一種抗血管新生化合物的制作方法
技術領域:
本發明涉及一種新化合物和用途。
背景技術:
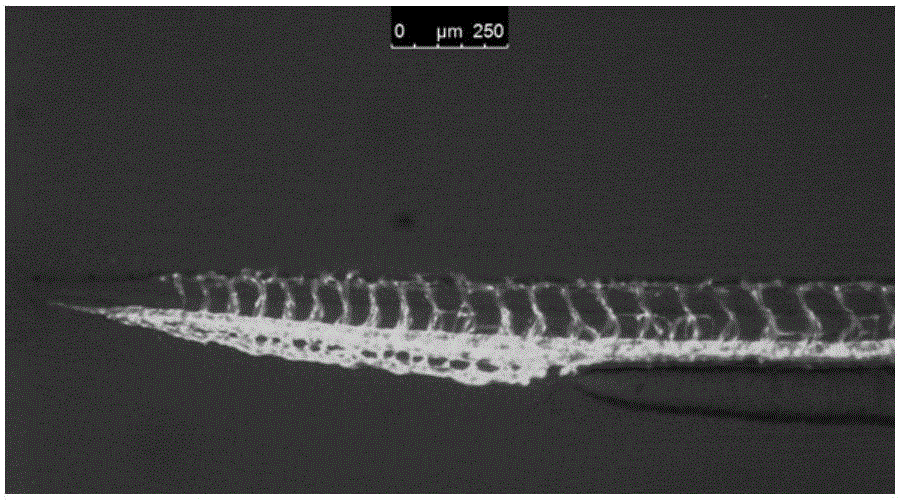
:血管新生(angiogenesis),是從已有血管發芽生成新血管的過程。這一過程與血管內皮細胞遷移和增殖相關。血管新生與多種人類重大疾病有關,如惡性腫瘤。目前發現,眼部血管新生性疾病,包括年齡相關性黃斑變性(AMD)、糖尿病視網膜病變、新生血管性青光眼等,這類疾病的共同特點都在于眼部新生血管的異常增生(金曉,等,抗VEGF藥物在眼科疾病中的應用及機制研究進展,中外醫療,2012年)。其中,黃斑變性主要有干性和濕性兩種,濕性黃斑變性(AMD)的特點是,脈絡膜的新生血管進入視網膜下以及繼而發生的出血、滲出及水腫等病理變化。濕性迅速喪失視力,較干性更為嚴重。目前,在濕性黃斑變性的治療方面已有較好的進展。早期的激光燒爍止血被血管內皮因子拮抗劑所代替,但因后者效果不佳,很快被光動力療法所取代。光動力療法雖然提高了療效,但仍不理想。近年又出現了新的血管內皮因子拮抗劑Lucentis(雷珠單抗),是一種人源性VEGF亞型單克隆抗體片段的重組體,可減少新生血管生成。2006年,該藥物被美國FDA批準用于治療濕性黃斑變性,療效良好;同時,目前也發現該類抗VEGF藥物對糖尿病視網膜病變、新生血管性青光眼有治療作用。但由于Lucentis為抗體藥,價格極高,它還不能在全世界普及。因此,研究療效良好、價格低廉的小分子新生血管抑制劑藥物是當今國際制藥界激烈競爭的焦點。技術實現要素:本發明的目的在于提供了一種新化合物,及其制備方法和用途。本發明提供了如式A所示的化合物及其藥學上可接受的鹽或前體藥物:其中,Z0為O或S;X為C,Y為N;;Z1、Z2分別獨立選自N或-CR8;r7選自氫或C1-C6烷基;r8選自氫或C1-C6烷基;r10選自H、氨基、羥基、巰基、C1-C6烷基或-(CH2)n1NHr1,其中,n1=1-5,r1為H或C1-3的烷基;r9選自H、氨基、羥基、巰基、(C-r11r12)nNr13r14、(C-r11r12)nOr15、(C-r11r12)nC(O)Er13、(C-r11r12)nS(O)mr17;或r8和r9與它們所連接的原子一起形成飽和的4-7元雜環或取代雜環,其中,雜環或取代雜環含有1或2個選自N,O或S的雜原子,其取代基為0,1,或2個,分別獨立地選自氧代基團,C1-C6烷基,C1-C6鹵代烷基,C1-C6烷氧基,C1-C6鹵代烷氧基,C1-C6烷酰基、C1-C6烷胺基、C3-C7環烷基、鹵素,羥基,氨基,羰基,氨基羰基,C1-C6氨烷基羰基,C1-C4酰基,C1-C6烷氧基羰基,C1-C6磺酰基,氨基磺酰基;Ar2選自苯基,萘基,單環或二環雜芳基,二環或三環雜環基,其中每個雜芳基或雜環基有1,2,3或4個選自N,O或S的雜原子;所述的苯基,萘基,雜芳基或雜環基團是未取代或取代的苯基,萘基,雜芳基或雜環基團,其取代基為1,2,或3個,分別獨立地選自C1-C8烷基,C1-C8鹵代烷基,羥基C1-C6烷基,C1-C8烷氧基,C1-C8鹵代烷氧基,鹵素,羥基,氨基,氨基C-C6烷基,C1-C6烷基氨基,苯基,C3-C7環烷基,螺環的C3-C7環烷基,氨基磺酰基,C1-C6烷基氨基磺酰基;m為0,1,或2;n為0,1,2,或3;E為空、氧或-Nr18;r11,r12和r18是相同或不同的,分別獨立選自氫或C1-C4烷基;r13,r14,r15,r16和r17獨立選自氫,C1-C6烷基,C1-C6酰基,苯基和雜環,或取代的C1-C6烷基,C1-C6酰基,苯基和雜環,其取代基為為0、1或2個,分別獨立選自C1-C4烷氧基,C1-C4烷基、鹵素、羥基、氨基、C1-C6氨烷基。進一步地,所述化合物結構式如下:其中,X為C,Y為N;R1選自H、氨基、羥基或巰基;R2選自H、氨基、羥基、巰基或-(CH2)nNHR8,其中,n=1-5,R8為H或C1-3的烷基;R3選自H或C1-6烷基;R4、R5、R6分別獨立選自H、鹵素、C1-6的烷基或鹵素取代烷基;R7選自H、C1-6的烷基或鹵素;或者,R2、R3與其相連的碳原子共同構成取代或非取代的含1-2個雜原子的五元或六元環,所述雜原子為N、O或S,其取代基為C1-6的烷基。進一步地,X、Y不相同;R1選自H或氨基;R2選自H、氨基或-(CH2)nNHR8,其中,n=1-3,R8為H或C1-2的烷基;R3選自H或C1-2烷基;R4、R5、R6分別獨立選自鹵素、C1-2的烷基或鹵素取代烷基;R7選自H或鹵素;或者,R2、R3與其相連的碳原子共同構成取代或非取代的含1個N的五元或六元環,取代基為C1-3的烷基。更進一步地,R2選自氨基或-(CH2)nNHR8;或者,R2、R3與其相連的碳原子共同構成取代或非取代的含1個N的五元或六元環,取代基為C1-3的烷基。進一步,所述化合物結構式如下:化合物為式II時,W1、W2分別獨立選自C或雜原子,n=1或2;R9為H、C1-C4烷基或鹵素;R10為鹵素;其中,n=1時,W1、W2不同時為C;優選地,n=1時,W1、W2同時為雜原子,雜原子優選為N;n=2時,W1、W2同時為C;化合物為式III時,W3選自C或雜原子,雜原子優選為N;R10為鹵素。更進一步地,所述鹵素為F或Cl。優選地,所述化合物為:本發明還提供了上述化合物及其藥學上可接受的鹽或前體藥物在制備血管新生抑制劑中的用途。進一步地,所述血管新生抑制劑為血管內皮生長因子受體2抑制劑。更進一步地,所述抑制劑是抗眼部血管新生的藥物。進一步地,所述藥物是脈絡膜新生血管抑制劑。更進一步地,所述藥物是治療或預防濕性黃斑變性、糖尿病視網膜病變或新生血管性青光眼的藥物。本發明還提供了上述化合物及其藥學上可接受的鹽或前體藥物在制備VEGFR2、PDGFR-β或/和KIT抑制劑類藥物中的用途。本發明還提供了一種藥物組合物,它是含有上述化合物及其藥學上可接受的鹽的眼用制劑。除了本發明提供的上述化合物以外,制劑中還可以包含其他已知具有相似治療用途的藥物。進一步地,所述眼用制劑為滴眼劑、眼膏劑或眼用注射液。在本領域中已知的方法可制備本發明化合物的鹽。用酸處理,或與合適的陰離子交換劑,與上述化合物可成鹽。本發明的化合物的藥學上可接受的鹽,可以從上述化合物具有堿性氮原子的有機或無機酸加成鹽。優選地,合適的無機酸包括但不限于,氫鹵酸,如鹽酸,硫酸,或者磷酸。優選地,合適的有機酸包括,但不限于,羧酸,磷酸,磺酸或氨基羧酸,例如乙酸,丙酸,辛酸,癸酸,十二烷酸,羥基乙酸,乳酸,富馬酸,琥珀酸,己二酸,庚二酸,辛二酸,壬二酸,蘋果酸,酒石酸,檸檬酸,氨基酸,如谷氨酸或天冬氨酸,馬來酸,羥基酸,甲基馬來酸,環己烷羧酸,金剛烷羧酸,苯甲酸酸,水楊酸,4氨基水楊酸,鄰苯二甲酸,苯乙酸,扁桃酸,肉桂酸,甲烷或乙烷磺酸磺酸,2-羥基乙磺酸,乙烷-1,2-二磺酸,苯磺酸,2-萘磺酸,1,5-萘二磺酸,2-甲基苯磺酸,對甲基苯磺酸,乙基硫酸,十二烷基硫酸的酸,N環己基氨基乙酸,N-甲基-N-乙基-N-丙基-氨基磺酸,或其它有機酸,如抗壞血酸。另外,它也可以藥學上不可接受的鹽用于分離或純化中,例如苦味酸鹽或高氯酸鹽。但是,用于治療用途的,只能是藥學上可接受的鹽或游離化合物,以適用的藥物制劑的形式。本發明所述藥學上可接受的前體藥物,是指所述化合物經過化學結構修飾后得到的在體內經酶或非酶的轉化釋放出活性成分而發揮藥效的化合物。本發明的一種實施方式中,還包括了同位素標記的上述化合物或其藥學上可接受的鹽,所述同位素標記化合物是指與本文中所列化合物相同,但是其中的一個或多個原子被另一個原子取代,該原子的原子質量或質量數不同于自然界中常見的原子質量或質量數。