一種反應性殼聚糖季銨鹽及其制備方法和應用與流程
本發明屬于紡織染色助劑技術領域,涉及一種紡織助劑及其制備方法和應用,具體涉及一種用于活性染料無鹽染色棉織物的助劑—反應性殼聚糖季銨鹽及其制備方法和應用。
背景技術:
活性染料染色方便,價格較低,色澤鮮艷,色譜齊全,勻染性好。在現今的紡織生產中,活性染料作為一種染棉織物的重要染料。但活性染料染色過程中最突出的問題是固色率低,在棉纖維傳統的染色工藝中,為了提高它的上染率,需要加入大量的無機鹽。但大量無機鹽的加入不僅加大成本,而且造成嚴重的環境污染,破壞生態平衡。隨著人類環保意識的增強,一種用于無鹽條件下的改性劑或助劑成為印染行業的研究熱點。
現有的改性劑或助劑能實現活性染料棉織物低鹽染色。如:1)小分子化合物,棉纖維經小分子改性劑后可以顯著提高對陰離子染料的結合能力,這類改性劑包括環氧類含氮化合物、氮雜環丁烷氯化物、氯三嗪型季銨化合物等,但存在對棉織物親水性差,活性染料上染率提高幅度有限,另外,還存在一定的毒副作用,不容易生物降解等的缺陷;2)交聯劑,合成的無鹽染色交聯劑分子中含有反應性或活性基團,在染色過程中,交聯劑與染料及纖維形成交聯共價鍵,提高纖維的直接性和染色牢度,并還能與水解染料交聯。通常這類交聯無鹽染色助劑大都含有甲醛,雖然反應性較好,但是含有毒物質,不符合綠色環保;3)低甲醛和超低甲醛交聯無鹽染色助劑,但其結構中含有N-羥甲基,在高溫條件下仍會分解釋放甲醛。
殼聚糖季銨鹽由于其具有可溶性好,無毒副作用,可降解等特點,被廣泛研究。在染整的應用性能體現在以下幾個方面:(一)促染:由于殼聚糖季銨鹽分子結構中季銨鹽里的正電性,處理棉織物后,可中和棉織物上的電荷負離子,提高染料與纖維的結合度。(二)固色:季銨鹽由于成膜性較好,能在纖維表面形成薄膜,從而對染料形成保護,而且結構中的陽離子可以提高纖維與陰離子染料之間的結合力,同時對環境無害,這不僅達到了固色效果,還滿足了環保要求。(三)抗菌:殼聚糖因其獨特的結構而具有廣泛抗菌性,棉織物經過殼聚糖季銨鹽整理后也具有抗菌性。(四)印染廢水處理:殼聚糖季銨鹽可以自身作為絮凝劑,也可以與其他物質一起生成符合絮凝劑,用在印染浮水絮凝脫色。
技術實現要素:
本發明的目的克服現有技術存在的缺陷,提供一種反應性殼聚糖季銨鹽及其制備方法,該反應性殼聚糖季銨鹽是用于活性染料棉織物無鹽染色中的一種新型助劑,可溶于水,無毒副作用,用于棉織物的活性染料無鹽染色時,有較好的染色效果,減少了廢水中鹽對環境的污染,大大降低了印染生產成本。
本發明的一種反應性殼聚糖季銨鹽的制備方法,包括以下步驟:
1)殼聚糖與2,3-環氧丙基三甲基氯化銨發生付-克反應,生成N-2-羥丙基三甲基氯化銨殼聚糖;
2)所述N-2-羥丙基三甲基氯化銨殼聚糖與化合物發生取代反應,生成含有活性基團的反應性殼聚糖季銨鹽;
所述化合物為三聚氯氰、四氯嘧啶或N-羥甲基丙烯酰胺。
如上所述的一種反應性殼聚糖季銨鹽的制備方法,所述殼聚糖的重均分子量為1~30萬,脫乙酰度40~90%;所述殼聚糖與所述2,3-環氧丙基三甲基氯化銨的摩爾比為1:0.1~10;所述付-克反應的反應溫度為40~100℃,反應時間為1~28小時。
如上所述的一種反應性殼聚糖季銨鹽的制備方法,所述活性基團為鹵代均三嗪活性基、鹵代嘧啶活性基或丙烯酰胺活性基。
如上所述的一種反應性殼聚糖季銨鹽的制備方法,所述N-2-羥丙基三甲基氯化銨殼聚糖與所述化合物的摩爾比為1:0.1~5;所述取代反應的反應溫度為0~130℃,反應時間為0.5~36小時。
如上所述的一種反應性殼聚糖季銨鹽的制備方法,所述反應性殼聚糖季銨鹽分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,具體為:
Re指活性基團,為鹵代均三嗪活性基、鹵代嘧啶活性基或丙烯酰胺活性基;
所述反應性殼聚糖季銨鹽分子中季銨鹽支鏈的含量為7~50wt%,活性基團支鏈的含量為5~46wt%。
