醚:乙酸乙酯=3:1的混合溶液作為洗脫液過硅膠柱進行提純,最后得到無色膠狀體,產率74%。1H NMR(400MHz,CDCl3):δ=8.61(d,J=4.59Hz,1H),8.36(d,J=7.92Hz,1H),7.85-7.80(m,2H),7.52-7.29(m,10H),4.73(t,J=7.38Hz,2H),1.90-1.84(m,4H),1.74-1.67(m,2H),1.25-1.04(m,14H),0.83(t,J=6.97Hz,3H),0.6-0.49(m,4H);13C NMR(100MHz,CDCl3):δ=152.48,150.81,149.96,148.70,142.72,139.16,136.86,136.67,130.33,126.20,124.76,123.77,123.32,122.58,121.61,121.31,120.17,110.30,55.70,45.34,40.30,40.17,31.88,30.03,29.95,29.27,26.52,23.69,22.72,14.21。
(4)化合物6a的制備:稱取化合物5(0.71g,1mmol)、9-己基咔唑-3-硼酸(1.0g,2.3mmol)和四(三苯基磷)鈀(0.05g,0.04mmol)加入到兩頸瓶中,并對反應裝置采取避光措施,然后在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將2M的碳酸鈉水溶液(2.5mL)、甲苯(5.0mL)及乙醇(2.5mL)分別用氮氣鼓泡除氧后,用注射器注入到反應體系中。升溫至70℃,攪拌反應36h。然后,將反應體系冷卻至室溫并倒入大量水中,用乙酸乙酯萃取。萃取液用飽和食鹽水洗后再用無水Na2SO4干燥。干燥后過濾除去無機鹽固體,將所得濾液用旋轉蒸發儀減壓下除去部分溶劑,濃縮到一定程度后用正己烷作溶劑進行重結晶純化,得白色固體(0.74g,70%)。1H NMR(400MHz,CDCl3):δ=8.54(d,J=4.37Hz,1H),8.39(d,J=1.26Hz,2H),8.19(d,J=7.70Hz,2H),7.83-7.78(m,5H),7.73-7.67(m,5H),7.55-7.39(m,7H),7.74-7.17(m,6H),4.67(t,J=7.70Hz,2H),4.35(t,J=7.13Hz,4H),1.95-1.88(m,4H),1.75-1.66(m,2H),1.53-1.44(m,2H),1.52-1.35(m,2H),1.46-1.28(m,16H),1.21-1.11(m,14H),0.88(t,J=6.91Hz,9H);13C NMR(100MHz,CDCl3,298K):δ=151.62,148.75,141.06,141.00,139.58,136.84,132.87,128.65,127.93,126.95,126.30,126.33,125.94,125.39,124.79,123.84,123.52,123.40,123.11,121.70,120.61,120.04,119.94,119.01,118.92,110.38,109.05,108.99,55.35,45.46,43.39,40.77,40.66,32.07,31.97,31.76,30.22,30.07,29.85,29.81,29.51,29.42,29.38,29.15,27.15,26.63,22.84,22.75,22.71,14.28,14.22,14.19。
(5)化合物1a的制備:稱取4-(2-苯并噻唑基)苯酚(1g),二苯基叔丁基氯硅烷2ml,咪唑200mg,加入到兩口瓶中,在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將純的N,N二甲基甲酰胺(5ml)加到反應瓶中,攪拌,并將反應體系的溫度升到40℃,反應約24h。處理:用水和乙酸乙酯萃取,真空旋干,乙醇重結晶,得到C^N配體。1H NMR(400MHz,DMSO)δ=6.91-6.86(dd,2H),7.51-7.39(m,8H),1.05(s,9H),7.71-7.66(m,4H),7.96-7.79(d,J=7.8Hz,1H),8.09-8.04(d,1H),7.91-7.86(m,2H).
