康普瑞汀藥物前體、藥物制劑和制備方法與流程
本發明屬于藥物合成領域,具體是涉及一種抗血管生成藥物康普瑞汀親脂性藥物前體、合成方法及其納米制劑和制劑的制備方法。
背景技術:
惡性腫瘤的生長和轉移依賴于腫瘤血管,腫瘤依靠血管為自身輸送氧氣和營養物質,供其細胞的快速增殖,并導致腫瘤細胞向遠端轉移。隨著腫瘤血管新生機制研究的深入,以及以腫瘤血管為靶點的藥物在臨床治療腫瘤時取得較好的療效,證實了通過抑制腫瘤血管新生來抑制腫瘤生長的理念。
康普瑞汀A4(Combretastatin,CA4)是從非洲灌木Combretum Caffrum的樹皮分離的一種順式二苯乙烯類天然物。20世紀80年代初,研究者在評估一系列Combretastatins化合物對體外細胞毒性時發現,CA4與秋水堿結構相似,能在遠小于其最大耐受劑量(MTD)時即發揮活性作用,系CA4與微管蛋白結合/解離快,半衰期較短,體內消除快所致(Pettit GR,Singh SB,Hamel E,Lin CM,Alberts DS,Garcia-Kendall D,Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A4.Experentia,1989,45:209-211)。但是CA4的水溶性極差,生物利用度較低,從而限制了其臨床應用。為了提高CA4的水溶性,使其更易于體內用藥,傳統方式是將CA4設計改造成磷酸二鈉前藥(CA4P),其磷酸鹽基團在體內需要在磷酸酯酶的作用下轉化成CA4,但是完全轉化尚需要較長的時間,另外,藥物動力學研究表明,CA4P的半衰期比較短,在體內消除較快,為維持阻斷腫瘤血管,必須短時間內反復用藥,副作用較大。
納米載藥體系具有良好的腫瘤靶向性,可改善藥物的溶解性,降低藥物的毒副作用,通過合理的抗腫瘤藥物納米給藥系統的設計,納米載藥系統已經在抗腫瘤研究中取得了良好的應用前景。我們前期的實驗中將CA4與兩親性高分子共組裝形成納米粒,但是結果顯示,直接包載CA4的納米粒存在藥物快速釋放的問題。
技術實現要素:
針對現有技術存在的問題,本發明提供了一種藥物釋放時間延長、藥物的療效提高的親脂性康普瑞汀藥物前體。
本發明同時提供了上述親脂性康普瑞汀藥物前體的制備方法,該方法操作簡單,易于實現大量生產。
本發明還提供了一種藥物釋放時間延長、藥物的療效提高的抗血管生成藥物納米粒康普瑞汀藥物制劑。
本發明還提供了一種上述康普瑞汀藥物制劑的制備方法,本發明將該藥物前體與先進納米技術結合,制備抗血管生成藥物的納米體系,延緩CA4的釋放,延長藥物的作用時間,提高藥物的臨床藥效。
為解決上述技術問題,技術方案分別如下:
一種康普瑞汀藥物前體,結構如下:
所述R為C1~C10的烷酰基。
作為優選,所述R為C1~C7的烷酰基。
作為進一步優選,所述康普瑞汀藥物前體結構如下:
上述兩種康普瑞汀藥物前體,藥物水解速率快,釋放速度大幅度延緩。
一種上述康普瑞汀藥物前體的制備方法,包括:康普瑞汀A4與所述烷酰基對應的羧酸、酰氯或者酸酐進行酯化反應,得到所述的康普瑞汀藥物前體。
作為優選,采用康普瑞汀A4與所述烷酰基對應的羧酸的進行酯化反應制備得到,反應過程中,可根據需要添加催化劑,縮合劑等,比如可采用1-(3-二甲氨基丙基)-3-乙基碳二亞胺的鹽酸鹽作為縮合劑,4-二甲氨基吡啶作為催化劑,同時可利用N,N-二異丙基乙胺中和1-(3-二甲氨基丙基)-3-乙基碳二亞胺的鹽酸鹽中的鹽酸。
作為進一步優選,所述康普瑞汀A4與羧酸、催化劑、縮合劑的摩爾比為1:1~1.5:0.5~1.5:1~1.5。