本發明的反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,將反應性殼聚糖季銨鹽用于活性染料棉織物的無鹽染色,具體步驟為:
1)棉織物的預處理:
棉織物常溫下浸入堿液中,然后進行升溫預處理;
2)棉織物的無鹽染色:
預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、堿性化合物和染料,進行無鹽染色;
3)棉織物染色后處理,得到活性染料棉織物。
如上所述的應用,所述的無鹽染色是指在不加鹽條件下對棉織物進行染色處理;所述步驟2)中無鹽染色的溫度為25-70℃;
所述步驟1)中的堿液為NaHCO3溶液或Na2CO3溶液,其濃度為20g/L;所述步驟1)中的常溫為25℃;所述升溫預處理的升溫速率為1.5-2.5℃/min,升溫預處理的溫度為50-70℃,所述升溫預處理的時間為25-35min;
所述步驟2)中,堿性化合物為NaHCO3或Na2CO3;所述反應性殼聚糖季銨鹽與堿性化合物的添加間隔為10-20min;所述堿性化合物與染料的添加間隔為20-40min;
所述步驟2)染色液中,反應性殼聚糖季銨鹽的濃度為0.1~8%owf,堿性化合物濃度為5~20g/L,染料的濃度為0.1~10%owf。
如上所述的應用,所述活性染料棉織物的上染率為70%-85%;固色率為47%-55%,干摩擦牢度為大于4級;濕摩擦牢度大于3級。
本發明反應性殼聚糖季銨鹽的制備方法中反應步驟1)對應的方程式如下:
本發明反應性殼聚糖季銨鹽的制備方法中反應步驟2)對應的方程式如下,其中Re指活性基團:
有益效果:
本發明制備了反應性殼聚糖季銨鹽,制備容易,操作簡單;該反應性殼聚糖季銨鹽是用于活性染料棉織物無鹽染色中的一種助劑,可溶于水,無毒副作用,環保性好,將其用于棉織物的活性染料無鹽染色時,有較好的染色效果,減少了廢水中鹽對環境的污染,大大降低了印染生產成本。
說明書附圖
圖1為本發明的無鹽染色工藝曲線;
圖2為現有技術中常規全鹽染色工藝曲線。
具體實施方式
下面結合具體實施方式,進一步闡述本發明。應理解,這些實施例僅用于說明本發明而不用于限制本發明的范圍。此外應理解,在閱讀了本發明講授的內容之后,本領域技術人員可以對本發明作各種改動或修改,這些等價形式同樣落于本申請所附權利要求書所限定的范圍。
實施例1
一種反應性殼聚糖季銨鹽的制備方法:首先是殼聚糖的預處理,將重均分子量為1萬,脫乙酰度40%的殼聚糖溶于1%的乙酸溶液中,攪拌使其逐漸溶解,然后逐滴加入35wt%的氫氧化鈉溶液,調節pH=9時,有白色沉淀析出,然后常溫浸泡8小時后真空抽濾,用去離子水洗至濾液呈中性后抽干水分,將產物置于真空干燥箱真空干燥。
然后將預處理后的殼聚糖溶于異丙醇中,加熱并攪拌,然后緩慢加入濃度為5wt%的2,3-環氧丙基三甲基氯化銨溶液,發生付-克反應,其中殼聚糖與2,3-環氧丙基三甲基氯化銨按摩爾比為1:0.1,付-克反應的溫度為40℃,時間為24小時,反應后生成N-2-羥丙基三甲基氯化銨殼聚糖;將得到的產物用適量無水乙醇洗滌3次,并離心過濾和真空烘干,得到N-2-羥丙基三甲基氯化銨殼聚糖粗產品,然后將粗產品溶于少量的蒸餾水中,離心過濾得濾液,用濾液體積的5倍的丙酮沉淀出濾液中淡黃色沉淀物,真空烘干得到精制的N-2-羥丙基三甲基氯化銨殼聚糖。
最后,配置濃度為0.124wt%的NaOH溶液和0.142wt%的三聚氯氰溶液,然后將三聚氯氰溶液分三次加入到NaOH溶液中,充分反應使溶液中呈中性,隨后將N-2-羥丙基三甲基氯化銨殼聚糖溶于濃度為0.062wt%的NaOH溶液中,在10℃的水浴溫度下,不斷攪拌逐滴緩慢滴加N-2-羥丙基三甲基氯化銨殼聚糖水溶液,使其與三聚氯氰發生取代反應,其中,N-2-羥丙基三甲基氯化銨殼聚糖與三聚氯氰反應的摩爾比為1:5,反應溫度為0℃,反應時間為36小時,反應后生成反應性殼聚糖季銨鹽,取反應液抽濾,取濾液倒入丙酮劇烈攪拌產生沉淀,靜置,倒出上層丙酮,如此反復洗三次后抽濾,取得固體在真空干燥機烘干,即得到反應性殼聚糖季銨鹽產品,產品分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,為:
制得的反應性殼聚糖季銨鹽分子中,季銨鹽支鏈的含量為7wt%,活性基團的含量為46wt%,Re為氯代均三嗪活性基。
實施例2
一種反應性殼聚糖季銨鹽的制備方法:首先是殼聚糖的預處理,將重均分子量為6萬,脫乙酰度50%的殼聚糖溶于1%的乙酸溶液中,攪拌使其逐漸溶解,然后逐滴加入35wt%的氫氧化鈉溶液,調節pH=9時,有白色沉淀析出,然后常溫浸泡8小時后真空抽濾,用去離子水洗至濾液呈中性后抽干水分,將產物置于真空干燥箱真空干燥。
然后將預處理后的殼聚糖溶于異丙醇中,加熱并攪拌,然后緩慢加入濃度為5wt%的2,3-環氧丙基三甲基氯化銨溶液,發生付-克反應,其中殼聚糖與2,3-環氧丙基三甲基氯化銨按摩爾比為1:2,付-克反應的溫度為55℃,時間為28小時,反應后生成N-2-羥丙基三甲基氯化銨殼聚糖;將得到的產物用適量無水乙醇洗滌3次,并離心過濾和真空烘干,得到N-2-羥丙基三甲基氯化銨殼聚糖粗產品,然后將粗產品溶于少量的蒸餾水中,離心過濾得濾液,用濾液體積的5倍的丙酮沉淀出濾液中淡黃色沉淀物,真空烘干得到精制的N-2-羥丙基三甲基氯化銨殼聚糖。
最后,配置濃度為0.124wt%的NaOH溶液和0.142wt%的三聚氯氰溶液,然后將三聚氯氰溶液分三次加入到NaOH溶液中,充分反應使溶液中呈偏堿性,隨后將N-2-羥丙基三甲基氯化銨殼聚糖溶于濃度為0.062wt%的NaOH溶液中,在10℃的水浴溫度下,不斷攪拌逐滴緩慢滴加N-2-羥丙基三甲基氯化銨殼聚糖水溶液,使其與三聚氯氰發生取代反應,其中,N-2-羥丙基三甲基氯化銨殼聚糖與三聚氯氰反應的摩爾比為1:3,反應溫度為15℃,反應時間為36小時,反應后生成反應性殼聚糖季銨鹽,取反應液抽濾,取濾液倒入丙酮劇烈攪拌產生沉淀,靜置,倒出上層丙酮,如此反復洗三次后抽濾,取得固體在真空干燥機烘干,即得到反應性殼聚糖季銨鹽產品,產品分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,具體為:
制得的反應性殼聚糖季銨鹽中季銨鹽分子中,季銨鹽支鏈的含量為15wt%,活性基團的含量為38wt%,Re為氯代均三嗪活性基。
實施例3
一種反應性殼聚糖季銨鹽的制備方法:首先是殼聚糖的預處理,將重均分子量為10萬,脫乙酰度60%的殼聚糖溶于1%的乙酸溶液中,攪拌使其逐漸溶解,然后逐滴加入35wt%的氫氧化鈉溶液,調節pH=9時,有白色沉淀析出,然后常溫浸泡8小時后真空抽濾,用去離子水洗至濾液呈中性后抽干水分,將產物置于真空干燥箱真空干燥。
然后將預處理后的殼聚糖溶于異丙醇中,加熱并攪拌,然后緩慢加入濃度為5wt%的2,3-環氧丙基三甲基氯化銨溶液,發生付-克反應,其中殼聚糖與2,3-環氧丙基三甲基氯化銨按摩爾比為1:4,付-克反應的溫度為65℃,時間為14小時,反應后生成N-2-羥丙基三甲基氯化銨殼聚糖;將得到的產物用適量無水乙醇洗滌3次,并離心過濾和真空烘干,得到N-2-羥丙基三甲基氯化銨殼聚糖粗產品,然后將粗產品溶于少量的蒸餾水中,離心過濾得濾液,用濾液體積的5倍的丙酮沉淀出濾液中淡黃色沉淀物,真空烘干得到精制的N-2-羥丙基三甲基氯化銨殼聚糖。
最后,配置濃度為0.124wt%的NaOH溶液和0.142wt%的四氯嘧啶溶液,然后將四氯嘧啶液分三次加入到NaOH溶液中,充分反應使溶液中偏堿性,隨后將N-2-羥丙基三甲基氯化銨殼聚糖溶于濃度為0.062wt%的NaOH溶液中,在10℃的水浴溫度下,不斷攪拌逐滴緩慢滴加N-2-羥丙基三甲基氯化銨殼聚糖水溶液,使其與四氯嘧啶發生取代反應,其中,N-2-羥丙基三甲基氯化銨殼聚糖與四氯嘧啶反應的摩爾比為1:1.5,反應溫度為10℃,反應時間為16小時,反應后生成反應性殼聚糖季銨鹽,取反應液抽濾,取濾液倒入丙酮劇烈攪拌產生沉淀,靜置,倒出上層丙酮,如此反復洗三次后抽濾,取得固體在真空干燥機烘干,即得到反應性殼聚糖季銨鹽產品,產品分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,具體為:
制得的反應性殼聚糖季銨鹽中季銨鹽的含量為26wt%,活性基團的含量為30wt%,Re為氯代嘧啶活性基。
實施例4
一種反應性殼聚糖季銨鹽的制備方法:首先是殼聚糖的預處理,將重均分子量為15萬,脫乙酰度70%的殼聚糖溶于1%的乙酸溶液中,攪拌使其逐漸溶解,然后逐滴加入35wt%的氫氧化鈉溶液,調節pH=9時,有白色沉淀析出,然后常溫浸泡8小時后真空抽濾,用去離子水洗至濾液呈中性后抽干水分,將產物置于真空干燥箱真空干燥。
然后將預處理后的殼聚糖溶于異丙醇中,加熱并攪拌,然后緩慢加入濃度為5wt%的2,3-環氧丙基三甲基氯化銨溶液,發生付-克反應,其中殼聚糖與2,3-環氧丙基三甲基氯化銨按摩爾比為1:6,付-克反應的溫度為80℃,時間為9小時,反應后生成N-2-羥丙基三甲基氯化銨殼聚糖;將得到的產物用適量無水乙醇洗滌3次,并離心過濾和真空烘干,得到N-2-羥丙基三甲基氯化銨殼聚糖粗產品,然后將粗產品溶于少量的蒸餾水中,離心過濾得濾液,用濾液體積的5倍的丙酮沉淀出濾液中淡黃色沉淀物,真空烘干得到精制的N-2-羥丙基三甲基氯化銨殼聚糖。
最后,配置濃度為0.124wt%的NaOH溶液和0.142wt%的四氯嘧啶溶液,然后將四氯嘧啶溶液分三次加入到NaOH溶液中,充分反應使溶液中呈中性,隨后將N-2-羥丙基三甲基氯化銨殼聚糖溶于濃度為0.062wt%的NaOH溶液中,在10℃的水浴溫度下,不斷攪拌逐滴緩慢滴加N-2-羥丙基三甲基氯化銨殼聚糖水溶液,使其與四氯嘧啶發生取代反應,其中,N-2-羥丙基三甲基氯化銨殼聚糖與四氯嘧啶反應的摩爾比為1:1,反應溫度為9℃,反應時間為20小時,反應后生成反應性殼聚糖季銨鹽,取反應液抽濾,取濾液倒入丙酮劇烈攪拌產生沉淀,靜置,倒出上層丙酮,如此反復洗三次后抽濾,取得固體在真空干燥機烘干,即得到反應性殼聚糖季銨鹽產品,產品分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,具體為:
制得的反應性殼聚糖季銨鹽中季銨鹽的含量為34wt%,活性基團的含量為20wt%,Re為氯代嘧啶活性基。
實施例5
一種反應性殼聚糖季銨鹽的制備方法:首先是殼聚糖的預處理,將重均分子量為20萬,脫乙酰度80%的殼聚糖溶于1%的乙酸溶液中,攪拌使其逐漸溶解,然后逐滴加入35wt%的氫氧化鈉溶液,調節pH=9時,有白色沉淀析出,然后常溫浸泡8小時后真空抽濾,用去離子水洗至濾液呈中性后抽干水分,將產物置于真空干燥箱真空干燥。
然后將預處理后的10g殼聚糖溶于異丙醇中,加熱并攪拌,然后緩慢加入濃度為5wt%的2,3-環氧丙基三甲基氯化銨溶液,發生付-克反應,其中殼聚糖與2,3-環氧丙基三甲基氯化銨按摩爾比為1:8,付-克反應的溫度為100℃,時間為10小時,反應后生成N-2-羥丙基三甲基氯化銨殼聚糖;將得到的產物用適量無水乙醇洗滌3次,并離心過濾和真空烘干,得到N-2-羥丙基三甲基氯化銨殼聚糖粗產品,然后將粗產品溶于少量的蒸餾水中,離心過濾得濾液,用濾液體積的5倍的丙酮沉淀出濾液中淡黃色沉淀物,真空烘干得到精制的N-2-羥丙基三甲基氯化銨殼聚糖。
最后,配置48wt%的N-羥甲基丙烯酰胺溶液,加入N-2-羥丙基三甲基氯化銨殼聚糖,使其完全溶解,然后加入0.2wt%對苯二酚和10wt%氯化銨,攪拌均勻,加熱至100℃,3小時結束反應。其中,N-2-羥丙基三甲基氯化銨殼聚糖與N-羥甲基丙烯酰胺反應的摩爾比為1:0.5,反應后生成反應性殼聚糖季銨鹽,先用甲醇溶解再用丙酮溶解,重復兩次。然后用體積比為1:1的乙醇和丙酮的混合溶劑洗滌2次,取得固體在真空干燥機烘干,即得到反應性殼聚糖季銨鹽產品,產品分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,具