化合物2a的制備:稱取IrCl3·3H2O(1mmol)和C^N配體(2.5mmol)加入到三口燒瓶中,在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將乙二醇乙醚和水的混合物(3:1v/v)用注射器注入到反應體系中,攪拌,并將反應體系升溫至110℃,反應約24h,反應過程中有沉淀生成。將反應體系冷卻至室溫,然后將沉淀過濾,并用水、乙醇洗,得到固體產物,即銥二氯橋化合物。產率:80%
(6)配合物Ir1的制備:稱取銥二氯橋化合物200mg和化合物6a(420mg)加入到三口瓶中,隨后加入15mL二氯甲烷和甲醇的混合溶劑(2:1,v/v),在磁力攪拌下回流。加入5倍當量的六氟磷酸鉀后繼續攪拌1h,然后除去溶劑,將所得固體混合物再溶于約10mL的二氯甲烷中,將不溶固體過濾除去,濾液除去溶劑后所得的固體物用柱層析(二氯甲烷/丙酮)分離得到純品。產率:61%。核磁數據:1H NMR(400MHz,CDCl3):δ=8.43(d,J=9.8Hz,2H),8.17-8.04(m,9H),7.98(d,J=7.8Hz,1H),7.91-7.75(m,11H),7.71-7.47(m,13H),7.45-7.29(m,15H),7.15-6.88(m,6H),6.81-6.66(dd,4H),4.19-4.62(m,6H),2.13-1.87(m,9H),1.44-1.12(m,32H),1.06(s,18H),0.85(t,J=7.1Hz,6H),0.78(t,J=6.9Hz,3H)。
探針Ir1的發射光譜對氟離子的響應性::
將銥配合物Ir1溶解在CH3OH/H2O(v/v,2:1,pH 7.2)混合溶液中,逐次加入氟離子的CH3OH/H2O混合溶液中,每次滴加氟離子后,在37℃恒溫水浴中加熱攪拌5min,使氟離子與銥配合物Ir1充分反應,隨后測試其熒光發射光譜,600nm處,發射強度由高到低依次對應的氟離子濃度為0eq、1eq、2eq、3eq、4eq、5eq、6eq、7eq、8eq、9eq、10eq、11eq、12eq。從圖1中可以看到,隨著體系中氟離子濃度的增加,化合物在600nm左右處的熒光強度逐漸變弱,而420nm左右處的熒光強度基本不變。
以上結果表明,本發明的化合物對氟離子具有較高的靈敏度,而且可以實現比率法的檢測。
探針Ir1在溶液中對氟離子的選擇性實驗:
配制10μM的配合物Ir1溶液(CH3OH/H2O(v/v,2:1,pH 7.2)),移取2.5mL所配化合物溶液于比色皿中,加入過量的(超過200倍當量)的含CO32-,HSO4-,NO2-,NO3-,CH3COO-,HSO3-,HCO3-,Cl-,Br-的水溶液,分別測其發射光譜。圖2為加入各種陰離子前后化合物在600nm處的熒光強度變化。從圖中可以看出,化合物對除氟離子之外的其它常見陰離子的熒光基本沒有響應。
配合物Ir1的MTT細胞毒性實驗:
將消化后的細胞接種在96孔板中,每孔的接種密度為104個/孔,在37℃,5%CO2的條件下繼續培養24h。吸除不新鮮的培養液后用不同濃度Ir1(1、5、10、25、50μM)的細胞培養液繼續培養細胞24h。每孔加入10μL MTT(5mg/mL)繼續培養4h后終止培養。吸除培養液,每孔加入150μL DMSO,搖床震蕩10min后使用酶標儀測試OD570。
MTT細胞毒性實驗結果如圖3所示,在配合物的濃度為1~50μM時,培養24h后的細胞存活率均大于80%,證明該配合物具有較低的細胞毒性,可用于細胞成像。
配合物Ir1活細胞成像實驗:
本發明采用的細胞均為人宮頸癌HeLa細胞。將消化后的細胞接種在培養皿中,在37℃,5%CO2的條件下繼續培養24h使之貼壁。將培養好的HeLa細胞用PBS緩沖溶液洗三次,然后用上述配好的化合物溶液培養15min后,加入NaF水溶液繼續培養15min。參照組則只用化合物溶液培養15min。明場成像和熒光成像用共聚焦顯微鏡觀測。共聚焦顯微鏡激發波長為405nm,收集波段通道為410-500nm和550-650nm區域。從圖4可以看出,其中圖4A、D是明場照片,圖4B、E是暗場照片,圖4C、F是疊加場照片,只用化合物溶液培養的HeLa細胞,在兩個波段區域都有很強的熒光信號。而加入氟離子之后,在410-500nm通道有很強的光信號,在550-650nm通道區域的熒光信號很弱。明場觀察可以看到很好的細胞形態,證明在整個實驗過程中細胞活性良好。
實施例2:探針Ir2,即當配體為的探針材料的制備及應用測驗:
探針Ir2的制備方法如下述方程式和步驟所示:
(1)化合物3的制備:在反應瓶中加入2,7-二溴-芴(10.0g,60.2mmol)、細粉狀的氫氧化鉀(1.69g,30.1mmol)和1-辛醇(40g,0.31mol),攪拌并加熱至150-220℃反應過夜。將反應混合物冷卻至室溫,減壓蒸餾出過量的1-辛醇。粗產物采用干的硅膠柱,以正己烷作為淋洗劑,過柱得到2,7-二溴-9-辛基-芴(86%)。
(2)化合物4的制備:在反應瓶中加入氫氧化鉀51mmol和2-吡啶基苯并咪唑10.2mmol,并加入1-丁基-3-甲基咪唑鎓六氟磷酸鹽(離子液體)20ml。混合物預先攪拌5min后,加入1,6-二溴己烷8ml,攪拌反應。反應完成后,用乙醚萃取三次,合并油相,采用旋轉蒸發儀除去溶劑。得到的粗產物用柱層析進行分離,淋洗液為體積比為石油醚:乙酸乙酯=3:1的混合溶液,最后得到無色油狀液體2.2g,產率60%。
(3)化合物5的制備:在氮氣保護下,化合物4(4.0mmol)、2,7-二溴-9-辛基芴(4.0mmol)、KOH(20mmol)和DMSO(10mL)的混合物加熱到30-110℃,攪拌反應。原料反應完全后,將混合物倒入大量飽和食鹽水中,用乙酸乙酯萃取三次。所得到的有機相用無水硫酸鈉干燥。干燥完全后,將固體過濾,所得濾液用旋轉蒸發儀在減壓下除去部分溶劑,然后以體積比為石油醚:乙酸乙酯=3:1的混合溶液作為洗脫液過硅膠柱進行提純,最后得到無色膠狀體,產率74%。
(4)化合物6b的制備:稱取化合物5(0.71g,1mmol)、4-硼酸三苯胺(0.67g,2.3mmol)和四(三苯基磷)鈀(0.05g,0.04mmol)加入到兩頸瓶中,并對反應裝置采取避光措施,然后在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將2M的碳酸鈉水溶液(2.5mL)、甲苯(5.0mL)及乙醇(2.5mL)分別用氮氣鼓泡除氧后,用注射器注入到反應體系中。升溫至30-110℃。然后,將反應體系冷卻至室溫并倒入大量水中,用乙酸乙酯萃取。萃取液用飽和食鹽水洗后再用無水Na2SO4干燥。干燥后過濾除去無機鹽固體,將所得濾液用旋轉蒸發儀減壓下除去部分溶劑,濃縮到一定程度后用正己烷作溶劑進行重結晶純化,得白色固體(0.74g,70%)。
(5)化合物1b的制備:稱取4-(2-吡啶基)苯酚(1g),二苯基叔丁基氯硅烷2ml,咪唑200mg,加入到兩口瓶中,在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將純的N,N二甲基甲酰胺(5ml)加到反應瓶中,攪拌,并將反應體系的溫度升到30-110℃。處理:用水和乙酸乙酯萃取,真空旋干,乙醇重結晶。
化合物2b的制備:稱取IrCl3·3H2O(1mmol)和C^N配體(2.5mmol)加入到三口燒瓶中,在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將乙二醇乙醚和水的混合物(3:1v/v)用注射器注入到反應體系中,攪拌,并將反應體系升溫至50-180℃,反應過程中有沉淀生成。將反應體系冷卻至室溫,然后將沉淀過濾,并用水、乙醇洗,得到固體產物,即。產率:80%