一種康普瑞汀納米制劑,包括康普瑞汀藥物前體和兩親性高分子材料,所述康普瑞汀藥物前體的結構如下:
所述R為C1~C10的烷酰基。
作為優選,所述R為C1~C10的烷酰基。
作為優選,所述康普瑞汀藥物前體的結構如下:
上述兩種康普瑞汀藥物制劑,藥物水解速率快,8h即可釋放約90%的活性藥物;同時藥物在納米粒中的釋放速度大幅度延緩,有利于藥物在體內的長循環,發揮長時間藥效。
作為優選,所述兩親性高分子材料選自聚乙二醇-聚乳酸(PEG-PLA)、聚乙二醇-聚乳酸-羥基乙酸(PEG-PLGA)、甲氧基聚乙二醇-聚乳酸(mPEG-PLA)或甲氧基聚乙二醇-聚乳酸-羥基乙酸(mPEG-PLGA)中的任意一種。
作為進一步優選,所述PEG-PLA、PEG-PLGA、mPEG-PLA或mPEG-PLGA中,PEG的分子量為2k~8k,mPEG的分子量為2k~8k,PLA的分子量為2k~16k,PLGA的分子量為2k~16k。
作為進一步優選,所述兩親性高分子材料為mPEG5k-PLA16k。
作為優選,所述康普瑞汀納米制劑為粉劑或片劑、注射劑、丸劑。
一種制備上述任一技術方案所述康普瑞汀納米制劑的方法,將康普瑞汀藥物前體和兩親性高分子材料溶解在有機溶劑中,然后加入到水中,除去有機溶劑,得到康普瑞汀納米制劑。
作為優選,所述有機溶劑為丙酮。
本發明提供了一種康普瑞汀親脂性藥物前體的合成及其納米制劑。本發明中提供的康普瑞汀親脂性藥物前體能夠快速水解釋放活性藥物成分,發揮藥效。相對于未改性康普瑞汀組成的納米粒的快速釋放,本發明中的納米粒可緩慢釋放內部藥物,有利于延長藥物在體內的循環,提高藥物的抗血管生成療效,具有良好的臨床應用前景和應用價值。
附圖說明
圖1為丁酸-CA4合成路線圖;
圖2為庚酸-CA4合成路線圖;
圖3為實施例4中納米粒電鏡圖;
圖4為實施例3、4、5中納米粒的粒徑圖;
圖5為測試例1中CA4藥物前體水解速率圖;
圖6為測試例2中納米粒體外釋放曲線;
圖中,CA4表示康普瑞汀A4,DCM表示二氯甲烷,EDC·HCl表示1-(3-二甲氨基丙基)-3-乙基碳二亞胺的鹽酸鹽,DMAP表示4-二甲氨基吡啶,DIEA表示N,N-二異丙基乙胺。
具體實施方式
下面結合附圖和具體實施方式對本發明作進一步詳細說明,但本發明并不受其限制。
實施例1
取丁酸(51.7μL,0.57mmol),CA4(化合物1,180mg,0.57mmol),EDC·HCl(163mg,0.855mmol),DMAP(76.6mg,0.627mmol),DIEA(188.9μL,1.14mmol)溶于4ml DCM中,室溫下攪拌過夜,加入乙酸乙酯,分別依次用5%檸檬酸、飽和NaHCO3、飽和食鹽水洗滌有機相,有機相用無水硫酸鈉干燥,過濾,收集濾液后減壓除去溶劑;固體用柱層析色譜分離純化(乙酸乙酯:正己烷=1:3),得到目標產物丁酸-CA4偶聯物化合物2(280mg,收率93%)。合成路線如圖1所示。
產物丁酸-CA4的1H NMR核磁數據以及質譜數據如下:
1H NMR(400MHz,CDCl3):δ1.00-1.04(t,3H),1.73-1.78(m,2H),2.49-2.53(t,2H),3.71(s,6H),3.80(s,3H),3.83(s,3H),6.45-6.45(d,2H,J=1.2),6.51(s,2H),6.83-6.85(d,1H,J=8.4),7.00-7.01(d,1H,J=1.6),7.10-7.12(q,1H).
