利用氟表面活性劑強化分子氧氧化醇反應的方法與流程
本發明屬于醇綠色氧化技術領域,具體涉及一種利用氟表面活性劑強化分子氧氧化醇反應的方法。
(二)
背景技術:
伯醇或仲醇選擇性氧化為相應的醛、酮等羰基化合物是實驗室研究和工業合成中最重要的有機反應之一。傳統的氧化方法要求使用化學計量的高價態金屬化合物(如KMnO4、MnO2、CrO3等)或者有機氧化物。然而,這些方法往往需要當量的或過量的較昂貴的氧化劑,并且這些過程往往伴生大量的廢棄污染物。再者,醛易被進一步氧化,如伯醇往往被氧化生成羧酸。因此,從環境保護和經濟性出發,開發綠色清潔及節能的醇類氧化工藝一直是研究的熱點(Advanced Synthesis&Catalysis,2004,346(9-10):1051-1071;化學進展,2007,19(11):1727-1735)。
完全清潔、節能的醇類氧化技術需從四個方面對反應工藝進行綠色化:一是采用綠色氧化試劑代替化學計量氧化試劑;二是采用綠色溶劑作反應介質代替有毒有機溶劑;三是降低反應條件,使得反應壓力和溫度溫和化;四是采用易于回收使用的高效綠色環保催化劑。
醇類清潔氧化技術的重點之一就是用純氧或空氣(分子氧)代替化學計量氧化試劑。對此,近年來開發綠色、溫和、經濟、高效的反應催化體系,以H2O2、O2或空氣作為清潔氧化劑液相催化氧化醇類至醛酮化合物成為研究熱點,特別是氧氣或空氣作為氧化劑更加是研究中的熱點,因為氧氣、空氣是所能得到的最豐富、廉價、安全及環境友好的氧化劑。因此,深入研究此類氧化反應不但對于實驗室范圍內很有價值,而且對于工業生產也有重要意義。
然而,采用氧氣為氧化劑的困難在于,大多數的化合物在室溫下與氧氣的反應很慢。對于基態氧分子,產生這種反應惰性有兩個原因:其一,氧氣參與的氧化還原反應往往是多電子反應,一般條件下不可能通過一步反應實現;其二,自旋守恒原理認為,產物自旋守恒的基元反應較容易進行,是自旋允許的反應,而自旋不守恒的基元反應需要附加的電子成對能,活化能較大,被稱為自旋禁阻反應,基態為三線態的氧分子與基態為單線態的醇在溫和條件下由于自旋禁阻作用很難反應,因而需要較高的活化能。另外,由于分子氧氧化醇本身是一個氣液多相反應,因而催化反應一般需要很長時間。其中氧氣分子的活化成為了一個研究的熱點,活化氧氣分子重點在于體系的篩選。醇的分子氧催化氧化體系可分為:均相催化體系中醇的分子氧氧化和非均相催化體系中醇的分子氧氧化。
對于醇的分子氧氧化,均相催化體系主要包括:金屬配合物催化體系、硝酸鹽/鹵素離子/氮氧自由基催化體系。對金屬配合物催化體系而言,金屬配合物主要由Ru、Pd、Cu、Co、V、Pt、Mn、Zn、Fe等金屬離子和不同配體組成。這類催化氧化反應以金屬離子為催化活性中心,應用堿為共催化劑,將醇選擇性氧化為醛、酮等羰基化合物。反應一般需要高溫、高壓以及有機溶劑的使用,使得反應過程并不綠色。對硝酸鹽/鹵素離子/氮氧自由基催化體系而言,一般認為這類催化氧化反應以硝酸鹽在酸性環境下產生的NO來活化分子氧產生NO2,在鹵素離子的作用下NO2將TEMPO氧化為TEMPO+,然后TEMPO+將醇選擇性氧化為醛、酮等羰基化合物。這類體系雖然避免了過渡金屬的使用,解決了金屬離子污染產物的問題。但是這類體系存在明顯的不足:相比于過渡金屬體系,其反應體系活化分子氧的能力較弱,反應一般在有機溶劑,高溫、高壓下進行。所以反應時間一般比過渡金屬反應體系要長。
