牛樟芝素的制備方法與流程
本發明涉及一種牛樟芝素的制備方法。
背景技術:
antrocin(牛樟芝素)是由姜宏哲(hung-chenchiang)教授和他的合作者于1994年從中國臺灣牛樟芝(又名牛樟菇,非褶菌目、多孔科、多年生蕈菌類,學名為antrodiacamphorata)中分離出來的,分子結構式為
牛樟芝的活性成分三萜類化合物除個別特殊外在其他薄孔菌屬的真菌中也均有發現。但是倍半萜化合物牛樟芝素卻唯有在野生牛樟芝的子實體中才能分離到,人工手段培養的牛樟芝尚未有報道分離得到牛樟芝素。
從結構上看,牛樟芝素是一種倍半萜稀內酯,具備一個6.6.5三環結構,包含三個手性中心及兩個全碳取代中心。2011年,臺灣朝陽科技大學的曾耀銘教授對牛樟芝素的生物活性進行研究。研究發現該分子具有選擇性抑制人轉移性乳腺癌細胞mda-mb-231細胞系的生理活性,其ic50值為0.6μm。2013年,曾耀銘教授又發現牛樟芝素在抑制人非小細胞肺癌細胞有很好的抑制增殖作用,對具野生型表皮生長因子受體(egfr)的肺癌細胞h441的ic50值為0.75μm,而對具突變型egfr的h1975肺癌細胞的ic50值0.83μm。
牛樟芝素具有良好的潛在抗癌活性,目前的人工合成報道,而且還未見有不對此合成的報道,因此目前的合成技術尚無法提供充足的牛樟芝素,以對其進行深入的活性及構效關系、毒理病理研究。因此,急需找到一種可以大量合成牛樟芝素的方法,以便提供足夠的牛樟芝素來研究其病理毒理、構效關系,并且通過結構修飾來闡明和改進它的生物活性。
技術實現要素:
基于此,有必要提供一種可以大量合成的牛樟芝素的牛樟芝素的制備方法,通過以化合合成手段大量的得到牛樟芝素,以便對牛樟芝素進行結構修飾和活性研究。
一種牛樟芝素的制備方法,包括如下步驟:
步驟一、將結構式為
步驟二、向所述第一反應液中加入硼氫化鈉,劇烈攪拌并充分反應,反應完成后純化得到結構式為
步驟三、在-78℃的條件下,將所述化合物a溶解于第二有機溶劑中,分批次投入碎鈉塊,攪拌并充分反應,得到含有結構式為
步驟四、除去所述第二反應液中的有機溶劑后酸化并萃取所得產物,接著除去萃取劑并加入第三有機溶劑,然后加入對甲苯磺酸,于60℃~100℃下充分反應,反應完成后純化得到結構式為
步驟五、將所述化合物d溶解于第四有機溶劑中,加入碘、三苯基膦和咪唑,攪拌并充分反應,反應完成后純化得到結構式為
步驟六、將所述化合物d1溶解于第五有機溶劑中,加入第一有機堿,在80℃下攪拌并充分反應,反應完成后純化得到牛樟芝素。
在一個實施例中,步驟一中,所述第一有機溶劑為二氯甲烷、甲醇或體積比為1~6:1的二氯甲烷和甲醇的混合液體;
步驟一中,所述鼓氣通入臭氧的操作的時間為1h。
在一個實施例中,所述第一有機溶劑為體積比為3:1的二氯甲烷和甲醇的混合液體、體積比為4:1的二氯甲烷和甲醇的混合液體或體積比為5:1的二氯甲烷和甲醇的混合液體。
在一個實施例中,步驟二中,所述劇烈攪拌并充分反應的操作的時間為1h。
在一個實施例中,步驟三中還包括在所述分批次投入碎鈉塊的操作之后,加入低元醇的操作,所述低元醇的碳原子數為1~4。
在一個實施例中,步驟三中,所述第二有機溶劑為液氨或體積比為5~10:1的液氨與四氫呋喃的混合液體;
步驟三中,所述劇烈攪拌并充分反應的操作的時間為1h。
在一個實施例中,步驟四中,所述酸化的操作所用的酸為鹽酸或醋酸,所述酸化的操作為酸化至ph為1~2;
步驟四中,所述萃取的操作中,所述萃取劑為乙酸乙酯或二氯甲烷。
在一個實施例中,步驟四中,所述第三有機溶劑為甲苯、苯或四氫呋喃;
步驟四中,所述100℃下充分反應的時間為1h~3h。
在一個實施例中,步驟五中,所述第四有機溶劑為四氫呋喃或二氯甲烷;
步驟五中,所述攪拌并充分反應的操作的時間為1h~3h。