體為:
制得的反應性殼聚糖季銨鹽中季銨鹽的含量為42wt%,活性基團的含量為13wt%,Re為丙烯酰胺活性基。
實施例6
一種反應性殼聚糖季銨鹽的制備方法:首先是殼聚糖的預處理,將重均分子量為30萬,脫乙酰度90%的殼聚糖溶于1%的乙酸溶液中,攪拌使其逐漸溶解,然后逐滴加入35wt%的氫氧化鈉溶液,調節pH=9時,有白色沉淀析出,然后常溫浸泡8小時后真空抽濾,用去離子水洗至濾液呈中性后抽干水分,將產物置于真空干燥箱真空干燥。
然后將預處理后的10g殼聚糖溶于異丙醇中,加熱并攪拌,然后緩慢加入濃度為5wt%的2,3-環氧丙基三甲基氯化銨溶液,發生付-克反應,其中殼聚糖與2,3-環氧丙基三甲基氯化銨按摩爾比為1:10,付-克反應的溫度為90℃,時間為13小時,反應后生成N-2-羥丙基三甲基氯化銨殼聚糖;將得到的產物用適量無水乙醇洗滌3次,并離心過濾和真空烘干,得到N-2-羥丙基三甲基氯化銨殼聚糖粗產品,然后將粗產品溶于少量的蒸餾水中,離心過濾得濾液,用濾液體積的5倍的丙酮沉淀出濾液中淡黃色沉淀物,真空烘干得到精制的N-2-羥丙基三甲基氯化銨殼聚糖。
最后,配置48wt%的N-羥甲基丙烯酰胺溶液,加入N-2-羥丙基三甲基氯化銨殼聚糖,使其完全溶解,然后加入0.2wt%對苯二酚和10wt%氯化銨,攪拌均勻,加熱至130℃,0.5小時結束反應。其中,N-2-羥丙基三甲基氯化銨殼聚糖與N-羥甲基丙烯酰胺反應的摩爾比為1:0.5,反應后生成反應性殼聚糖季銨鹽,先用甲醇溶解再用丙酮溶解,重復兩次。然后用體積比為1:1的乙醇和丙酮的混合溶劑洗滌2次,取得固體在真空干燥機烘干,即得到反應性殼聚糖季銨鹽產品,產品分子結構中含有殼聚糖主鏈結構、季銨鹽支鏈結構和活性基團支鏈結構,具體為:
制得的反應性殼聚糖季銨鹽中季銨鹽的含量為50wt%,活性基團的含量為5wt%,Re為丙烯酰胺活性基。
實施例7-9
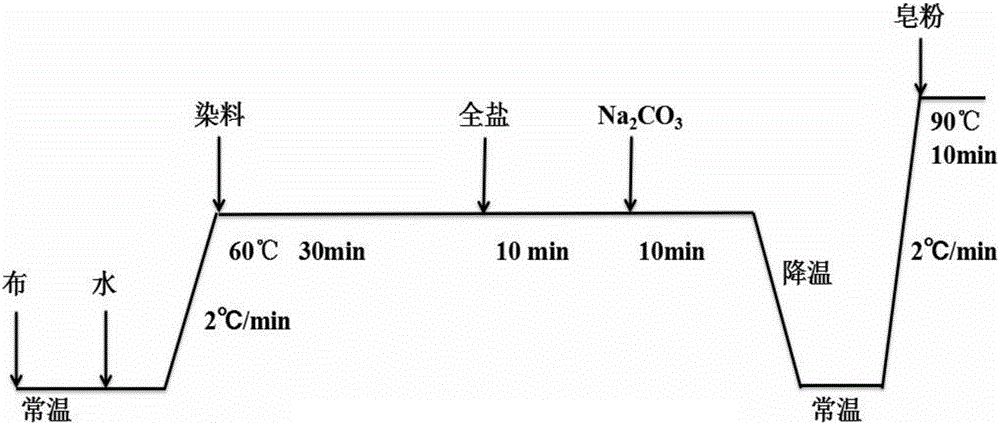
一種反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,步驟為:1)預處理棉織物:棉織物在25℃下浸入NaHCO3溶液中,然后進行升溫預處理;2)棉織物的無鹽染色:預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、碳酸鈉和染料,進行無鹽染色;3)棉織物染色后處理:染色后自然冷卻降溫至25℃,然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物,具體工藝流程如圖1所示。
本實施例中步驟1)和步驟2)采用的具體工藝配方和條件如下:
1)預處理棉織物:
2)染色液中添加反應性殼聚糖季銨鹽,對棉織物進行無鹽染色:
反應性殼聚糖季銨鹽與堿性化合物的添加間隔為15min;
堿性化合物與染料的添加間隔為30min;
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,具體過程為:
1)對本實施例制得的活性染料棉織物進行上染率的測定,根據染料的標準曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本實施例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本實施例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本實施例中采用DyStar公司生產的紅、黃、藍三種染料,采用的反應性殼聚糖季銨鹽的制備方法與實施例1的制備方法相同,采用本實施例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。由表中的數據可以看出,采用反應性殼聚糖季銨鹽作為活性染料無鹽染色助劑染色效果良好,得到的織物上染率(>70%)和固色率(>47%)較高,耐摩擦色牢度較高,能夠滿足生產和加工的需求。