可以引入化合物中的同位素包括氫、碳、氮、氧、硫,即2H,3H、13C、14C、15N、17O、18O、35S。含有上述同位素和/或其它原子同位素的化合物及其立體異構體,以及該化合物、立體異構體的可藥用的鹽均應包含在本發明范圍之內。本發明中的關鍵中間體和化合物進行分離和純化,所使用的方式是有機化學中常用的分離和純化方法且所述方法的實例包括過濾、萃取、干燥、旋干和各種類型的色譜。可選擇地,可以使中間體不經純化即進行下一步反應。在某些實施方式中,本發明的一種或多種化合物可以彼此聯合使用。也可選擇將本發明的化合物與任何其它的活性試劑結合使用,用于制備調控細胞功能或治療疾病的藥物或藥物組合物。如果使用的是一組化合物,則可將這些化合物同時、分別或有序地對受試對象進行。本發明化合物具有良好的抗血管新生作用,初步認為該類化合物是通過抑制VEGFR2(即KDR)產生活性的。該類化合物可用于對新生血管、VEGFR2等蛋白激酶異常所致疾病的治療,如濕性黃斑變性、惡性腫瘤等。附圖說明圖1本發明化合物對斑馬魚血管發育的抑制作用圖2陰性對照結果圖,其中,斑馬魚血管發育正常圖3藥物組結果圖,其中,斑馬魚血管發育部位被本發明化合物部分抑制圖4藥物組結果圖,其中,斑馬魚血管發育部位被本發明化合物完全抑制圖5本發明化合物對VEGFR2體外抑制作用圖6本發明化合物KDR4脈絡膜新生血管的抑制作用,其中,A:陰性對照,B:KDR4圖7本發明化合物V01脈絡膜新生血管的抑制作用,其中,A:陰性對照,B:V01圖8化合物V01的核磁圖圖9化合物V3的質譜圖圖10化合物V4的質譜圖圖11化合物V5的質譜圖圖12化合物V5的核磁圖圖13化合物V6的質譜圖圖14化合物KDR3的質譜圖圖15KDR4化合物質譜圖圖16KDR6的質譜圖圖17KDR6A的質譜圖圖18KDR8的質譜圖圖19KDR8A的核磁圖圖20KDR8A的質譜圖圖21KDR8B的質譜圖圖22化合物V01的劑量效應曲線圖23化合物V01對小鼠眼部新生血管和出血的影響,A:右眼V01處理,B:左眼PBS對照具體實施方式實施例1本發明中間體的制備步驟如下:第1步:混和米氏酸(75.3g、0.522mol)和原甲酸三乙酯(92mL,0.55mol),加熱至55℃,維持90分鐘,然后冷卻到45℃。化合物1(92g、0.5mol)溶于甲醇(200mL),并加入到反應混合物中反應45分鐘,同時保持反應混合物的溫度低于50℃,然后在室溫下攪拌過夜,薄層色譜(PE/EA=3:1)監控至反應完成。反應混合物被冷卻到0℃,沉淀物過濾,干燥后得到白色固體純化合物2(155.9g,產量:96%)。第2步:化合物2(155.9g、0.441mol)在DowthermA熱導管中(1.1L)被加熱到100℃,然后慢慢添加到連接有DowthermA熱導管的燒瓶中(500mL,預熱到210℃)中,同時保持溫度高于207℃。反應物在210℃下攪拌反應一個小時,然后冷卻到室溫。過濾收集的沉淀,用乙醚、丙酮洗后,干燥后得到棕色的固體純化合物3(67g、產量:61%)。第3步:在室溫下,伴隨攪拌并往三氯氧磷P℃l3(35mL)中添加化合物3(10g、0.0398mol)。添加后,反應混合物回流攪拌3個小時,薄層色譜(二氯甲烷DCM/甲醇=10:1)跟蹤反應完成,再將反應混合物注入冰水,碳酸鈉堿化調pH=8,然后以乙酸乙酯萃取,收集有機相,用鹽水洗滌,以Na2SO4干燥后,粗產物再由硅膠柱色譜(石油醚PE/乙酸乙酯EA=20:1,1:1),得到淡黃色固體純化合物4(6.0g,產量:56%)。第4步:將化合物4(10.0g,37mmol)、鋅粉(0.36g、5.55mmol)、Zn(CN)2(2.69g、22.9Zn),dppf(8.82g、15.9mmol)和Pd2(dba)3(7.8g、8.51mmol)混合于DMA(50mL)中,80℃攪拌過夜。薄層色譜(PE/EA=5:1)跟蹤至幾乎所有的化合物4被消耗。將反應混合物冷卻到室溫后倒入水中,過濾,濾液以乙酸乙酯提取,收集有機相,用鹽水洗滌,在Na2SO4上干燥濃縮獲得粗產物,硅膠柱色譜純化(PE/EA=50:1,10:1)獲得淡黃色固體純化合物5(6.0g,產量:62.3%)。第5步:混合化合物5(6.0g、23.08mmol)、氫氧化鈉(10g、250mmol)在乙二醇(70mL)和水(10mL)在回流蒸餾器攪拌3小時。薄層色譜(DCM/甲醇=10:1)跟蹤至反應完成。將反應混合物冷卻到室溫,加入檸檬酸水溶液并酸化到pH=4,過濾,沉淀干燥后得到淡黃色固體純化合物6(6.0g,產量:93.2%)。第6步:攪拌混和化合物6(6.0g、21.5mmol)于甲醇(100mL)中,在0℃逐滴添加SOCl2(4.7mL,64.5mmol)。添加后,反應混合物在室溫下攪拌1個小時,然后加熱回流過夜。薄層色譜(DCM/甲醇=10:1)跟蹤至反應完成。將反應混合物蒸發除去大部分溶劑,殘留物注入冰水,添加固體碳酸鈉調節pH=8,并用乙酸乙酯提取,收集有機相,用鹽水洗滌后,在Na2SO4上干燥,濃縮獲得褐黃色固體化合物7(3.9g,產量:61.9%)。第7步:混和化合物7(3.9g、13.3mmol)和10%Pd/C(1.0g)在甲醇(150mL)中,在室溫下H2氣中攪拌2小時。薄層色譜(PE/EA=2:1)跟蹤至反應完成。將反應混合物過濾,濾液濃縮后得到棕色的固體化合物8(2.6g,產量:96.3%)。第8步:攪拌混和化合物8(0.5g、2.46mmol)于甲醇(5mL)中,室溫下添加2NNaOH(2.5mL,5mmol。添加后,反應混合物在室溫下攪拌夜。薄層色譜(DCM/甲醇=10:1)指示反應完成。將反應混合物蒸發,殘留物用水稀釋,并用乙酸乙酯提取,棄去乙酸乙酯層,水相添加檸檬酸水溶液并調節pH=2,過濾,沉淀干燥后得到純化合物9(0.4g,產量:86%)。第9步:混和化合物9(0.4g、2.11mmol)、3-三氟甲基苯胺(375mg,2.32mmol),HATU(960mg,2.53mmol)和DIPEA(820mg,6.33mmol)在DMF(5mL)室溫下于N2中攪拌過夜。薄層色譜(DCM/甲醇=10:1)顯示反應完成。將反應混合物用水稀釋后乙酸乙酯提取,收集有機相,用鹽水洗滌,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化后獲得黃色的固體純化合物10(0.4g,產量:57%)。實施例2化合物V01(本發明中亦可記為V01)的制備步驟1:化合物11A(20g、0.15mol)、DMAP(1.9g、15.4mmol)和(BoC)2O(75g,0.34mol),在四氫呋喃(750mL)中混合,反應混合物在室溫攪拌過夜。薄層色譜(PE/EA=3:1)顯示反應完成。將反應混合物濃縮后懸浮于PE/EA(10:1,200mL),過濾后得到白色固體純化合物11(50g,100%)。步驟2:混合化合物10(50mg,0.15mmol)和Cs2CO3(150mg,0.45mmol)于DMSO(1mL)中,在N2中室溫攪拌半個小時,然后加入化合物11(60mg)。由此產生的反應混合物攪拌半個小時,用薄層色譜(DCM/甲醇=10:1)檢測至反應完成。將反應混合物加水稀釋后,用EA提取,收集有機相,用鹽水洗滌,以Na2SO4干燥后,色譜純化后制備黃色的固體純化合物12(30mg,32%)。室溫下N2中攪拌混合化合物12(20mg,0.032mmol)和TFA(0.1mL)半個小時。薄層色譜(DCM/甲醇=10:1)顯示至反應完成。