一種3?正丁胺基?4?溴?N?苯基馬來酰亞胺的制備方法與流程
本發明屬于馬來酰亞胺類化合物的制備領域,特別涉及一種3-正丁胺基-4-溴-N-苯基馬來酰亞胺的制備方法。
背景技術:
3-胺基-4-溴馬來酰亞胺及其衍生物是重要的藥物合成中間體,利用該中間體可以合成3-胺基-4-芳基馬來酰亞胺類化合物,在抗菌、抗病毒以及抑制蛋白激酶等方面有著重要的應用(Budke,B.;Kalin,J.H.;Pawlowski,M.;Zelivianskaia,A.S.;Wu,M.;Kozikowski,A.P.;Connell,P.P.J.Med.Chem.,2013,56:254-263.和Awuah,E.;Capretta,A.J.Org.Chem.,2011,76:3122-3130.)。另外,近年來也有關研究其在熒光材料、熒光探針等方面的應用(Mabire,A.B.;Robin,M.P.;Quan,W.D.;Willcock,H.;Stavros,V.G.;O’Reilly,R.K.Chem.Commun.,2015,51:9733-9736.)。文獻報道3-胺基-4-溴馬來酰亞胺類化合物的合成方法主要包括兩種:方法一,馬來酰亞胺類化合物經液溴溴化,制得3,4-二溴基馬來酰亞胺類化合物,然后經脫溴化氫氧化、胺化,再與液溴反應,最終合成3-胺基-4-溴-馬來酰亞胺類化合物(Patil,N.S.;Deshmukh,G.B.;Makale,K.A.;Gosavi,K.S.;Patil,S.V.Indian.J.Chem.,2015,54B:272-278);方法二,馬來酸酐為原料,在三氯化鋁和溴素的參與下,120℃回流反應16h,然后在苯胺的參與下在醋酸中125℃反應3h,最后再與正丁胺在室溫中反應1.5h,合成3-胺基-4-溴-馬來酰亞胺類化合物(Mabire,A.B.;Robin,M.P.;Quan,W.D.;Willcock,H.;Stavros,V.G.;O’Reilly,R.K.Chem.Commun.,2015,51:9733-9736.)。上述兩種合成方法中,方法一需要多次的溴化、氧化脫溴、胺化等過程,步驟多,反應時間長;方法二反應時間長,反應溫度高,醋酸的參與使原料容易聚合,副產物較多等。
綜上所述,上述方法存在反應路線長、收率低和反應條件要求苛刻等缺點,在大規模生產中成本相對較高。
合成方法一的反應式:
合成方法二的反應式:
技術實現要素:
本發明所要解決的技術問題是提供一種3-正丁胺基-4-溴-N-苯基馬來酰亞胺的制備方法,該方法以N-苯基馬來酰亞胺和正丁胺為原料,以溴化亞銅為溴化劑,反應得到3-正丁胺基-4-溴-N-苯基馬來酰亞胺,收率達90%。該方法制備工藝簡單、反應時間短、成本低、環境友好、純度和收率高、適合工業化生產。具體的制備反應式如下:
本發明的一種3-正丁胺基-4-溴-N-苯基馬來酰亞胺的制備方法,包括:
將N-苯基馬來酰亞胺、正丁胺和溴化劑加入到溶劑中,室溫~100℃下反應1~3小時,反應結束后,加水攪拌,乙酸乙酯萃取、無水硫酸鈉干燥、濃縮蒸去有機溶劑得黃棕色固體,而后經重結晶得到黃色固體3-正丁胺基-4-溴-N-苯基馬來酰亞胺。
所述N-苯基馬來酰亞胺、正丁胺和溴化劑摩爾比為1.0:1.0~1.2:1.0~2.0。
所述溴化劑為溴化銅。
所述N-苯基馬來酰亞胺與溶劑的比例為1克:1毫升~100毫升。