(6)配合物Ir2的制備:稱取銥二氯橋化合物200mg和化合物6b(433mg)加入到三口瓶中,隨后加入15mL二氯甲烷和甲醇的混合溶劑(2:1,v/v),在30-110℃下磁力攪拌下回流。加入5倍當量的六氟磷酸鉀后繼續攪拌,然后除去溶劑,將所得固體混合物再溶于約10mL的二氯甲烷中,將不溶固體過濾除去,濾液除去溶劑后所得的固體物用柱層析(二氯甲烷/丙酮)分離得到純品。產率:60%。核磁數據
探針Ir2的發射光譜對氟離子的響應性::
將銥配合物Ir2溶解在CH3OH/H2O(v/v,2:1,pH 7.2)混合溶液中,逐次加入氟離子的CH3OH/H2O混合溶液中,每次滴加氟離子后,在37℃恒溫水浴中加熱攪拌5min,使氟離子與銥配合物Ir2充分反應,隨后測試其熒光發射光譜,從圖1中可以看到,隨著體系中氟離子濃度的增加,化合物在592nm左右處的熒光強度逐漸變弱,而415nm左右處的熒光強度基本不變。
以上結果表明,本發明的化合物對氟離子具有較高的靈敏度,而且可以實現比率法的檢測。
探針Ir2在溶液中對氟離子的選擇性實驗:
配制10μM的配合物Ir2溶液(CH3OH/H2O(v/v,2:1,pH 7.2)),移取2.5mL所配化合物溶液于比色皿中,加入過量的(超過200倍當量)的含CO32-,HSO4-,NO2-,NO3-,CH3COO-,HSO3-,HCO3-,Cl-,Br-的水溶液,分別測其發射光譜。圖2為加入各種陰離子前后化合物在592nm處的熒光強度變化。從圖中可以看出,化合物對除氟離子之外的其它常見陰離子的熒光基本沒有響應。
配合物Ir2的MTT細胞毒性實驗:
將消化后的細胞接種在96孔板中,每孔的接種密度為104個/孔,在37℃,5%CO2的條件下繼續培養24h。吸除不新鮮的培養液后用不同濃度Ir2(1、5、10、25、50μM)的細胞培養液繼續培養細胞24h。每孔加入10μL MTT(5mg/mL)繼續培養4h后終止培養。吸除培養液,每孔加入150μL DMSO,搖床震蕩10min后使用酶標儀測試OD570。
MTT細胞毒性實驗結果如圖3所示,在配合物的濃度為1~50μM時,培養24h后的細胞存活率均大于80%,證明該配合物具有較低的細胞毒性,可用于細胞成像。
配合物Ir2活細胞成像實驗:
本發明采用的細胞均為人宮頸癌HeLa細胞。將消化后的細胞接種在培養皿中,在37℃,5%CO2的條件下繼續培養24h使之貼壁。將培養好的HeLa細胞用PBS緩沖溶液洗三次,然后用上述配好的化合物溶液培養15min后,加入NaF水溶液繼續培養15min。參照組則只用化合物溶液培養15min。明場成像和熒光成像用共聚焦顯微鏡觀測。共聚焦顯微鏡激發波長為405nm,收集波段通道為410-500nm和550-650nm區域。從圖4可以看出,只用化合物溶液培養的HeLa細胞,在兩個波段區域都有很強的熒光信號。而加入氟離子之后,在410-500nm通道有很強的光信號,在550-650nm通道區域的熒光信號很弱。明場觀察可以看到很好的細胞形態,證明在整個實驗過程中細胞活性良好。
實施例3:探針Ir2,即當配體為的探針材料的制備及應用測驗:
探針Ir3的制備方法如下述方程式和步驟所示:
化合物3、4和5的制備過程和實施例2中的一致。
化合物6c的制備:稱取化合物5(0.71g,1mmol)、苯硼酸(0.28g,2.3mmol)和四(三苯基磷)鈀(0.05g,0.04mmol)加入到兩頸瓶中,并對反應裝置采取避光措施,然后在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將2M的碳酸鈉水溶液(2.5mL)、甲苯(5.0mL)及乙醇(2.5mL)分別用氮氣鼓泡除氧后,用注射器注入到反應體系中。升溫至30-110℃。然后,將反應體系冷卻至室溫并倒入大量水中,用乙酸乙酯萃取。萃取液用飽和食鹽水洗后再用無水Na2SO4干燥。干燥后過濾除去無機鹽固體,將所得濾液用旋轉蒸發儀減壓下除去部分溶劑,濃縮到一定程度后用正己烷作溶劑進行重結晶純化,得白色固體(0.74g,70%)。