對均相催化體系中醇的氧分子氧化而言,因為反應物和催化劑在同一相中,這類體系表現出了均相催化,因而具有較高的活性和選擇性。但具有不易分離及催化劑不便循環利用等諸多缺點。因此與均相催化相比,非均相催化的優點在于產物分離提純簡單、催化劑易于分離回收使用。非均相催化氧化醇大多是按照氧化脫氫機理進行的,反應底物被吸附到催化劑表面并脫氫,生成相應的羰基化合物,被脫除的氫再被氧氣氧化生成水。所以隨著人們對環境問題的日益關注,非均相催化氧化的研究進展很快。
非均相催化氧化使用比較多的是將Ru、Pd、Au等絡合物或者其納米粒子通過物理吸附或者化學鍵合的形式固載在載體表面或孔洞結構中。載體一般為SiO2、介孔分子篩、水滑石、羥磷灰石等無機材料,以及交聯聚苯乙烯等有機載體。反應結束后,催化劑可以通過離心分離來循環使用。但是由于催化劑負載后催化活性位點表面受限,導致在相同條件下相比于均相催化體系,其催化活性有所降低。
除了使用清潔的氧化劑分子氧外,醇類清潔氧化技術的另一途徑是采用水、離子液體、超臨界二氧化碳等作為反應溶劑。在水相中進行的醇的氧分子催化氧化的方法,主要集中在使用納米金、鈀固載在無機/有機載體上以及近年來使用銅鹽/含N配體/TEMPO體系。對納米貴金屬而言,可以在較溫和條件下催化氧化醇。該體系的催化活性與貴金屬納米粒子的尺寸及載體的性質有關。和過渡金屬絡合體系相同,其反應體系也需要外加弱堿才能使醇順利地氧化為相應的醛、酮。相比于過渡金屬體系,納米貴金屬催化體系簡單,但是催化劑在水相中容易失活以及循環使用后納米粒子容易團聚等催化劑失活問題。近年來有使用銅鹽/含N配體/TEMPO催化體系在室溫常壓下,以空氣中氧分子為氧化劑,水相中將醇選擇性氧化為相應的醛、酮等羰基化合物。雖然該體系反應條件溫和,反應介質環保、反應過程節能,但是催化劑體系對脂肪醇、雜原子醇催化活性偏低(RSC Advances,2014(4):61907-61911)。
對于采用離子液體為反應介質的體系,表現出了有別于傳統分子溶劑的反應歷程和結果,傳統有機溶劑的揮發和污染問題因為離子液體的使用而得到一定程度的改善,產物的分離也因為離子液體和部分作為萃取劑的非極性有機溶劑不混溶而大大簡化。離子液體的可設計性使離子液體取代固相合成的固體載體成為可能。但是由于離子液體價格一般較昂貴,這將限制其工業化應用(J.Org.Chem.,2007,72(2):517-524)。
由于采用超臨界二氧化碳為溶劑既能消除水溶劑的不利因素,又能排除有機溶劑和表面活性劑的使用,而且工藝更加安全和環境友好。在有些情況下能比傳統溶劑獲得更高的反應速率和選擇性,并能延長催化劑的使用壽命。它的優勢不僅體現在其對反應物為惰性和難燃性以及與產品、催化劑易于分離,更突出的優點在于其溶劑強度的可調性和有利的傳質特性,因而更有利于反應。但是由于超臨界二氧化碳為溶劑需要在很高的壓力下進行,給工業化帶來一定的難度(Encyclopedia of Sustainability Science&Technology.,2012:10169-10188)。
因此,現有技術均不能完全滿足清潔、節能的醇類氧化技術條件,即實現常溫、常壓及使用綠色溶劑,反應時間短(<1h)。
(三)
技術實現要素:
針對現有的醇類清潔氧化技術存在的缺陷,本發明的目的在于提供一種在常溫常壓下、水性膠束溶液中醇的分子氧氧化的方法。本發明方法中使用了氟表面活性劑,其目的在于:第一,構建氟環境對氧氣的吸附,提高膠束環境的局部氧氣濃度;第二,通過構建膠束環境來增加有機底物的溶解性,從而提高膠束環境的局部底物濃度;第三,通過構建膠束表面的異性電荷層對反應性離子的吸引環境來提高反應活性離子的反應活性;從而強化醇的分子氧氧化。
為實現上述目的,本發明采用如下技術方案:
一種利用氟表面活性劑強化分子氧氧化醇反應的方法,所述的方法包括如下步驟:
(1)將底物醇類化合物(伯醇或仲醇)、水(優選去離子水)、氟表面活性劑混合均勻,得到體系I;