所述溶劑為1,4-二氧六環、DMF、DMSO或THF。
所述重結晶溶劑為95%(v/v)乙醇。
本發明的3-正丁胺基-4-溴-N-苯基馬來酰亞胺的結構式:
熔點:78~80℃;
性狀:黃色固體;
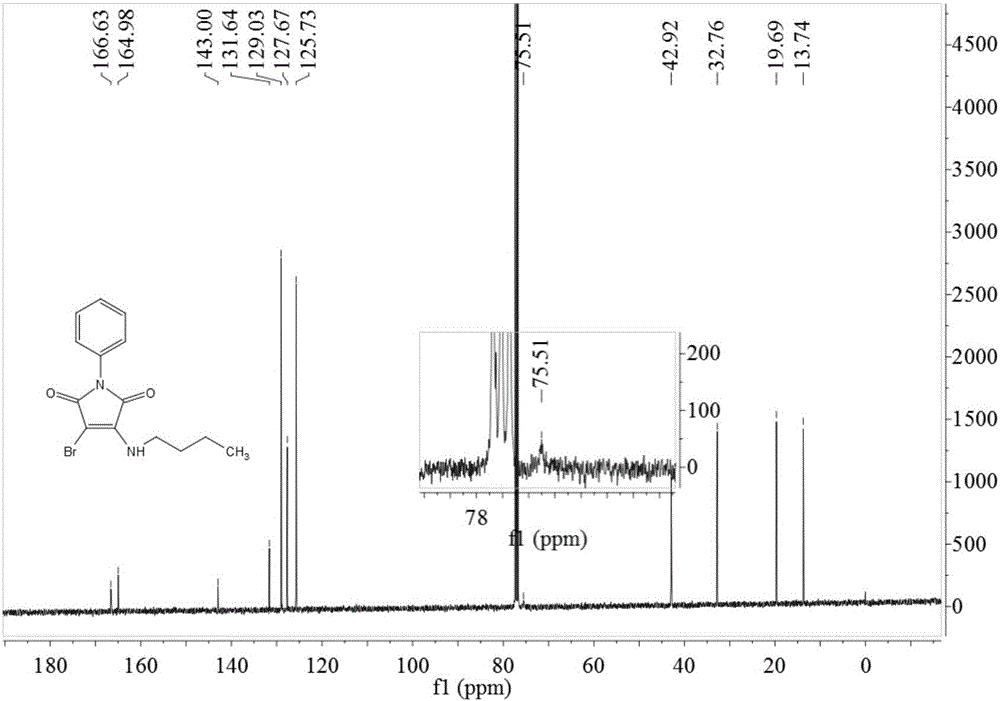
3-正丁胺基-4-溴-N-苯基馬來酰亞胺的核磁數據如下:
1H NMR(400MHz,CDCl3)δ0.98(t,J=7.2Hz,3H),1.49–1.39(m,2H),1.68(p,J=7.0Hz,2H),3.69(q,J=6.7Hz,2H),5.62(s,1H),7.40–7.28(m,3H),7.44(t,J=7.3Hz,2H);
13C NMR(101MHz,CDCl3)δ13.74,19.69,32.76,42.92,75.51,125.73(2C),127.67,129.03(2C),131.64,143.00,164.98,166.63。
有益效果
本發明在制備3-正丁胺基-4-溴-N-苯基馬來酰亞胺的過程中,以溴化銅作為催化劑和溴化劑,縮短了反應時間和反應步驟,反應收率達90%以上,產率高,降低了三廢處理,該方法同樣收率較高;該制備方法起始原料易得,成本低,反應操作簡單,反應路線短,易于工業化生產。
附圖說明
圖1為實施例1中化合物3-正丁胺基-4-溴-N-苯基馬來酰亞胺的核磁共振氫譜;
圖2為實施例1中化合物3-正丁胺基-4-溴-N-苯基馬來酰亞胺的核磁共振碳譜。
具體實施方式
下面結合具體實施例,進一步闡述本發明。應理解,這些實施例僅用于說明本發明而不用于限制本發明的范圍。此外應理解,在閱讀了本發明講授的內容之后,本領域技術人員可以對本發明作各種改動或修改,這些等價形式同樣落于本申請所附權利要求書所限定的范圍。
實施例1
取N-苯基馬來酰亞胺17.3g(0.1mol)、正丁胺8.0g(0.11mol)和溴化銅22.3g(0.1mol)加入至500mL圓底燒瓶中,加入1,4-二氧六環200mL,加熱100℃攪拌2h,反應畢,加水100mL,攪拌10分鐘,乙酸乙酯萃取(200mL×3)、無水硫酸鈉干燥有機相、濃縮蒸去有機溶劑得黃棕色固體,而后經95%(v/v)乙醇重結晶得到黃色固體27.1g,收率84%,mp:78~80℃。3-正丁胺基-4-溴-N-苯基馬來酰亞胺的核磁共振氫譜和碳譜如圖1和圖2所示。
實施例2
取N-苯基馬來酰亞胺1.73g(0.01mol)、正丁胺0.8g(0.011mol)和溴化銅3.35g(0.015mol)加入至250mL圓底燒瓶中,加入1,4-二氧六環80mL,加熱100℃攪拌2h,反應畢,加水100mL,攪拌10分鐘,乙酸乙酯萃取(100mL×3)、無水硫酸鈉干燥有機相、濃縮蒸去有機溶劑得黃棕色固體,而后經95%(v/v)乙醇重結晶得到黃色固體2.97g,收率92%,mp:78~80℃。
實施例3
取N-苯基馬來酰亞胺1.73g(0.01mol)、正丁胺0.8g(0.011mol)和溴化銅3.35g(0.015mol)加入至250mL圓底燒瓶中,加入1,4-二氧六環80mL,加熱60℃攪拌2h,反應畢,加水100mL,攪拌10分鐘,乙酸乙酯萃取(100mL×3)、無水硫酸鈉干燥有機相、濃縮蒸去有機溶劑得黃棕色固體,而后經95%(v/v)乙醇重結晶得到黃色固體2.74g,收率85%,mp:78~80℃。
實施例4