在一個實施例中,步驟六中,所述第一有機堿為1,8-二氮雜二環十一碳-7-烯或乙醇鈉;步驟六中,所述在80℃下攪拌并充分反應的操作的時間為3h。
這種牛樟芝素的制備方法操作簡單,步驟簡短,易于工業化生產,通過步驟一的臭氧氧化切斷反應、步驟三的伯奇還原反應、步驟四酯縮合反應以及步驟五和通過步驟六的碘代-消除反應,快速地構建了分子的骨架結構,從而完成了牛樟芝素的不對稱全合成。這種牛樟芝素的制備方法可以大量合成的牛樟芝素,合成的牛樟芝素可以用來研究其病理毒理、構效關系,并且通過結構修飾來闡明和改進它的生物活性。
附圖說明
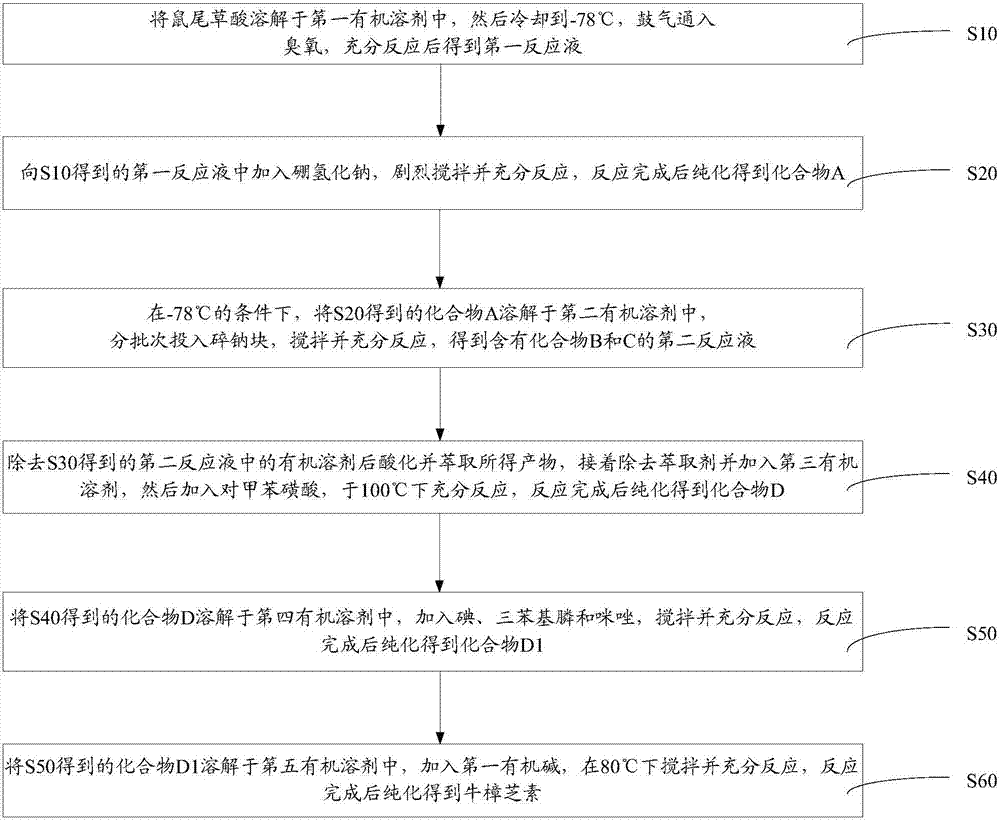
圖1為一實施方式的牛樟芝素的制備方法的制備方法的流程圖。
具體實施方式
為使本發明的上述目的、特征和優點能夠更加明顯易懂,下面結合附圖和具體實施例對本發明的具體實施方式做詳細的說明。在下面的描述中闡述了很多具體細節以便于充分理解本發明。但是本發明能夠以很多不同于在此描述的其它方式來實施,本領域技術人員可以在不違背本發明內涵的情況下做類似改進,因此本發明不受下面公開的具體實施的限制。
本發明的牛樟芝素的制備方法,其各步反應均為在氮氣保護下、干燥的溶劑中、無水的條件下進行。
本發明的牛樟芝素的制備方法中,所有使用的化學試劑都是商業可得的原料,無須進一步處理。
如圖1所示的一實施方式的牛樟芝素的制備方法,包括如下步驟:
s10、將鼠尾草酸溶解于第一有機溶劑中,然后冷卻到-78℃,鼓氣通入臭氧,充分反應后得到第一反應液。
鼠尾草酸的結構式為
s10中,第一有機溶劑可以為二氯甲烷、甲醇或體積比為2~6:1的二氯甲烷和甲醇的混合液體。
s10中,鼓氣通入臭氧的操作的時間為1h。
優選的,第一有機溶劑為體積比為3:1的二氯甲烷和甲醇的混合液體、體積比為4:1的二氯甲烷和甲醇的混合液體或體積比為5:1的二氯甲烷和甲醇的混合液體。
特別優選的,第一有機溶劑為體積比為3:1的二氯甲烷和甲醇的混合液體。
s20、向s10得到的第一反應液中加入硼氫化鈉,劇烈攪拌并充分反應,反應完成后純化得到化合物a。
化合物a的結構式為
s20中,鼠尾草酸與硼氫化鈉的摩爾比為1:3~10。
s20中,劇烈攪拌并充分反應的操作的時間為1h。
s20中,反應完成后純化得到化合物a的操作為:加入飽和的氯化銨溶液淬滅反應,用乙酸乙酯萃取,有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮,然后用硅膠柱層析純化,得到化合物a。
s30、在-78℃的條件下,將s20得到的化合物a溶解于第二有機溶劑中,分批次投入碎鈉塊,攪拌并充分反應,得到含有化合物b和c的第二反應液。
化合物b結構式為
s30中,化合物a與碎鈉塊的摩爾比為1:5~20;
s30中,第二有機溶劑為液氨或體積比為5:1~10:1的液氨與四氫呋喃的混合液體。
優選的,第二有機溶劑為液氨。
s30中,劇烈攪拌并充分反應的操作的時間為1h。
s30中還包括在分批次投入碎鈉塊的操作之后,加入低元醇的操作。低元醇的碳原子數為1~4。
低元醇與化合物a的摩爾比為5~10:1。
低元醇可以為甲醇、乙醇、叔丁醇或異丙醇。
優選的,低元醇可以為乙醇。
s40、除去s30得到的第二反應液中的有機溶劑后酸化并萃取所得產物,接著除去萃取劑并加入第三有機溶劑,然后加入對甲苯磺酸,于100℃下充分反應,反應完成后純化得到化合物d。
化合物d的結構式為
s40中,對甲苯磺酸和化合物a的摩爾比為0.1~0.5:1。
s40中,酸化的操作所用的酸為鹽酸或醋酸,酸化的操作為酸化至ph為1~2。
優選的,酸化的操作所用的酸為鹽酸。
s40中,萃取的操作中,萃取劑為乙酸乙酯或二氯甲烷。
優選的,萃取劑為乙酸乙酯。