實施例10-12
一種反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,步驟為:1)預處理棉織物:棉織物在25℃下浸入碳酸鈉溶液中,然后進行升溫預處理;2)棉織物的無鹽染色:預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、碳酸氫鈉和染料,進行無鹽染色;3)棉織物染色后處理:染色后自然冷卻降溫至25℃,然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物。
本實施例中步驟1)和步驟2)采用的具體工藝配方和條件如下:
1)預處理棉織物:
2)染色液中添加反應性殼聚糖季銨鹽,對棉織物進行無鹽染色:
反應性殼聚糖季銨鹽與堿性化合物的添加間隔為10min;
堿性化合物與染料的添加間隔為20min;
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,具體過程為:
1)對本實施例制得的活性染料棉織物進行上染率的測定,根據染料的標準
曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本實施例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本實施例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本實施例中采用DyStar公司生產的紅、黃、藍三種染料,采用的反應性殼聚糖季銨鹽的制備方法與實施例2的制備方法相同,采用本實施例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。分析表中的數據得到的結論同實施例7~9,采用反應性殼聚糖季銨鹽作為活性染料無鹽染色助劑,在無鹽條件下,能夠獲得優良的染色效果。
實施例13-15
一種反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,步驟為:1)預處理棉織物:棉織物在25℃下浸入碳酸氫鈉溶液中,然后進行升溫預處理;2)棉織物的無鹽染色:預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、堿性化合物和染料,進行無鹽染色;3)棉織物染色后處理:染色后自然冷卻降溫至25℃,然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物。
本實施例中步驟1)和步驟2)采用的具體工藝配方和條件如下:
1)預處理棉織物:
2)染色液中添加反應性殼聚糖季銨鹽,對棉織物進行無鹽染色:
反應性殼聚糖季銨鹽與堿性化合物的添加間隔為20min;
堿性化合物與染料的添加間隔為40min;
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,具體過程為:
1)對本實施例制得的活性染料棉織物進行上染率的測定,根據染料的標準曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本實施例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本實施例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本實施例中采用DyStar公司生產的紅、黃、藍三種染料,采用的反應性殼聚糖季銨鹽的制備方法與實施例3的制備方法相同,采用本實施例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。分析表中的數據得到的結論同實施例7~9,采用反應性殼聚糖季銨鹽作為活性染料無鹽染色助劑,在無鹽條件下,能夠獲得優良的染色效果。