將反應混合物由Na2CO3堿化后,用DCM提取,收集有機相,用鹽水洗滌,以Na2SO4上干燥,濃縮后獲得粗產物,由硅膠柱色譜純化后獲得黃色的固體純化合物V01(4.5mg,33%),結構鑒定參見圖8。實施例3化合物V3(本發明中亦可記為V03)的制備步驟1:攪拌混和化合物13(5.0g,45mmol)和吡啶(200mL),65℃下將(Boc)2O(14.7g,67.5mmol)滴加到上述混合物中。添加后,在85℃攪拌反應4小時。薄層色譜(DCM/甲醇=10:1)跟蹤至反應完成后,將反應混合物冷卻到0℃,加入濃HCl(100mL)和H2O(50mL),EA提取后,收集有機相,用NaHCO3水溶液洗滌,以Na2SO4干燥后,得到黃色的油狀物,將其懸浮于Et2O,過濾,獲得白色固體化合物14(4.3g,產量:45.2%)。攪拌混和化合物14(2.4g、11.4mmol)和N,N二甲基苯胺(6.6mL)于DCM(84mL)中,0℃N2中,滴加POCl3(3.2mL,34.2mmol)。添加后,反應混合物在室溫下攪拌2小時。薄層色譜(PE/EA=2:1)跟蹤至反應完成后,將反應混合物注入冰水。有機相用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化,獲得白色固體純化合物15(1.8g,70%)。在THF(1mL)中攪拌混和化合物15(100mg,0.47mmol)和DMAP(12mg,0.09mmol),室溫下加入(BOC)2O(124mg,0.57mmol),然后室溫下攪拌過夜。薄層色譜(PE/EA=2:1)表明反應完成后,將反應混合物用水稀釋,EA提取,收集有機相,用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化,獲得白色固體純化合物16(70mg,44.8%)。室溫下,在N2中DMSO(1mL)中攪拌混和化合物10(50mg,0.15mmol)和Cs2CO3(150mg,0.45mmol)半小時,然后加入化合物16(60mg,0.18mmol),再攪拌半小時。薄層色譜(PE/EA=2:1)表明反應完成后,將反應混合物用水稀釋,EA提取,收集有機相,用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化,獲得黃色固體純化合物17(50mg,53.3%)。步驟2:室溫下,在N2中DMSO(1mL)中攪拌混和化合物17(50mg,0.08mmol)和TFA(0.2mL)半小時。薄層色譜(DCM/甲醇=10:1)表明反應完成后,這個反應混合物由Na2CO3堿化后,用DCM提取。有機相是用鹽洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化后獲得黃色的固體純化合物V3(15mg,44%),其結構鑒定數據參見圖9。實施例3化合物V4(本發明中亦可記為V04)的制備步驟1:攪拌混和化合物18(50g,0.47mol)于DCM(600mL)中,加入TEA(94g,0.93mol),然后在0℃,N2中,滴加溴乙酸乙酯(94g,0.56mol)。反應混合物在室溫下攪拌過夜。薄層色譜(PE/EA=1:1)表明反應完成后,反應混合物用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化(用PE/EA=20:1to10:1to5:1洗脫),獲得黃色油狀純化合物19(46g,51%)。95℃下,在甲苯(800mL)攪拌混和化合物19(38g,196.65mmol)和TEA(29.9g,295mmol),逐滴加入4-溴丁酸乙酯(72.9g,373.65mmol).加完后回流加熱過夜。薄層色譜(PE/EA=5:1)表明反應完成后,反應混合物濃縮,由硅膠柱色譜純化(用PE/EA=20:1to5:洗脫),獲得黃色油狀純化合物20(32g,yield:53%)。在甲苯(800mL)中攪拌混和化合物20(32g,104.3mmol),0℃下,加入t-BuOK(51.2g,456.3mmol)。添加后,反應混合物在室溫下反應1小時。薄層色譜(PE/EA=5:1)表明反應完成后,添加2NHCl調節pH=6,然后EA提取,收集有機相,用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物21(17g,yield:62.5%),為暗黃色油,可直接用于下一步反應,不需進一步純化。MeONa((11g,161.65mmol)溶于甲醇(280mL),反應混合物被冷卻到5℃,再加入乙酸甲脒(3.0g,29.15mmol)。攪拌反應混合物半個小時,再加入化合物21(17g、65.1mmol)。將反應混合物40℃攪拌過夜。薄層色譜(DCM/甲醇=10:1)表明反應完成后,將反應混合物冷卻到室溫,蒸發除去大部分的溶劑。殘留物水處理和EA提取,有機相用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化獲得淡黃色固體純化合物22(2.5g,產量:16%)。在壓力為0.5MPaH2中,攪拌過夜混和化合物22(2.5g,10.4mmol),10%Pd/C(0.5g)和(B℃)2O(2.7g,12.4mmol)于MeOH(40mL)中。薄層色譜(DCM/甲醇=10:1)表明反應完成后,將反應混合物過濾濃縮得到黃色固體純化合物23(2.6g,100%)。在1,2-DCE(64mL)中混和化合物23(1.6g,6.3mmol),PPh3(3.33g,12.6mmol)和CCl4(2.93g,18.9mmol),70℃加熱1小時。薄層色譜(DCM/甲醇=10:1)表明反應完成后,將反應混合物濃縮后,由硅膠柱色譜純化獲得淡黃色固體純化合物24(1.38g,81%)。室溫N2中,在DMSO(2mL)中攪拌混和化合物10(100mg,0.3mmol)和Cs2CO3(300mg,0.9mmol)半個小時,再加入化合物24(97mg,0.36mmol)。反應混合物攪拌混合半小時。薄層色譜(DCM/MeOH=10:1)表明反應完成后,反應物用水稀釋,然后EA提取。有機相用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,將反應混合物濃縮后由硅膠柱色譜純化獲得黃色固體純化合物25(21mg,12.4%)。步驟2:室溫N2中,中攪拌混和化合物25(21mg,0.037mmol)和TFA(0.2mL)半個小時。薄層色譜(DCM/MeOH=10:1)表明反應完成。將反應混合物由Na2CO3堿化后,用DCM提取。有機相用鹽水洗,在Na2SO4上干燥濃縮后獲得粗產物,由硅膠柱色譜純化后獲得黃色的固體純化合物V4(10mg,57.9%),其結構鑒定數據參見圖10。實施例4化合物V5(本發明中亦可記為V05)的制備步驟1:(S)-1-苯基乙胺(10g,8.26mmol)和乙基乙酰丙酸(11.9g,8.26mmol)的1,2-DCE(200mL)溶液中,分批加入NaBH(OAc)3(35g,16.52mmol)。加完后,將反應混合物在室溫下攪拌4小時。然后將反應混合物加入NaHCO3水溶液堿化,并以DCM萃取,分離有機相,并用飽和食鹽水洗滌,在Na2SO4上干燥,并濃縮得到粗的化合物34(20.5g,收率:100%),可不經進一步純化直接用于下一步驟中。在1,2-DCE(200mL)中攪拌混和化合物34(20.5g,82mmol)和乙醛酸乙酯((33mL,165mmol,50%的甲苯溶液),NaBH(OAc)3(35g,165mmol),在室溫下攪拌4小時。然后將反應混合物用NaHCO3水溶液堿化,并以DCM萃取。