s40中,第三有機溶劑為甲苯、苯或四氫呋喃;
s40中,100℃下充分反應的時間為1h~3h。
s40中的反應,在得到化合物d的同時還會得到結構式為
通過在s30中加入低元醇,可以增加s40得到的反應物中化合物d的比例,提高了產率。
s40中,反應完成后純化得到化合物d的操作為:加入飽和碳酸氫鈉溶液,用乙酸乙酯萃取,有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮,然后用硅膠柱層析純化,得到化合物d。
s50、將s40得到的化合物d溶解于第四有機溶劑中,加入碘、三苯基膦和咪唑,攪拌并充分反應,反應完成后純化得到化合物d1。
化合物d1的結構式為
s50中,碘與化合物d的摩爾比為1.1~1.6:1,三苯基膦與化合物d的摩爾比為1~1.3:1,咪唑與化合物d的摩爾比為1.2~1.7:1。
s50中,第四有機溶劑為四氫呋喃或二氯甲烷。
優選的,第四有機溶劑為四氫呋喃。
s50中,攪拌并充分反應的操作的時間為3h。
反應完成后純化得到化合物d1的操作為:加入飽和氯化銨溶液淬滅反應,用乙酸乙酯萃取,有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮,然后用硅膠柱層析純化,得到化合物d1。
s60、將s50得到的化合物d1溶解于第五有機溶劑中,加入第一有機堿,在80℃下攪拌并充分反應,反應完成后純化得到牛樟芝素。
s60中,第五有機溶劑為甲苯、苯或乙醇。
s60中,第一有機堿為1,8-二氮雜二環十一碳-7-烯或乙醇鈉。
優選的,第一有機堿為1,8-二氮雜二環十一碳-7-烯。
s60中,在80℃下攪拌并充分反應的操作的時間為3h。
這種牛樟芝素的制備方法操作簡單,步驟簡短,易于工業化生產,通過s10的臭氧氧化切斷反應、s30的伯奇還原反應、s40酯縮合反應以及s50和s60的碘代-消除反應,快速地構建了分子的骨架結構,從而完成了牛樟芝素的不對稱全合成。這種牛樟芝素的制備方法可以大量合成的牛樟芝素,合成的牛樟芝素可以用來研究其病理毒理、構效關系,并且通過結構修飾來闡明和改進它的生物活性。
以下為具體實施例。
實施例中,反應檢測時使用的是0.25mm煙臺銀龍硅膠有限公司生產的薄層色譜硅膠板(60f-254);柱層析使用青島譜科分離材料有限公司生產的200-300目硅膠,使用石油醚沸程為60~90℃;核磁共振數據由以下儀器測得:brükeradvance500或brükeradvance400或brükeradvance300,使用tms或氘代溶劑中殘留的未氘代溶劑做內標,解釋多重裂分時所用到的簡寫為s單峰,d雙重裂分,t三重裂分,q四重裂分,m多重裂分,br寬峰;質譜由absciex
實施例1
在無水的條件下,將結構式為
在第一反應液中加入硼氫化鈉(12.6g,450.0mmol),劇烈攪拌1小時后,加入飽和的氯化銨溶液淬滅反應,用乙酸乙酯萃取(3×400ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到11.5g結構式為
在-78℃的條件下,將化合物a(9.0g,34.1mmol)溶解于液氨(500ml)中,分批次投入剪碎的鈉塊(11.8g,511.5mmol),攪拌2小時,得到包括結構式為
小心緩慢的往第二反應液中滴加氯化銨溶液(250ml),淬滅反應后多余的液氨在真空下除去,加入適量鹽酸,調節溶液ph=1~2,所得的反應液用乙酸乙酯(3×300ml)萃取,有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。濃縮產物溶解于甲苯中(100ml),加入對甲苯磺酸(1.44g,10.2mmol),于100℃攪拌2小時,冷卻到室溫后小心的加入飽和碳酸氫鈉溶液(100ml),用乙酸乙酯萃取(3×300ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到3.53g結構式為