步驟(1)中,所述的醇類化合物與氟表面活性劑的物質的量之比為1:0.0025~0.1;所述的醇類化合物與水的質量比為1:10~30;
所述的底物可以為:芳香伯醇、芳香仲醇、脂肪伯醇、脂肪仲醇、脂環醇、烯丙基醇、雜原子醇等醇類化合物;具體例如:苯甲醇、對硝基苯甲醇、對甲氧基苯甲醇等;
所述的氟表面活性劑可以為:全氟辛基磺酸四乙基銨、全氟辛基羧酸鉀、全氟辛基硫酸鈉、全氟辛基磷酸鈉、全氟辛基磺酸鈉等含氟陰離子表面活性劑;
(2)惰性氣體(如:氮氣)保護下,將配體、銅鹽、水(優選去離子水)混合均勻,得到體系II;
步驟(2)中,所述的銅鹽與配體的物質的量之比為1:1.5~3.5;所述的銅鹽與水的質量比為1:300~600;
所述的配體可以為:4-二甲氨基吡啶、聯吡啶等含N化合物;
所述的銅鹽可以為:CuCl、CuCl2、CuBr、CuBr2、CuI、CuI2、CuOTf、Cu(OTf)2、Cu(NO3)2、Cu(OAc)2、CuSO4等;
(3)在步驟(1)所得體系I中,加入步驟(2)所得體系II、氮氧自由基化合物的水溶液,在空氣或氧氣氛圍中,于20~30℃下反應30min~5h(優選30min~60min),之后反應液經后處理,得到氧化產物(當底物為伯醇時,所得氧化產物為醛類化合物;當底物為仲醇時,所得氧化產物為酮類化合物);
步驟(3)中,所述氮氧自由基化合物的水溶液的濃度為0.001~0.05mol/L;所述的體系I與體系II、氮氧自由基化合物的水溶液的體積比為1:1~3:0.5~2;
所述的氮氧自由基化合物可以為:2,2,6,6-四甲基哌啶-氮氧化物、4-羥基-2,2,6,6-四甲基哌啶-氮氧化物、4-甲氧基-2,2,6,6-四甲基哌啶-氮氧化物、4-氨基-2,2,6,6-四甲基哌啶-氮氧化物、4-羰基-2,2,6,6-四甲基哌啶-氮氧化物、9-氮雜雙環(3,3,1)壬烷-氮氧化物、3-羥基-9-氮雜雙環(3,3,1)壬烷-氮氧化物、3-環氧基-9-氮雜雙環(3,3,1)壬烷-氮氧化物、3-羰基-9-氮雜雙環(3,3,1)壬烷-氮氧化物或2-氮雜雙環金剛烷-氮氧化物等氮氧自由基;
通常,所述反應液后處理的方法為:反應結束后,將反應液進行分液,下層油相為粗產物,上層水相為溶有氟表面活性劑的水溶液;分液所得油相經飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,得到氧化產物;水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
本發明中,所述的體系I、體系II沒有特殊的含義,標記為“I”、“II”只是用于區分不同步驟中的反應體系。
與現有技術相比,本發明的有益效果在于:本發明所述的利用氟表面活性劑強化分子氧氧化醇反應的方法,在室溫、無需加壓的條件下即可進行,反應條件溫和;以空氣為氧化劑、水為溶劑,反應過程環保;并且,相比于國內外的現有技術,反應時間短,效率高;是一種清潔、節能的醇類氧化技術。
(四)附圖說明
圖1是本發明實施例1~6與對比例1的苯甲醇有氧氧化動力學曲線,圖1中(1)為對比例1,(2)~(7)依次為實施例1~6;
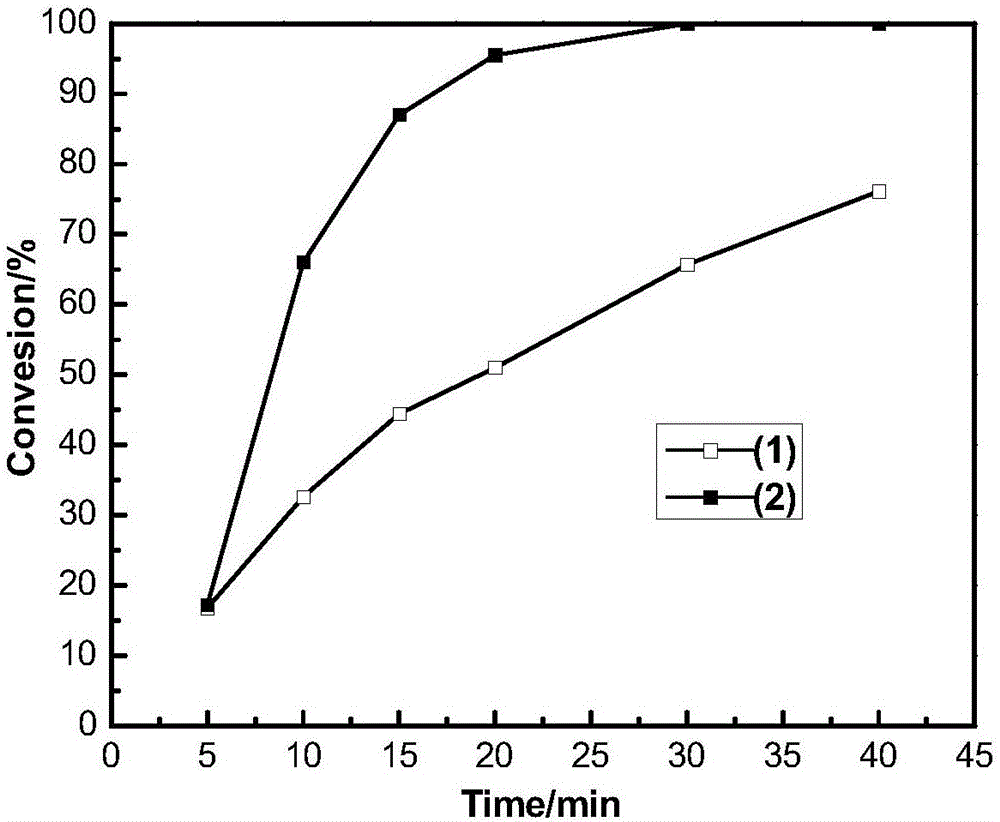
圖2是本發明實施例4與對比例2的苯甲醇有氧氧化動力學曲線,圖2中(1)為對比例2,(2)為實施例4。
(五)具體實施方式
下面通過具體實施例對本發明作進一步的說明,但本發明的保護范圍并不僅限于此。