取N-苯基馬來酰亞胺3.46g(0.02mol)、正丁胺1.6g(0.022mol)和溴化銅6.7g(0.03mol)加入至250mL圓底燒瓶中,加入DMF 80mL,加熱100℃攪拌2h,反應畢,加水100mL,攪拌10分鐘,乙酸乙酯萃取(100mL×3)、無水硫酸鈉干燥有機相、濃縮蒸去有機溶劑得黃棕色固體,而后經95%(v/v)乙醇重結晶得到黃色固體5.36g,收率83%,mp:77~80℃。
實施例5
取N-苯基馬來酰亞胺8.65g(0.05mol)、正丁胺4.0g(0.055mol)和溴化銅16.75g(0.075mol)加入至500mL圓底燒瓶中,加入DMSO 200mL,加熱100℃攪拌2h,反應畢,加水100mL,攪拌10分鐘,乙酸乙酯萃取(200mL×3)、無水硫酸鈉干燥有機相、濃縮蒸去有機溶劑得黃棕色固體,而后經95%(v/v)乙醇重結晶得到黃色固體13.25g,收率82%,mp:78~80℃。
實施例6
取N-苯基馬來酰亞胺1.73g(0.01mol)、正丁胺0.8g(0.011mol)和溴化銅3.35g(0.015mol)加入至250mL圓底燒瓶中,加入THF 80mL,加熱100℃攪拌2h,反應畢,加水100mL,攪拌10分鐘,乙酸乙酯萃取(100mL×3)、無水硫酸鈉干燥有機相、濃縮蒸去有機溶劑得黃棕色固體,而后經95%(v/v)乙醇重結晶得到黃色固體2.42g,收率75%,mp:76~78℃。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 3-(4-氰基苯基)-4-(3-溴苯胺甲基)異噁唑及其制備方法
- 化合物(2z)-2-((4-溴苯基)亞肼基)苊-1-酮及其制備方法與應用
- 一種2-(4-溴甲基苯基)丙酸制備方法
- 溴化2-(2-甲基苯基)-3-甲氧基-丙烯酸甲酯的化學方法
- 4-[(z)-(4-溴苯基)(乙氧基亞氨基)甲基]-1′-[(2
- (4-溴苯基)(4-哌啶)亞甲基-(z)-o-乙基肟及其鹽的合成的制作方法
- N-(4-三氟甲基苯基)-5-甲基-異噁唑-4-酰胺的結晶方法
- 2-(4-溴甲基苯基)丙酸的合成的制作方法
- 殺菌劑中間體(e)-2-(2'-溴甲基)苯基-3-甲氧基丙烯酸甲酯的制備方法
- 9-(4-溴苯基)-9h-咔唑和4-(9h-9-咔唑基)苯基硼酸及其合成方法
- 一種基于咔唑衍生物的雙羥基熒光擴鏈劑及其制備與應用的制作方法與工藝
- 基于苯基苯并咪唑單極熱激發延遲熒光主體、電子傳輸材料的制備方法及應用與流程
- 基于2?(2’?羥基苯基)苯并噻唑衍生物的pH熒光探針及其制備方法和應用與流程
- 取代的5?羥基?2?雜芳基?3?苯基戊腈衍生物、其制備方法及其作為除草劑和/或植物生長調節劑的用途與流程
- 1?[(2?溴苯基)磺酰基]?5?甲氧基?3?[(4?甲基?1?哌嗪基)甲基]?1H?吲哚二甲磺酸鹽一水合物的活性代謝物和活性代謝物的二甲磺酸二水合物鹽的制作方法與工藝
- 一種3?正丁胺基?4?溴?N?苯基馬來酰亞胺的制備方法與流程
- 2,5?二溴?1,4?苯二氧基二乙酸鋅配合物單晶與應用的制作方法與工藝
- 三羥基咪唑?熒光素pH熒光探針及其制備方法與流程
- 6?羥甲基?1?苯基吡啶?2?酮及其制備方法與應用與流程
- 一種合成2?(2,4?二羥基苯基)?4,6?二苯基?1,3,5?均三嗪的方法與流程