分離有機相并用飽和食鹽水洗滌,在Na2SO4干燥并濃縮得到粗的化合物35(26g,100%),可不經進一步純化直接用于下一步驟中。化合物35(26g,78mmol)和t-BuOK(22g,156mmol)在甲苯(600mL)中的混合,攪拌回流過夜。然后將反應混合物用NaHCO3水溶液堿化并以DCM萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗化合物36。取粗化合物36通過硅膠色譜法(PE/EA=20:1洗脫)純化,得到純的化合物37(3g)。在乙醇(50mL)中的混合化合物37(3.0g,0.01mol)和甲脒乙酸鹽(3.24g,0.03mol),混合物中溶液中加入NaOEt(2.38g,0.035mol)。攪拌該反應混合物于90℃過夜,然后蒸發以除去大部分的溶劑。殘余物用H2O稀釋,用2NHCl酸化至pH為6,然后用DCM萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過硅膠色譜法純化,得到純的化合物38(1.0g,收率:35.8%),為黃色固體。將化合物38(1.0g,10.4mmol),10%的Pd/C(0.1g)和(B℃)2O(2.7g,12.4mmol)混合于MeOH(40mL)中,在壓力為0.5兆帕的氫氣中攪拌過夜。TLC(DCM/甲醇=10:1)表明反應完成。將反應混合物過濾,將濾液濃縮,得到粗產物,將其通過硅膠色譜法純化,得到純的化合物39(0.7g,產率:71%),為黃色固體。取化合物39(0.7g,6.3mmol)和三苯基膦PPh3(3.33g,12.6mmol)在四氯化碳(10mL)中混合,于80℃過夜攪拌。TLC(DCM/甲醇=15:1)表明反應完成。將反應混合物濃縮,并將殘余物通過硅膠色譜法純化,得到純的化合物40(0.4g,53.4%),為淺黃色油狀物。在氮氣下,化合物10(150mg,0.45mmol)和Cs2CO3(440mg,1.35mmol)于DMSO(2ml)中混合,在室溫下攪拌半小時,然后加入化合物40(153mg,0.54mmol)。將所得的反應混合物攪拌半小時,TLC(DCM/甲醇=10:1)表明反應完成。將反應混合物用水稀釋并用EA萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過制備的TLC純化,得到純的化合物41(60mg,23%),為黃色固體。步驟2:取化合物41(60mg,0.104mmol)和TFA(0.3mL)的混合物在氮氣下,室溫攪拌半小時。TLC(DCM/甲醇=10:1)表明反應完成。將反應混合物NaHCO3水溶液堿化并以DCM萃取。有機相用鹽水洗滌,在Na2SO4上干燥濃縮,得到粗產物,將其通過制備的TLC純化,得到純化合物V5(20mg,40.3%),為黃色固體,其結構鑒定數據參見圖11、12。實施例5化合物V6(本發明中亦可記為V06)的制備步驟1:配備有機械攪拌器和氯化鈣管的3L的燒瓶中,裝入化合物26(100g,0.716mol)和乙醇(1.0L)。將該混合物攪拌20分鐘,然后滴加三乙胺(72.5g,0.716mol)。將所得的混合物攪拌10分鐘,然后,加入丙烯酸乙酯(61.6g,0.716mol)。將反應混合物在室溫下攪拌17小時。(BOC)2O(234.5g,1.08mol)在室溫下逐滴加入。滴加結束后,將反應混合物在室溫下攪拌過夜。將反應混合物濃縮以除去大部分EtOH中。將殘余物溶解在水(3L)中,并用Et2O(1L×3)萃取。合并的有機相用氯化銨(500L×3)水溶液和鹽水(500L×3)洗滌,無水Na2SO4干燥,并濃縮得到粗的化合物28(300g,92%),為黃色油狀物,其用于下一步驟而無需額外的純化。鈉(27.6g,0.765mol)加入無水乙醇(1.5L)中。當固體鈉完全消失,將化合物28(300g,1.04mol)加入熱溶液中。反應混合物回流過夜。薄層色譜法(石油醚/乙酸乙酯=4:1)表明起始原料完全被消耗。將反應混合物蒸發,將殘余物溶解在水(1L)中,然后檸檬酸水溶液酸化至pH為6。然后將混合物用EtOAc(1升×3)萃取。將萃取液合并,并用鹽水(1L×3)洗滌,在Na2SO4干燥并蒸發,得到化合物29(169g,63.4%),為棕色油狀物。該粗產物無需進一步純化用于下一步驟。甲醇鈉(2.6g,48.58mmol)溶解于MeOH(50mL)中,并將該溶液冷卻至5℃。加入醋酸甲脒(3.0g,29.15mmol),將反應混合物攪拌半小時。再加入化合物29(5.0g,19.43mmol),將反應混合物攪拌回流過夜。TLC(DCM/甲醇=10:1)表明反應完成。將反應混合物冷卻至室溫,并蒸發以除去大部分溶劑。將殘余物用水處理并用EA萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過硅膠色譜法純化,得到純的化合物30(680mg,收率:14.7%),為淺黃色固體。在0℃氮氣下,三氯氧磷(P℃l3,174mg,1.26mmol)加入到攪拌混合有化合物30(100mg,0.42mmol)和N,N-二甲基苯胺(0.28mL)的DCM(4mL)中,加完后,將反應混合物傾入冰水中,通過加入固體碳酸鈉堿化,然后用DCM萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過制備TLC純化,得到純的化合物31(60mg,55.6%),為白色固體。在氮氣下,化合物10(50mg,0.15mmol)和Cs2CO3(150mg,0.45mmol)混合于DMSO(1mL)中,混合物在室溫下攪拌半小時,然后加入化合物31(46mg,0.18mmol)。將所得的反應混合物攪拌半小時,TLC(DCM/甲醇=10:1)表明反應完成。將反應混合物用水稀釋并用EA萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過制備的TLC純化,得到純的化合物32(13mg,15.7%),為黃色固體。步驟2:氮氣下,化合物32(13mg,0.038mmol)和TFA(0.1mL)的混合物室溫下攪拌半小時。TLC(DCM/甲醇=10:1)表明反應完成。將反應用混合物用Na2CO3水溶液堿化和DCM萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過制備TLC純化,得到純的化合物,V6(6mg,收率:56.3%),為黃色固體,其結構鑒定數據參見圖13。實施例6KDR3的制備方法第1步:將化合物1(1.4克,0.01mmol),4-(芐氧基甲基)-6-氯嘧啶(4.7克,0.02)和K2CO3(4.2克,0.03摩爾)在DMF(50mL)中混合,在100℃、N2下攪拌為5小時。TLC(PE/EA=4:1)表明反應完成。將反應混合物冷卻至室溫并用水稀釋,用EA萃取。收集有機相,用鹽水洗滌,用Na2SO4干燥并濃縮,得到粗產物,將其通過柱層析純化,得到純的化合物2(3.0克,產率:90%),為黃色固體。步驟2:在N2氣氛下,化合物2(1.2克,3.56毫摩爾),水合肼(0.36克,7.12毫摩爾)和RaneyNi(0.2g)的THF(20mL)中的混合物回流攪拌1小時。TLC(PE/EA=2:1)表明反應完成。將反應混合物過濾,過濾濃縮,得到粗產物,將其通過柱層析純化,得到純的化合物3(0.8克,產率:73%),為黃色固體。步驟3:在0℃在氮氣下,將TCDI(0.78克,4.4mmol)和化合物3(0.9克,2.93mmol)在干燥的DCM(10mL)中緩慢混合。