化合物1c的制備:稱取4-(2-喹啉基)苯酚(1g),二苯基叔丁基氯硅烷2ml,咪唑200mg,加入到兩口瓶中,在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將純的N,N二甲基甲酰胺(5ml)加到反應瓶中,攪拌,并將反應體系的溫度升到30-110℃。處理:用水和乙酸乙酯萃取,真空旋干,乙醇重結晶。
化合物2c的制備:稱取IrCl3·3H2O(1mmol)和C^N配體(2.5mmol)加入到三口燒瓶中,在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將乙二醇乙醚和水的混合物(3:1v/v)用注射器注入到反應體系中,攪拌,并將反應體系升溫至50-180℃,反應過程中有沉淀生成。將反應體系冷卻至室溫,然后將沉淀過濾,并用水、乙醇洗,得到固體產物,即。產率:80%
配合物Ir3的制備:稱取銥二氯橋化合物200mg和化合物6c(400mg)加入到三口瓶中,隨后加入15mL二氯甲烷和甲醇的混合溶劑(2:1,v/v),在磁力攪拌下回流。加入5倍當量的六氟磷酸鉀后繼續攪拌,然后除去溶劑,將所得固體混合物再溶于約10mL的二氯甲烷中,將不溶固體過濾除去,濾液除去溶劑后所得的固體物用柱層析(二氯甲烷/丙酮)分離得到純品。產率:61%。核磁數據
探針Ir3的發射光譜對氟離子的響應性::
將銥配合物Ir3溶解在CH3OH/H2O(v/v,2:1,pH 7.2)混合溶液中,逐次加入氟離子的CH3OH/H2O混合溶液中,每次滴加氟離子后,在37℃恒溫水浴中加熱攪拌5min,使氟離子與銥配合物Ir3充分反應,隨后測試其熒光發射光譜,從圖1中可以看到,隨著體系中氟離子濃度的增加,化合物在588nm左右處的熒光強度逐漸變弱,而416nm左右處的熒光強度基本不變。
以上結果表明,本發明的化合物對氟離子具有較高的靈敏度,而且可以實現比率法的檢測。
探針Ir3在溶液中對氟離子的選擇性實驗:
配制10μM的配合物Ir3溶液(CH3OH/H2O(v/v,2:1,pH 7.2)),移取2.5mL所配化合物溶液于比色皿中,加入過量的(超過200倍當量)的含CO32-,HSO4-,NO2-,NO3-,CH3COO-,HSO3-,HCO3-,Cl-,Br-的水溶液,分別測其發射光譜。圖2為加入各種陰離子前后化合物在588nm處的熒光強度變化。從圖中可以看出,化合物對除氟離子之外的其它常見陰離子的熒光基本沒有響應。
配合物Ir3的MTT細胞毒性實驗:
將消化后的細胞接種在96孔板中,每孔的接種密度為104個/孔,在37℃,5%CO2的條件下繼續培養24h。吸除不新鮮的培養液后用不同濃度Ir3(1、5、10、25、50μM)的細胞培養液繼續培養細胞24h。每孔加入10μL MTT(5mg/mL)繼續培養4h后終止培養。吸除培養液,每孔加入150μL DMSO,搖床震蕩10min后使用酶標儀測試OD570。
MTT細胞毒性實驗結果如圖3所示,在配合物的濃度為1~50μM時,培養24h后的細胞存活率均大于80%,證明該配合物具有較低的細胞毒性,可用于細胞成像。
配合物Ir3活細胞成像實驗:
本發明采用的細胞均為人宮頸癌HeLa細胞。將消化后的細胞接種在培養皿中,在37℃,5%CO2的條件下繼續培養24h使之貼壁。將培養好的HeLa細胞用PBS緩沖溶液洗三次,然后用上述配好的化合物溶液培養15min后,加入NaF水溶液繼續培養15min。參照組則只用化合物溶液培養15min。明場成像和熒光成像用共聚焦顯微鏡觀測。共聚焦顯微鏡激發波長為405nm,收集波段通道為410-500nm和550-650nm區域。從圖4可以看出,只用化合物溶液培養的HeLa細胞,在兩個波段區域都有很強的熒光信號。而加入氟離子之后,在410-500nm通道有很強的光信號,在550-650nm通道區域的熒光信號很弱。明場觀察可以看到很好的細胞形態,證明在整個實驗過程中細胞活性良好。
實施例4:探針Ir4,即當配體為的探針材料的制備及應用測驗:
探針Ir4的制備方法如下述方程式和步驟所示:
化合物1a和2a的制備過程和實施例1中的一致,化合物3、4和5的制備過程和實施例2中的一致。
化合物6d的制備:稱取化合物5(0.71g,1mmol)、(9,9-二辛基)-芴-2-硼酸(1.0g,2.3mmol)和四(三苯基磷)鈀(0.05g,0.04mmol)加入到兩頸瓶中,并對反應裝置采取避光措施,然后在雙排管上抽真空-充氮氣-抽真空,循環三次,最后用氮氣保護反應體系。將2M的碳酸鈉水溶液(2.5mL)、甲苯(5.0mL)及乙醇(2.5mL)分別用氮氣鼓泡除氧后,用注射器注入到反應體系中。升溫至30-110℃。然后,將反應體系冷卻至室溫并倒入大量水中,用乙酸乙酯萃取。萃取液用飽和食鹽水洗后再用無水Na2SO4干燥。干燥后過濾除去無機鹽固體,將所得濾液用旋轉蒸發儀減壓下除去部分溶劑,濃縮到一定程度后用正己烷作溶劑進行重結晶純化,得白色固體(0.79g,74.7%)。
配合物Ir4的制備:稱取銥二氯橋化合物200mg和化合物6d(450mg)加入到三口瓶中,隨后加入15mL二氯甲烷和甲醇的混合溶劑(2:1,v/v),在磁力攪拌下回流。加入5倍當量的六氟磷酸鉀后繼續攪拌,然后除去溶劑,將所得固體混合物再溶于約10mL的二氯甲烷中,將不溶固體過濾除去,濾液除去溶劑后所得的固體物用柱層析(二氯甲烷/丙酮)分離得到純品。產率:65%。
探針Ir4的發射光譜對氟離子的響應性:
將銥配合物Ir4溶解在CH3OH/H2O(v/v,2:1,pH 7.2)混合溶液中,逐次加入氟離子的CH3OH/H2O混合溶液中,每次滴加氟離子后,在37℃恒溫水浴中加熱攪拌5min,使氟離子與銥配合物Ir4充分反應,隨后測試其熒光發射光譜,從圖1中可以看到,隨著體系中氟離子濃度的增加,化合物在587nm左右處的熒光強度逐漸變弱,而411nm左右處的熒光強度基本不變。
以上結果表明,本發明的化合物對氟離子具有較高的靈敏度,而且可以實現比率法的檢測。
探針Ir4在溶液中對氟離子的選擇性實驗:
配制10μM的配合物Ir4溶液(CH3OH/H2O(v/v,2:1,pH 7.2)),移取2.5mL所配化合物溶液于比色皿中,加入過量的(超過200倍當量)的含CO32-,HSO4-,NO2-,NO3-,CH3COO-,HSO3-,HCO3-,Cl-,Br-的水溶液,分別測其發射光譜。圖2為加入各種陰離子前后化合物在600nm處的熒光強度變化。從圖中可以看出,化合物對除氟離子之外的其它常見陰離子的熒光基本沒有響應。
配合物Ir4的MTT細胞毒性實驗:
將消化后的細胞接種在96孔板中,每孔的接種密度為104個/孔,在37℃,5%CO2的條件下繼續培養24h。吸除不新鮮的培養液后用不同濃度Ir4(1、5、10、25、50μM)的細胞培養液繼續培養細胞24h。每孔加入10μL MTT(5mg/mL)繼續培養4h后終止培養。吸除培養液,每孔加入150μL DMSO,搖床震蕩10min后使用酶標儀測試OD570。
MTT細胞毒性實驗結果如圖3所示,在配合物的濃度為1~50μM時,培養24h后的細胞存活率均大于80%,證明該配合物具有較低的細胞毒性,可用于細胞成像。
配合物Ir4活細胞成像實驗:
本發明采用的細胞均為人宮頸癌HeLa細胞。將消化后的細胞接種在培養皿中,在37℃,5%CO2的條件下繼續培養24h使之貼壁。將培養好的HeLa細胞用PBS緩沖溶液洗三次,然后用上述配好的化合物溶液培養15min后,加入NaF水溶液繼續培養15min。參照組則只用化合物溶液培養15min。明場成像和熒光成像用共聚焦顯微鏡觀測。共聚焦顯微鏡激發波長為405nm,收集波段通道為410-500nm和550-650nm區域。從圖4可以看出,只用化合物溶液培養的HeLa細胞,在兩個波段區域都有很強的熒光信號。而加入氟離子之后,在410-500nm通道有很強的光信號,在550-650nm通道區域的熒光信號很弱。明場觀察可以看到很好的細胞形態,證明在整個實驗過程中細胞活性良好。
- 還沒有人留言評論。精彩留言會獲得點贊!