HR-ESI Qq-LTMS:計算值:[C22H26O6]+[M+H]+=387.1802;檢測值:387.1811。
實施例2
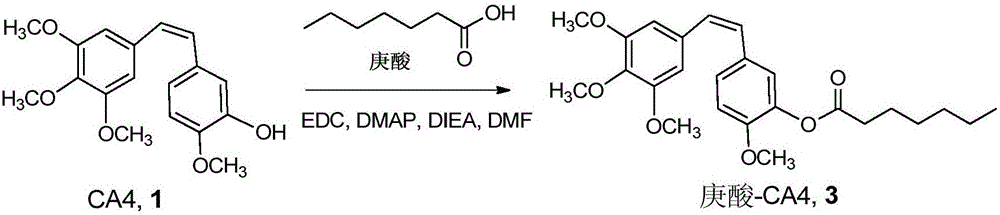
取庚酸(80.8μL,0.57mmol),CA4(180mg,0.57mmol),EDC·HCl(163mg,0.855mmol),DMAP(76.6mg,0.627mmol),DIEA(188.9μL,1.14mmol)溶于4mL DCM中,室溫下攪拌過夜,加入乙酸乙酯,分別依次用5%檸檬酸、飽和NaHCO3、飽和食鹽水洗滌有機相,有機相用無水硫酸鈉干燥,過濾,收集濾液后減壓除去溶劑;固體用柱層析色譜分離純化(乙酸乙酯:正己烷=1:3),得到目標產物庚酸-CA4偶聯物化合物3(260mg,收率85%)。合成路線如圖2所示。
1H NMR(400MHz,CDCl3):δ0.88-0.91(t,3H),1.26-1.41(m,6H),1.70-1.74(t,2H),2.51-2.54(t,2H),3.71(s,6H),3.80(s,3H),3.84(s,3H),6.45-6.45(d,2H,J=1.6),6.51(s,2H),6.83-6.85(d,1H,J=8.4),7.00-7.00(d,1H,J=1.6),7.10-7.12(q,1H).
HR-ESI Qq-LTMS:計算值[C25H32O6]+[M+H]+=429.2212;檢測值:429.2282。
實施例3
將CA4(CA4含量0.5mg)和mPEG5k-PLA16k(藥物質量的20倍)溶解于1mL丙酮中,均勻后滴加到10mL水中,滴加完畢后,減壓去除丙酮,即可得到CA4納米藥物(記為1-NP)。
實施例4
將實施例1制備的丁酸-CA4偶聯物2(按照CA4計,CA4的量為0.5mg)和mPEG5k-PLA16k(CA4前藥質量的20倍)溶解于1mL丙酮中,均勻后滴加到10mL水中,滴加完畢后,減壓去除丙酮,即可得到丁酸-CA4納米藥物(2-NP),2-NM電鏡下形貌如圖3,觀察到納米粒呈較好的圓形,大小均勻。
實施例5
將實施例2制備的庚酸-CA4偶聯物3(按照CA4,CA4的量0.5mg)和mPEG5k-PLA8k(CA4前藥質量的20倍)溶解于1mL丙酮中,均勻后滴加到10mL水中,滴加完畢后,減壓去除丙酮,即可得到庚酸-CA4納米藥物(3-NP)。
DLS(動態光散射,Dynamic Light Scattering)測得各實施例1、2、3中制備的納米粒的粒徑如圖4和表1所示。結果顯示實施例3、4、5中制備的1-NP、2-NP、3-NP納米粒的粒徑均在30nm左右,并且粒徑分布較窄,呈現較好的均勻性。
表1粒徑大小
以下通過測試例進一步闡明本發明所述康普瑞汀前體及其納米粒的體外特性:
測試例1:藥物水解速率
取實施例1、2中制備的CA4藥物前體化合物2、化合物3,分別溶于DMSO(CA4等同量2mg/mL),再用HEPES(4-羥乙基哌嗪乙磺酸,pH 8.0)稀釋至0.5mg/mL(CA4等同量),置于37℃,每隔一定時間,取出100μL液體分析,繪制藥物水解曲線。如圖5所示,CA4藥物前體化合物2、化合物3能夠在體外環境中快速水解,8h即可釋放約90%的活性藥物。
測試例2:體外釋放實驗
取實施例3~5中制備的1-NP、2-NP、3-NP各10mL置于分子量為14kDa的透析袋中,釋放條件為50mL、pH 7.4的磷酸緩沖液(含0.2%的吐溫80),37℃,不同時間點取出外界磷酸緩沖液,用HPLC測得CA4含量,從而得到納米粒中藥物的體外釋放情況。藥物的釋放曲線如圖6所示,結果顯示與自由藥物CA4組成的納米粒1-NP相比,對CA4進行結構改性后的2-NP和3-NP釋放速度明顯減緩,24h,1-NP釋放率達91%,而2-NP和3-NP的釋放率分別為37%和4.6%。實驗證實,對CA4的結構改造,有效延緩藥物在納米粒中的釋放速度,有利于藥物在體內的長循環,發揮長時間藥效。
- 還沒有人留言評論。精彩留言會獲得點贊!