加完后,將反應混合物在室溫下攪拌半小時。TLC(PE/EA=1:1)表明反應混合物中完成。將反應混合物濃縮,得到粗產物,將其用閃蒸色譜法純化,得到粗化合物4(1.0克,產率:100%),為灰白色固體。步驟4:將化合物4(1.1克,3.15毫摩爾),4-甲基-5-(三氟甲基)苯1,2-二胺(0.6克,3.15毫摩爾)和碳二亞胺(1.2克,6.30毫摩爾)的THF(50毫升)在氮氣下,回流下攪拌2小時。TLC(DCM/甲醇=10:1)表明,反應混合物中完成。與EA稀釋該反應混合物,并過濾沉淀物。將濾液蒸發,得到粗產物,將其用柱色譜法(PE/EA=10:1至4:1)純化,得到純的化合物5(0.8克,產率:50%),為棕色油狀物。步驟5:化合物5(0.7克,1.38毫摩爾)和TFA(15ml)中的混合物于60℃過夜攪拌。將反應混合物升溫至70℃,并在此溫度下攪拌1小時。TLC(DCM/甲醇=10:1)表明,反應混合物中完成。將反應混合物冷卻至室溫并傾入冰水中,用2N的NaOH堿化至pH為10,然后用EA萃取。有機相用鹽水洗滌,用Na2SO4干燥并濃縮,得到化合物6的粗產物(0.4g,收率:70%),為黃色油狀物,其不經進一步純化直接用于下一個步驟。步驟6:在攪拌著的化合物6(60mg,每日0.1445mmol)的DCM(5ml)中,加入TEA(30毫克,0.289mmol)和MSCL(25毫克,0.2168mmol)在0℃氮氣保護下。滴加結束后,反應結束。將該混合物用DCM稀釋,用鹽水洗滌,在Na2SO4干燥并濃縮,得到化合物7的粗產物,它不經進一步純化直接用于下一步驟中。第7步:化合物7(60毫克,0.12毫摩爾)和MeNH2(0.6毫升,1.2毫摩爾,在MeOH中的2N)的無水THF(5ml)中的混合物于室溫過夜攪拌。TLC(DCM/甲醇=10:1)表明,反應混合物中完成。將反應混合物濃縮并純化殘余物,TLC純化三次,得到純KDR3(6毫克,產率:12%),為白色固體,其結構鑒定數據參見圖14。實施例7KDR4化合物制備第一步:將化合物1(1.0克,4.5毫摩爾)在ACN(20毫升)中混合2-氯-5-硝基吡啶2-氯-5-硝基吡啶(0.8克,5毫摩爾)和碳酸銫(3.3克,10毫摩爾),在室溫下攪拌過夜。TLC(PE/EA=1:1)表明反應混合物中完成。將反應混合物用水稀釋,用EA萃取。有機相用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過快速色譜純化,得到純的化合物2(0.4克,收率:24.5%),為黃色固體。第二步:向化合物2(0.3克,0.83毫摩爾)的THF(5mL)中的攪拌混合物中加入阮內鎳(0.1克),然后加熱至回流。H2N-NH2H2O(1.66毫摩爾)的THF(5mL)中的溶液逐滴加入。將所得的反應混合物攪拌10分鐘。TLC(DCM/甲醇=15:1)表明,反應混合物中完成。將反應混合物過濾,過濾濃縮,得到粗產物,將其通過快速色譜純化,得到純的化合物3(0.16克,產率:50%),為黃色固體。第三步:在化合物3(0.16克,0.483mmol)的攪拌溶液中,在0℃氮氣保護下,在DCM(10mL)中加入TCDI(95mg,0.531mmol)。滴加結束后,將反應混合物在室溫下攪拌。過夜。TLC(DCM/甲醇=10:1)表明,反應混合物中完成。將反應混合物濃縮,得到粗產物,將其通過快速色譜純化,得到純的化合物4(50毫克,產率:28%)為灰白色固體。第四步:將化合物4(50毫克,0.134mmol),4-甲基-5-(三氟甲基)-苯-1,2-二胺(25.5mg,0.134毫摩爾)和EDCI(51米克,0.268毫摩爾)在無水THF(10mL)中,氮氣下回流攪拌1小時。TLC(DCM/MEOH=15:1)表明,反應混合物中完成。稀釋反應混合物,用EA和過濾沉淀物,得到粗產物,將其由預TLC純化,得到純的化合物5(40毫克,收率:56%),為白色固體。第五步:化合物5(20毫克,0.038毫摩爾)和三氟乙酸(0.2毫升)的混合物,在室溫下攪拌半小時。TLC(DCM/甲醇=5:1)表明反應混合物中完成。將反應混合物蒸發以除去TFA,將殘余物溶解在THF中,由固體K2CO3堿化,過濾并濃縮濾液,得到純化合物KDR4(10毫克,產率:62.5%),為白色固體,其結構鑒定數據參見圖15。實施例10KDR6化合物制備第一步:7-甲氧基喹啉-4-醇(10.0克,57.1mmol)溶解在50ml三氯氧磷,然后將反應混合物在120℃加熱3小時。然后將混合物冷卻至室溫,并倒入冰水(50毫升)。用EtOAc萃取該混合物(100mLx3),將有機層用飽和食鹽水洗滌,用無水硫酸鈉干燥并濃縮,得到粗產物,將其用閃式色譜法(與PE/EA=30:1洗脫)純化,得到的產物(8.5克,收率:76.9%),為白色固體。第二步:4-氯-7-甲氧基喹啉(5.5G,28.4mmol),鋅(276.9mg,4.26mmol),Zn(CN)2(2.07克,17.6mmol),DPPF(6.65克,12.21mmol),PD2(DBA)3(5.98克,6.53mmol)溶解在100mL的DMAc中,將混合物用氬氣覆蓋并加熱在160℃過夜,然后將混合物冷卻至室溫,并通過硅藻土過濾,將濾液用H2O處理,然后用EA萃取將有機層用飽和食鹽水洗滌,用無水硫酸鈉干燥,并在真空下濃縮。通過閃式色譜法(與PE/EA=10:1洗脫)純化,得到產物7-甲氧基喹啉-4-甲腈(4.38克,收率:83.7%),為棕色固體。第三步:7-甲氧基喹啉-4-甲腈(4.38克,23.78mmol)溶解在30mL的H2O中,加入氫氧化鈉(2.38克,59.45mmol)。然后將混合物加熱至100℃過夜。將反應混合物冷卻至室溫,并用EA萃取,以除去雜質。將水相酸化,通過加入6N鹽酸至pH為2。將黃色的沉淀物通過過濾收集并干燥,得到2.5g的7-甲氧基喹啉-4-羧酸,產率52%,為棕色固體。第四步:7-甲氧基喹啉-4-羧酸(2.0克,9.84mmol)和DIPEA(3.82克,29.52mmol)溶解在DMF中。然后將反應混合物冷卻至0℃,加入HATU(4.49克,11.81mmol)和3-(三氟甲基)苯胺(1.74克,10.82mmol)。該混合物用氬氣覆蓋,然后在室溫下攪拌過夜。該混合物用水稀釋并用EtOAc(3×50毫升)萃取。洗滌有機層,用鹽水洗滌,用無水硫酸鈉干燥,并在真空下濃縮,得到粗產物,將其通過快速色譜純化,與PE/EA=1:1洗脫,得到產物(2.5g,收率:79.6%)為淺黃色固體。第五步:7-甲氧基-N-(3-(三氟甲基)苯基)喹啉-4-甲酰胺(0.6克,1.733mmol)于DCM(50毫升)溶液中,10℃氮氣下逐滴加入BBr3(1.73mL,6.93毫摩爾。加完后,將反應混合物在室溫下攪拌100小時。TLC(DCM/甲醇=15:1)表明反應完成。將反應混合物傾入冰-水中并用DCM萃取。洗滌有機層,用鹽水洗滌,無水Na2SO4干燥,濃縮的產物,為棕色固體(0.5克,產率:87%),將其用于下一步,無需進一步純化。第六步:將7-羥基-N-(3-(三氟甲基)苯基)喹啉-4-甲酰胺(50毫克,0.151mmol),叔丁基(6-氯嘧啶-4-基)甲基(甲基)氨基甲酸叔丁酯(46.6mg,0.181mmol)和K2CO3(41.7mg,0.302mmol)在DMSO(2ml)溶液中混合,在90℃攪拌過夜。TLC(PE/EA=1:2)表明反應完成。將反應混合物用水稀釋并用EA萃取。洗滌有機層,用鹽水洗滌,無水Na2SO4干燥,濃縮,粗品,這是通過預-TLC純化,得到純的產物(25毫克,產率:30%),為白色固體。