實施例16-18
一種反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,步驟為:1)預處理棉織物:棉織物在25℃下浸入碳酸鈉溶液中,然后進行升溫預處理;2)棉織物的無鹽染色:預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、碳酸氫鈉和染料,進行無鹽染色;3)棉織物染色后處理:染色后自然冷卻降溫至25℃,然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物。
本實施例中步驟1)和步驟2)采用的具體工藝配方和條件如下:
1)預處理棉織物:
2)染色液中添加反應性殼聚糖季銨鹽,對棉織物進行無鹽染色:
反應性殼聚糖季銨鹽與堿性化合物的添加間隔為18min;
堿性化合物與染料的添加間隔為35min;
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,具體過程為:
1)對本實施例制得的活性染料棉織物進行上染率的測定,根據染料的標準曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本實施例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本實施例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本實施例中采用DyStar公司生產的紅、黃、藍三種染料,采用的反應性殼聚糖季銨鹽的制備方法與實施例4的制備方法相同,采用本實施例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。分析表中的數據得到的結論同實施例7~9,采用反應性殼聚糖季銨鹽作為活性染料無鹽染色助劑,在無鹽條件下,能夠獲得優良的染色效果。
實施例19-21
一種反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,步驟為:1)預處理棉織物:棉織物在25℃下浸入碳酸氫鈉溶液中,然后進行升溫預處理;2)棉織物的無鹽染色:預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、碳酸氫鈉和染料,進行無鹽染色;3)棉織物染色后處理:染色后自然冷卻降溫至25℃,然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物。
本實施例中步驟1)和步驟2)采用的具體工藝配方和條件如下:
1)預處理棉織物:
2)染色液中添加反應性殼聚糖季銨鹽,對棉織物進行無鹽染色:
反應性殼聚糖季銨鹽與堿性化合物的添加間隔為20min;
堿性化合物與染料的添加間隔為30min;
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,
具體過程為:
1)對本實施例制得的活性染料棉織物進行上染率的測定,根據染料的標準曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本實施例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本實施例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本實施例中采用DyStar公司生產的紅、黃、藍三種染料,采用的反應性殼聚糖季銨鹽的制備方法與實施例5的制備方法相同,采用本實施例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。分析表中的數據得到的結論同實施例7~9,采用反應性殼聚糖季銨鹽作為活性染料無鹽染色助劑,在無鹽條件下,能夠獲得優良的染色效果。