將化合物d(1.0g,3.97mmol)溶解于四氫呋喃(30ml)中,依次加入碘(1.51g,5.95mmol)、三苯基膦(1.35g,5.16mmol)、咪唑(405mg,5.95mmol),室溫攪拌3小時,加入飽和氯化銨溶液(100ml)淬滅反應,用乙酸乙酯萃取(3×300ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到1.44g結構式為
將化合物d1(2.1g,5.80mmol)溶解于甲苯(50ml)中,加入1,8-二氮雜二環十一碳-7-烯(8.6ml,58.0mmol),在80℃攪拌反應3小時,加入飽和氯化銨溶液(100ml)淬滅反應,用乙酸乙酯萃取(3×300ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到0.68g牛樟芝素(產率50%)。牛樟芝素的檢測數據如下:
實施例2
在無水的條件下,將結構式為
在第一反應液中加入硼氫化鈉(12.6g,450.0mmol),劇烈攪拌1小時后,加入飽和的氯化銨溶液淬滅反應,用乙酸乙酯萃取(3×400ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到11.5g結構式為
在-78℃的條件下,將化合物a(9.0g,34.1mmol)溶解于液氨(500ml)中,分批次投入剪碎的鈉塊(11.8g,511.5mmol)后加入乙醇(10ml),攪拌2小時,得到包括結構式為
小心緩慢的往第二反應液中滴加乙醇(100ml),淬滅反應后多余的液氨在真空下除去,加入適量鹽酸,調節溶液ph=1~2,所得的反應液用乙酸乙酯(3×300ml)萃取,有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。濃縮產物溶解于甲苯中(100ml),加入對甲苯磺酸(1.44g,10.2mmol),于60℃攪拌1小時,冷卻到室溫后小心的加入飽和碳酸氫鈉溶液(100ml),用乙酸乙酯萃取(3×300ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到4.76g結構式為
將化合物d(1.0g,3.97mmol)溶解于四氫呋喃(30ml)中,依次加入碘(1.51g,5.95mmol)、三苯基膦(1.35g,5.16mmol)、咪唑(405mg,5.95mmol),室溫攪拌3小時,加入飽和氯化銨溶液(100ml)淬滅反應,用乙酸乙酯萃取(3×300ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到1.44g結構式為
將化合物d1(2.1g,5.80mmol)溶解于甲苯(50ml)中,加入1,8-二氮雜二環十一碳-7-烯(8.6ml,58.0mmol),在80℃攪拌反應3小時,加入飽和氯化銨溶液(100ml)淬滅反應,用乙酸乙酯萃取(3×300ml),有機相用飽和氯化鈉溶液洗滌、無水硫酸鈉干燥,過濾后真空濃縮。然后用硅膠柱層析純化,得到0.68g牛樟芝素(產率50%)。
以上所述實施例僅表達了本發明的幾種實施方式,其描述較為具體和詳細,但并不能因此而理解為對本發明專利范圍的限制。應當指出的是,對于本領域的普通技術人員來說,在不脫離本發明構思的前提下,還可以做出若干變形和改進,這些都屬于本發明的保護范圍。因此,本發明專利的保護范圍應以所附權利要求為準。
- 還沒有人留言評論。精彩留言會獲得點贊!