第七步:叔丁基甲基((6-(4-(3-(三氟甲基)苯基氨基甲酰基)喹啉-7-基氧基)嘧啶-4-基)甲基)氨基甲酸叔丁酯(25毫克,0.045mmol)和TFA(0.25毫升)的混合物,在0℃下攪拌半小時。TLC(DCM/甲醇=5:1)表明反應完成。蒸發TFA,并將殘余物通過水溶液堿化。Na2CO3和用DCM萃取。洗滌有機相,用鹽水洗滌,用Na2SO4干燥并濃縮,得到粗產物,將其通過預-TLC純化,得到純的化合物06(7毫克,收率:21%),為白色固體,其結構鑒定數據參見圖16。實施例11KDR6A化合物制備第一步:7-甲氧基喹啉-4-醇(10.0克,57.1mmol)溶解在50ml三氯氧磷,然后將反應混合物在120℃加熱3小時。然后將混合物冷卻至室溫,并倒入冰水(50毫升)。用EtOAc萃取該混合物(100mLx3),將有機層用飽和食鹽水洗滌,用無水硫酸鈉干燥并濃縮,得到粗產物,將其用閃式色譜法(與PE/EA=30:1洗脫)純化,得到的產物(8.5克,收率:76.9%),為白色固體。第二步:4-氯-7-甲氧基喹啉(5.5G,28.4mmol),鋅(276.9mg,4.26mmol),鋅(CN)2(2.07克,17.6mmol),DPPF(6.65克,12.21mmol),PD2(DBA)3(5.98克,6.53mmol)溶解在100mL的DMAc中,將混合物用氬氣覆蓋并加熱在160℃過夜,然后將混合物冷卻至室溫,并通過硅藻土過濾,將濾液用H2O處理,然后用EA萃取將有機層用飽和食鹽水洗滌,用無水硫酸鈉干燥,并在真空下濃縮。通過閃式色譜法(與PE/EA=10:1洗脫)純化,得到產物7-甲氧基喹啉-4-甲腈(4.38克,收率:83.7%),為棕色固體。第三步:7-甲氧基喹啉-4-甲腈(4.38克,23.78mmol)溶解在30mL的H2O中,加入氫氧化鈉(2.38克,59.45mmol)。然后將混合物加熱至100℃過夜。將反應混合物冷卻至室溫,并用EA萃取,以除去雜質。將水相酸化,通過加入6N鹽酸至pH為2。將黃色的沉淀物通過過濾收集并干燥,得到2.5g的7-甲氧基喹啉-4-羧酸,產率52%,為棕色固體。第四步:7-甲氧基喹啉-4-羧酸(406mg,2毫摩爾)和DIPEA(775mg,6毫摩爾)溶解在DMF(5mL)中。將反應混合物冷卻至5℃,然后加入HATU(912mg,2.4mmol)和4-氟-3-(三氟甲基)苯胺(394mg,2.2毫摩爾)。該混合物在環境溫度下用氬氣和覆蓋,過夜。TLC(PE/EA=1:2)表明反應完成。將反應混合物用水稀釋并用EA萃取。洗滌有機層,用鹽水洗滌,無水Na2SO4干燥,并濃縮,粗產物,將其由CCTO純化,得到純的產物(220毫克,收率:30%),為棕色固體。第五步:在N-(4-氟-3-(三氟甲基)苯基)-7-甲氧基喹啉-4-甲酰胺(0.2克,0.549mmol)的DCM(10ml)溶液中,0℃下N2氣氛下攪拌加入4NBBr3(0.68毫升,2.75mmol)的DCM溶液。加完后,將反應混合物在室溫下攪拌為20小時。然后將反應混合物在-30℃攪拌4小時。TLC(DCM/甲醇=10:1)表明起始原料依然。將反應混合物傾入冰-水中并用DCM萃取。將有機層用飽和食鹽水洗滌,用無水硫酸鈉干燥并濃縮,得到粗產物,將其由預TLC純化,得到的原料(120毫克)和純的產物N-(4-氟-3-(三氟甲基基)苯基)-7-羥基喹啉-4-甲酰胺(60毫克,78%),為白色固體。第六步:將N-(4-氟-3-(三氟甲基)苯基)-7-羥基喹啉-4-甲酰胺(30毫克,0.086mmol),叔丁基(6-氯嘧啶-4-基)甲基(甲基)氨基甲酸叔丁酯(26.5mg,0.103mmol)和K2CO3(23.8mg,0.172mmol)在DMF(1mL)中,50℃下攪拌15小時。TLC(DCM/甲醇=10:1)表明反應完成。將反應混合物用水稀釋。并提取EA。將有機層用飽和食鹽水洗滌,用無水硫酸鈉干燥并濃縮,得到粗產物,將其通過預-TLC純化,得到純的產物(6-(4-(4-氟-3-(三氟甲基叔丁基)苯基氨基甲酰基)喹啉-7-基氧基)嘧啶-4-基)甲基(甲基)氨基甲酸叔丁酯(20毫克,收率:41%),為黃色固體。第七步:甲叔丁酯的混合物(6-(4-(4-氟-3-(三氟甲基)苯基氨基甲酰基)喹啉-7-基氧基)嘧啶-4-基)甲基(甲基)氨基甲酸叔丁酯(20毫克,0.035mmol)和三氟乙酸(0.3毫升)在室溫下攪拌一小時。TLC(DCM/MEOH=5:1)表明反應完成。蒸發TFA,并將殘余物由Na2CO3堿化并用DCM萃取。洗滌有機相,用鹽水洗滌,用Na2SO4干燥并濃縮,得到粗產物,將其通過預-TLC純化,得到純的化合物06A(5毫克,收率:30%),為黃色固體,其結構鑒定數據參見圖17。實施例12KDR8化合物制備第1步:草酸二乙酯(70毫升)的混合物加熱到120℃,加入化合物1(50克,0.4mol),并將混合物加熱到180℃的,維持5分鐘。將混合物冷藏過夜,過濾收集白色沉淀物,干燥,從而得到白色粉末狀的化合物2(75.0克,收率:59.11%)。步驟2:將化合物2(25克,0.11摩爾)溶解在沸騰的二甲苯(1L)中,緩慢地加入P2S5(9.5克,0.0385摩爾),回流,直到反應完成(約5小時),再繼續回流。酰胺TLC(PE/EA=5/1)表明該反應完成后,將反應混合物冷卻,并用1NNaOH萃取,水相用鹽酸酸化,收集黃色沉淀物,然后將其溶解在EtOAc中,用H2O洗滌,用Na2SO4干燥,并濃縮,得到的化合物3(19克,收率:70.9%)為一種紅色油狀物。步驟3:化合物3(30克,0.142摩爾溶于1N氫氧化鈉(500mL)中,用鐵氰化鉀(135克,0.416mol,在340ml水中)氧化,通過緩慢加入硫代酰胺到鐵氰化物溶液來進行,使溫度保持低于10℃。在1小時后,TLC(DCM/MeOH中=3/1)表明反應完全。將反應混合物在與水中稀釋,酸化(由2N的HCl至pH為6),然后過濾,得到的化合物4(15g,收率:50.48%)。步驟4:化合物4(1克,4.34毫摩爾)在DMF(20mL)中攪拌溶解,加入1-methyl-5-(trifluoromethyl)-1H-pyrazol-3-amine(0.717g,4.35mmol),隨后加入在室溫氮氣中加入HATU(2.47g,6.5mmol)和DIEA(1.12g,8.6mmol)。在相同的的溫度下下攪拌過夜后,TLC(PE/EA=2/1)表明該反應完成。該反應混合物被傾入水中,并用EtOAc萃取。洗滌將有機的相,用鹽水洗滌,在Na2SO4上干燥并濃縮,得到粗的的產品,由硅膠色譜法(PE/EA洗脫=20/1-5/1)進行純化,,得到純的化合物5(900毫克,收率:75.5%)。步驟5:-10℃氮氣中,在化合物5(0.4g,1.12mmol)的DCM(10mL)溶液中,加入BBr3(10mL,4NinDCM)。4小時后,TLC(PE/EA=1/1)表明反應完成了。然后反應中加入冰,過濾除去未溶解的材料。分離濾液,用DCM萃取。將有機相用鹽水洗滌,用Na2SO4干燥并濃縮,得到粗的的產品,這進行純化,由TLC得到純的化合物6的化合物(100mg,收率:26.02%)。步驟6:化合物6的溶液(50mg,0.146mmol)的DMF(10mL)溶液中加入K2CO3(70mg,0.500mmol)和tert-butyl(6-chloropyrimidin-4-yl)methyl(methyl)carbamate(41.41mg,0.161mmol)。將反應混合物在50℃氮氣下攪拌過夜。