實施例22-24
一種反應性殼聚糖季銨鹽在活性染料棉織物無鹽染色中的應用,步驟為:1)預處理棉織物:棉織物在25℃下浸入碳酸鈉溶液中,然后進行升溫預處理;2)棉織物的無鹽染色:預處理后的棉織物浸入染色液中,然后往染色液中按一定時間間隔依次加入反應性殼聚糖季銨鹽、碳酸鈉和染料,進行無鹽染色;3)棉織物染色后處理:染色后自然冷卻降溫至25℃,然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物。
本實施例中步驟1)和步驟2)采用的具體工藝配方和條件如下:
1)預處理棉織物:
2)染色液中添加反應性殼聚糖季銨鹽,對棉織物進行無鹽染色:
反應性殼聚糖季銨鹽與堿性化合物的添加間隔為10min;
堿性化合物與染料的添加間隔為40min;
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,具體過程為:
1)對本實施例制得的活性染料棉織物進行上染率的測定,根據染料的標準曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本實施例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本實施例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本實施例中采用DyStar公司生產的紅、黃、藍三種染料,采用的反應性殼聚糖季銨鹽的制備方法與實施例6的制備方法相同,采用本實施例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。分析表中的數據得到的結論同實施例7~9,采用反應性殼聚糖季銨鹽作為活性染料無鹽染色助劑,在無鹽條件下,能夠獲得優良的染色效果。
對比例1-3
為了對比考察棉織物經反應性殼聚糖季銨鹽處理后的棉織物無鹽染色的染色效果,將棉織物進行全鹽染色,步驟為:1)預處理棉織物;2)對棉織物進行全鹽染色;3)染色后自然降溫至25℃;4)然后以2℃/min的升溫速率升至90℃,進行皂洗10min,得到活性染料棉織物,具體工藝流程如圖2所示。
本對比例中步驟2)采用的具體工藝配方和條件如下:
將制得的活性染料棉織物分別進行上染率、固色率和耐摩擦色牢度的測定,具體過程為:
1)對本對比例制得的活性染料棉織物進行上染率的測定,根據染料的標準曲線,計算出染色前后染料的濃度,通過下式計算染料的上染率E%:
式中:C0為染前濃度,V0為染前體積,a為染后殘液測吸光度時稀釋倍數,C1為染后殘液濃度,V1為染后體積。
2)對本對比例制得的活性染料棉織物進行固色率的測定,根據染料的標準曲線,計算皂洗前后染料的濃度,通過下式計算染料的固色率F%:
式中:C0—染色前的染液濃度,V0—染色前的染液體積,a—測吸光度時染色后的殘液稀釋倍數,C1—染色后的殘液濃度,V1—染色后的染液體積。b—皂洗液測吸光度時稀釋倍數,C2—皂洗后殘液濃度,V2—皂洗液體積。
3)對本對比例制得的活性染料棉織物進行耐摩擦色牢度的測試,檢測耐摩擦牢度時必須嚴格按照標準GB/T3920—2008中的檢測方案對已染織物進行檢測,測試完成后,用對應的沾色用灰色樣卡進行觀察評級,得出結果。
本對比例中采用DyStar公司生產的紅、黃、藍三種染料,采用本對比例中制備流程,制得的活性染料棉織物進行上染率、固色率和耐摩擦色牢度的測定結果見下表。
通過對比可看出加入反應性殼聚糖季銨鹽后無鹽染色得到的棉織物的染色性能與全鹽染色織物接近,全鹽染色織物的上染率和固色率較高,雖然能夠滿足生產和加工的需要,但是鹽分加入量過大,提高了生產成本,同時造成印染廢水處理困難,環境污染嚴重等問題,加入反應性殼聚糖季銨鹽作為染色助劑能夠達到較好的染色效果,滿足生產和加工需要的同時實現了無鹽染色,避免了全鹽染色存在的問題。
- 還沒有人留言評論。精彩留言會獲得點贊!