實施例1:全氟辛基磺酸四乙基銨對苯甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.0032g(0.005mmol)全氟辛基磺酸四乙基銨(TEAPOSF),攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(2,2,6,6-四甲基哌啶-氮氧化物,0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.08g,收率37%,GC含量96%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例2:全氟辛基磺酸四乙基銨對苯甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.0057g(0.009mmol)全氟辛基磺酸四乙基銨(TEAPOSF),攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.09g,收率42%,GC含量96%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例3:全氟辛基磺酸四乙基銨對苯甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.017g(0.027mmol)全氟辛基磺酸四乙基銨(TEAPOSF),攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.19g,收率87%,GC含量97%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例4:全氟辛基磺酸四乙基銨對苯甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.03g(0.047mol)全氟辛基磺酸四乙基銨(TEAPOSF),攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應30min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.19g,收率88%,GC含量99%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例5:全氟辛基磺酸四乙基銨對苯甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.042g(0.065mmol)全氟辛基磺酸四乙基銨(TEAPOSF),攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.206g,收率97%,GC含量99%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例6:全氟辛基磺酸四乙基銨對苯甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.06g(0.094mmol)全氟辛基磺酸四乙基銨(TEAPOSF),攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.208g,收率98%,GC含量99%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例7:全氟辛基磺酸鉀對1-苯乙醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.244g(2mmol)1-苯乙醇、3ml去離子水、0.03g(0.056mmol)全氟辛基磺酸鉀,攪拌5min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0312g(0.2mmol)聯吡啶、0.025g(0.1mmol)五水硫酸銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml 9-氮雜雙環(3,3,1)壬烷-氮氧化物(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應5h,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物1-苯乙醛0.233g,收率97%,GC含量99%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
實施例8:全氟辛基硫酸鈉對吡啶甲醇有氧氧化過程的強化
(1)在50ml燒瓶①中加入0.218g(2mmol)吡啶甲醇、3ml去離子水、0.06g(0.12mmol)全氟辛基硫酸鈉,攪拌10min至表面活性劑溶解,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.0362g(0.1mmol)Cu(OTf)2、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml 2-氮雜雙環金剛烷-氮氧化物(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應5h,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物吡啶甲醛0.205g,收率96%,GC含量98%;分液所得上層水相用乙醚萃取,萃取液蒸除溶劑即可回收氟表面活性劑。
對比例1:
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水,混合均勻,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃條件下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.007g,收率3%,GC含量84%。
對比例2:
(1)在50ml燒瓶①中加入0.216g(2mmol)苯甲醇、3ml去離子水、0.03g(0.1042mmol)十二烷基硫酸鈉(SDS),混合均勻,待用。
(2)在50ml燒瓶②中加入0.0244g(0.2mmol)4-二甲氨基吡啶、0.01g(0.1mmol)氯化亞銅、4.5ml去離子水,在氮氣氛圍下溶解好固體后,停止通氮氣,得到絡合溶液,待用。
(3)將4.5ml步驟(2)所得絡合溶液和2.5ml TEMPO(0.04M)溶液加入步驟(1)燒瓶①中,在空氣氛圍、20℃下反應40min,反應結束后,將反應液進行分液,分液所得下層油相經飽和氯化鈉溶液洗滌,無水硫酸鈉干燥,得到氧化產物苯甲醛0.16g,收率75%,GC含量94%。
附圖1為對比例1與實施例1-6的苯甲醇有氧氧化動力學曲線;附圖2為對比例2與實施例4的苯甲醇有氧氧化動力學曲線;由圖1可以看出,通過在反應體系中添加少量的氟表面活性劑就可以極大提高苯甲醇有氧氧化的反應速率,從而說明了本發明的技術先進性;由圖2可以看出,通過在反應體系中加入不同類型的表面活性劑,也可以提高苯甲醇有氧氧化的反應速率,但是在表面活性劑相同質量條件下氟表面活性劑效果優于烷烴類表面活性劑。
- 還沒有人留言評論。精彩留言會獲得點贊!