TLC(PE/EA=1/1)表明反應完成。將反應混合物用水洗滌,用EA萃取。有機相用鹽洗滌4次,用Na2SO4干燥并濃縮,得到粗的的產品,由TLC純化得到純化合物7(40mg,收率:48.59%)。第7步:化合物7(40mg,0.071mmol)和TFA(0.4mL)在氮氣下室溫混合,攪拌半小時。TLC(DCM/甲醇=10/1)表明反應完成。將反應混合物蒸發以除去TFA。將殘余物堿化,用Na2CO3堿化至pH9,然后用DCM萃取。將有機相用鹽水洗滌,硫酸鈉干燥,濃縮,得到粗產物,將其通過TLC純化,得到純的化合物8(20mg,收率:44%)。其結構鑒定數據參見圖18。實施例13KDR8A化合物制備第1步:草酸二乙酯(70毫升)加熱到120℃,加入化合物1(50克,0.4mol),并將混合物加熱到180℃,加熱時間為:5分鐘。冷卻后,混合物在冰箱中過夜,加入50毫升水,然后形成白色沉淀物。收集該固體,干燥,得到白色粉末狀的化合物的2(75.0克,收率:59.11%)。步驟2:將化合物2(25克,0.11摩爾)溶解在沸騰的二甲苯(1L)中,緩慢加入P2S5(9.5克,0.0385摩爾),回流,直到反應完成(約5小時),再繼續回流。TLC(PE/EA=5/1)表示反應完成后,將反應混合物冷卻,并用1NNaOH萃取,水相用鹽酸酸化,收集黃色沉淀物,然后將其溶解在EtOAc中,用H2O洗滌,用Na2SO4干燥,并濃縮,得到的化合物3(19克,收率:70.9%),為紅色油狀物。步驟3:粗化合物3(30g,0.142mol)溶解在1N氫氧化鈉(500mL)中,用鐵氰化鉀(135克,0.416mol,在340ml水中)氧化,通過緩慢加入硫代酰胺到鐵氰化物溶液來進行,使溫度保持低于10℃。在1小時后,TLC(DCM/MeOH中=3/1)表明反應完全。將反應混合物在水中稀釋,由2N的HCl調節至pH為6,然后過濾,得到的化合物4(15g,收率:50.48%)步驟4:化合物4(1克,4.78毫摩爾)在DMF(40毫升)溶液中加入3-(三氟甲基)苯胺,隨后加入HATU(2.73克,7.17毫摩爾)和DIEA(1.24克,9.56毫摩爾),在室溫氮氣下攪拌。在相同的溫度下攪拌過夜后TLC(PE/EA=2/1)表明反應完成后,將反應混合物傾入水中,并用EtOAc萃取。洗滌有機相,用鹽水洗滌,在Na2SO4干燥并濃縮,得到粗產物,將其通過硅膠色譜(與PE/EA=20/1-5/1洗脫)純化,得到純的化合物5(1.68克,收率:99.0%)。步驟5:-10℃在氮氣下,化合物5(0.1克,0.283毫摩爾)的DCM(10mL)溶液中加入4NBBr3(0.5mL)。4小時后,TLC(PE/EA的=1/1)表明反應完成后,加入冰,然后過濾以除去所述未溶解的材料。濾液用DCM萃取。用鹽水洗滌有機相,用Na2SO4干燥并濃縮,得到粗產物,將其通TLC純化,得到純的化合物6(64mg,收率:66.65%)。步驟6:化合物6(64毫克,0.189毫摩爾)DMF(10毫升)溶液中,加入K2CO3(78毫克,0.567毫摩爾)和叔丁基(6-氯嘧啶-4-基)甲基(甲基)氨基甲酸叔丁酯(tert-butyl(6-chloropyrimidin-4-yl)methyl(methyl)carbamate,53毫克,0.208毫摩爾)。將反應混合物在氮氣下50℃攪拌過夜,TLC(PE/EA=1/1)表明反應完成。將反應混合物用水洗滌,EA萃取。洗滌有機相,用鹽水洗滌四次,用Na2SO4干燥并濃縮,得到粗產物,將其通過預-TLC純化,得到純的化合物7(20mg,收率:19.68%)。第7步:化合物7(20毫克,0.035毫摩爾)和TFA(0.2mL)的混合物,在氮氣下室溫攪拌半小時。TLC(DCM/MeOH中=10/1)表明反應完成。將反應混合物蒸發除去TFA。將殘余物由sNa2CO3調節pH為9,然后用DCM萃取。洗滌有機相,用鹽水洗滌,用Na2SO4干燥并濃縮,得到粗產物,將其通過預-TLC純化,得到純的化合物KDR08A(12mg,收率:73.07%),其結構鑒定數據參見圖19、22。實施例14KDR8B化合物制備第1步:草酸二乙酯(70毫升)的混合物加熱到120℃,加入化合物1(50克,0.4mol),并將混合物加熱到180℃,維持5分鐘。將混合物后冷藏過夜,形成白色沉淀物。收集該固體被通過過濾中,并干燥,從而得到為白色粉末狀的化合物的2(75.0克,收率:59.11%)。步驟2:將化合物2(25克,0.11摩爾)溶解在沸騰的二甲苯(1L)中,緩慢地加入P2S5(9.5克,0.0385摩爾),回流,直到將反應物完成(約5小時).TLC(PE/EA=5/1)表明反應完成。該反應混合物冷卻,并用1NNaOH的萃取,水相用鹽酸酸化,收集黃色沉淀物,然后將其溶解在EtOAc中,用H2O洗滌,用Na2SO4干燥,并濃縮,得到的化合物3(19克,收率:70.9%)為紅色油狀物。步驟3:化合物3(30克,0.142mol)溶解在1N氫氧化鈉(500毫升)中,用鐵氰化鉀(135克,0.416mol,在340ml水中)氧化,通過緩慢加入硫代酰胺到鐵氰化物溶液來進行,使溫度保持低于10℃。在1小時后,TLC(DCM/MeOH中=3/1)表明反應完成。將反應混合物在水中稀釋,酸化(由2NHCl至pH為6,),然后過濾,得到的化合物4(15g,收率:50.48%)。步驟4:化合物4(1克,4.78毫摩爾)的DMF(20mL)溶液中,室溫氮氣下,攪拌加入4-氟-3-(三氟甲基)苯胺鹽酸鹽(4-fluoro-3-(trifluoromethyl)anilineHydrochloride,1.24克,5.74毫摩爾),隨后加入HATU(2.73克,7.17毫摩爾)和DIEA(1.85克,14.34毫摩爾)。在相同的的溫度下攪拌過夜后,TLC(PE/EA=2/1)表明反應完成。將反應混合物傾入水中,并用EtOAc萃取。用鹽水洗滌有機相,在Na2SO4干燥并濃縮,得到粗產物,將其通過硅膠色譜(與洗脫)純化,得到純的化合物5(1.3克,收率:73.4%)。步驟5:化合物5(0.5克,1.35毫摩爾)DCM(10mL)溶液中,在-10℃氮氣下,加入4NBBr3(2.5mL),反應4小時后,TLC(PE/EA的=1/1)表明反應完成。反應中加入冰,然后過濾以除去未溶解的的材料。分離濾液,用DCM萃取。鹽水洗滌有機相,用Na2SO4干燥并濃縮,得到粗產物,將其通過預-TLC純化,得到純的化合物6(250mg,收率:51.97%)。步驟6:化合物6(70毫克,0.196毫摩爾)的DMF(10mL)溶液中,加入K2CO3(81.46毫克,0.589毫摩爾)和叔丁基(6-氯嘧啶-4-基)甲基(甲基)氨基甲酸叔丁酯(tert-butyl(6-chloropyrimidin-4-yl)methyl(methyl)carbamate,55.70毫克,0.216毫摩爾),將反應混合物在50℃氮氣下攪拌過夜。TLC(PE/EA=1/1)表明反應完成。將反應混合物用水洗滌,與EA萃取。洗滌有機相,用鹽水洗滌4次,在Na2SO4干燥并濃縮,得到粗產物,將其通過TLC純化,得到純的化合物7(40毫克,39.51%)。第7步:化合物7(38毫克,0.071毫摩爾)和TFA(0.4毫升)的混合物,在氮氣室溫下攪拌半小時。TLC(DCM/MeOH中=10/1)表明反應完成。將反應混合物蒸發除去TFA。將殘余物由Na2CO3調節至pH為9,然后用DCM萃取。洗滌有機相,用鹽水洗滌,用Na2SO4干燥并濃縮,得到粗產物,將其通過TLC純化,得到純的化合物KDR8B(15毫克,47.75%),其結構鑒定數據參見圖21。以下通過試驗例具體說明本發明的有益效果。試驗例1斑馬魚血管發育抑制試驗斑馬魚(Daniorerio)為鯉科(Cyprinidae)短擔尼魚屬(Danio)的一種硬骨魚。其基因與人類基因的相似度高達85%,雌魚一次可產卵200~300枚,其受精和胚胎發育過程均在體外進行,24小時內就可發育成形,且胚胎通體透明,便于觀察體內器官組織的變化。諸多特點使其成為了是國際標準化組織認可的5種魚類實驗動物之一。目前,斑馬魚在人類疾病研究中得到廣泛應用,用于心血管系統方面的研究尤為多。實驗方法:該實驗使用通常作為篩查化合物對血管形成作用影響的斑馬魚為動物模型。將出生后的斑馬魚胚胎經挑選后,放于培養皿中并置于培養箱中撫育3-5天,本發明化合物在斑馬魚出生后按不同濃度(1μM,10μM,30μM,100μM)直接加入培養液中。24小時后檢查脊椎血管的發育情況并用熒光顯微鏡照相。130B為帕唑帕尼(Pazopanib),作為陽性對照,DMSO作為陰性對照。試驗結果參見圖1-4、表1a,1b。表1a:KDR系列小分子對FLK-1轉基因斑馬魚血管發育的抑制作用濃度(uM)KDR3KDR4KDR6KDR8KDR8A10%0%0%0%0%100%0%0%0%0%300%10%0%0%0%10050%100%10%5%10%表1bKDR系列小分子對FLK-1轉基因斑馬魚血管發育的抑制作用AI:血管抑制率由圖1-4可知,本發明化合物KDR4及V01-v6均能抑制斑馬魚血管發育,尤其是V01,v3,v6,其藥效活性顯著。由表1b可知,化合物DKR4的活性明顯優于KDR3、KDR6、KDR8和KDR8A,化合物V01,V03的活性明顯優于KDR4。試驗例2本發明化合物的藥效實驗1.對VEGFR2體外抑制試驗檢測本發明化合物抑制VEGFR2激酶磷酸化的實驗方法如下:1)將培養至P3-P5HUVEC細胞轉移至6孔板,每孔約2*105細胞2)待細胞生長至70-80%,加入候選小分子抑制劑(10nM,100nMor1μM),孵育30min3)加入VEGF50ng/ml進行刺激,持續10min4)加入非變性裂解液終止反應、收集細胞裂解液、進行蛋白定量5)SDS-PAGE電泳,轉膜,將膜切下后用5%脫脂奶配制的TTBS緩沖液(Tris-bufferedsaline/0.01%Tween20)封閉2小時6)洗膜,用抗磷酸化VEGFR2抗體(1:1000稀釋)4℃封閉過夜7)第2天洗膜后用辣根過氧化物酶標記的山羊抗兔二抗與膜共孵育1小時8)洗膜后用化學發光試劑盒(Millipore)顯色,照相。試驗結果參見圖5。由圖5可知,本發明化合物KDR4及V01、V3均能抑制VEGFR2,其中,化合物V01、V3效果較KDR4更好。試驗例3本發明化合物對脈絡膜新生血管的抑制作用小鼠為c57/BL,采用激光誘導的脈絡膜新生血管(ChoroidalNeovascularization,CNV)動物模型進行實驗。此模型目前也是應用最廣的一種被用來研究藥物對CNV形成影響的動物模型。采用532激光在C57/bl6小鼠(每個視網膜4個激光點)行激光視網膜光凝來誘導的CNV形成。激光實施后立即進行注射:一只小鼠右眼注射濃度150uM的KDR4,另一只小鼠右眼注射濃度1uM的V01,兩小鼠左眼注射均注射與藥物相同體積的PBS作為對照。注射后5天將動物處死并獲取眼球。去除眼前節、玻璃體和視網膜后,制作脈絡膜鋪片并行Isolectin染色。采用Zeiss熒光顯微鏡系統進行拍照和CNV面積的測量。結果見圖6、圖7。根據圖6可知,化合物KDR4(150uM)、V01(1uM)均能顯著抑制脈絡膜新生血管形成,可有效治療或緩解濕性黃斑變性。試驗例4本發明化合物對激酶的影響蛋白激酶(proteinkinases)又稱蛋白質磷酸化酶(proteinphosphakinase)。一類催化蛋白質磷酸化反應的酶。它能把腺苷三磷酸(ATP)上的γ-磷酸轉移到蛋白質分子的氨基酸殘基上某些絲氨酸、蘇氨酸或酪氨酸殘基的羥基上,從而改變蛋白質、酶的構象和活性。蛋白質的磷酸化是多種信號傳導途徑的重要環節,細胞內大部分重要的生命活過程都離不開蛋白質磷酸化。蛋白激酶分為5類:蛋白絲氨酸/蘇氨酸激酶、蛋白酪氨酸激酶、蛋白組氨酸激酶、蛋白色氨酸激酶和蛋白天冬氨酰基/谷氨酰基激酶。蛋白激酶在細胞過程的調節和維持起到了重要作用,在許多疾病狀態中都觀察到了激酶活性異常,所述疾病狀態包括:惡性腫瘤,免疫性疾病、心血管疾病、糖尿病、感染性疾病、關節炎和其它免疫紊亂、神經系統如老年性癡呆癥,阿默海茨癥AD等,現已發現與超過400種人類疾病與蛋白激酶相關。其中,VEGFR(血管內皮細胞生長因子受體)家族成員為受體酪氨酸激酶,如VEGFR1、VEGFR2等,該類受體在惡性腫瘤的生長、轉移中以及血管增生性疾病(如黃斑變性、腫瘤)等疾病的發展過程中有重要影響。PDGFR(血小板衍生生長因子受體)家族成員為受體酪氨酸激酶,如PDGFRα和PDGFRβ以及集落刺激因子1受體、干細胞生長因子受體KIT等,目前也發現該類激酶與腫瘤的發生、發展有密切聯系。如PDGFR的異常表達已在黑色素瘤、腦膜瘤、神經內分泌腫瘤、卵巢癌、前列腺癌、肺癌和胰腺癌中有發現,KIT的異常活化則是許多腫瘤發生、發展的直接誘因。1)抑制劑量效應研究化合物V01溶解于100%DMSO中,稀釋為3組系列濃度,DMSO在最后測試中均保持1%。測試最高濃度為50uM。用非選擇性蛋白激酶的活性抑制劑星形孢菌素(Staurosporine)作為參照,最高濃度為1uM。結果見表3、圖22。表2目標供應商[酶],nM[ATP],μM孵育時間,hrKDRInvitrogen0.25803表3對KDR的抑制活性化合物IC50(μM)95%置信率HillStaurosporine0.009420.001211.002392V010.02070.002871.0079532)抑制特異性研究對化合物VO1,測試了22種激酶活性抑制試驗,測試濃度為5mM,重復兩次,在ATPKm測試。V01首先溶于100%DMSO中,溶解濃度為最終測試濃度的100倍,所有最終測試時的DMSO都為1%。非選擇性蛋白激酶的活性抑制劑星形孢菌素(Staurosporine)作為對照,測試時用最大濃度10mM.具體試驗方法及試劑如下表所示:表4V01對激酶兩次試驗平均抑制率如下表:表5從試驗結果可知,V01對蛋白激酶KDR的抑制率高達100%,對其他兩種與腫瘤相關的蛋白激酶也有一定抑制作用,其中,PDGFR-β抑制率為52.7%,KIT抑制率為68%,但V01對其余蛋白激酶抑制活性絕大部分小于5%。由此可見,和現有研發的小分子激酶抑制劑相比,本發明化合物對KDR具有高特異性和高效率的抑制作用,且對PDGFR-β和KIT這兩種與腫瘤有密切關聯的蛋白激酶也有一定的抑制作用,這就表明化合物V01對KDR、PDGFR-β和KIT這三種蛋白激酶異常活化相關的各種腫瘤及老年濕性黃斑變性等疾病均有潛在治療活性。3)新生血管抑制的研究共測試了5只鼠.在眼角膜中央,用75%硝酸銀溶液和25%硝酸鉀棒誘導化學燒傷造模。化學燒傷之后立即在右眼結膜下注射0.2ml化合物V01,然后每天滴0.2ml化合物V012次。7天后眼角膜拍照比較。由圖23所示,化合物V01能顯著減少了新生血管和出血。綜上所述,本發明化合物具有良好的抗血管新生作用,初步認為該類化合物是通過抑制VEGFR2(即KDR)產生活性的。該類化合物可用于對新生血管、VEGFR2、PDGFR-β、KIT等蛋白激酶異常所致疾病的治療,如濕性黃斑變性、惡性腫瘤等。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1