含籠狀磷酸酯結構的咪唑離子液體型阻燃劑及其制備方法與流程
本發明屬于咪唑離子液體型阻燃劑及其制備方法,具體來說涉及一種含籠狀磷酸酯結構的咪唑離子液體型阻燃劑的制備方法。
背景技術:
離子液體作為一類環境友好的化合物,因具有獨特而優異的物理化學性質在近年來得到了飛速的發展和廣泛的應用,如其幾乎無蒸汽壓,不易揮發、液程范圍寬、熱穩定性好、熱容量大、電化學窗口寬、不可燃等特點,被稱為可以替代傳統有機溶劑的“綠色溶劑”。更重要的是離子液體的分子可設計性強,可通過調節陽離子的烷基鏈長、改變烷基取代基種類、選擇適當的陰離子來調節離子液體的物理性質和化學性質,這使得我們可以根據具體的需要來設計“功能性離子液體”。目前離子液體在合成、催化、分離技術、電化學、分析化學及納米材料科學方面已有很多的研究,而離子液體作為高分子材料的增塑劑、分散劑、阻燃劑等功能性助劑的報道也日漸活躍。專利CN102924749A中報道了研究合成的一種離子液體型磷酸酯類阻燃劑,并將其復配為膨脹阻燃劑添加到聚丙烯(PP)中,取得較好的阻燃效果。但是,在該專利中,所合成的離子液體型阻燃劑需要引用一定量的鹵素才能夠達到相應的效果,而隨著高分子材料應用領域的擴大,人們對阻燃劑和阻燃材料的要求越來越高,鹵系阻燃劑因在燃燒的過程中會釋放出大量濃煙和二噁英、苯呋喃等腐蝕性有毒氣體,目前已逐漸被限制使用。且該專利所報道的阻燃劑需要配合多聚磷酸銨、季戊四醇和三聚氰胺復配成膨脹型阻燃劑添加到材料中才發揮較好的阻燃效果,單獨使用效果的并不理想,這使其在應用中具有一定的限制性。
技術實現要素:
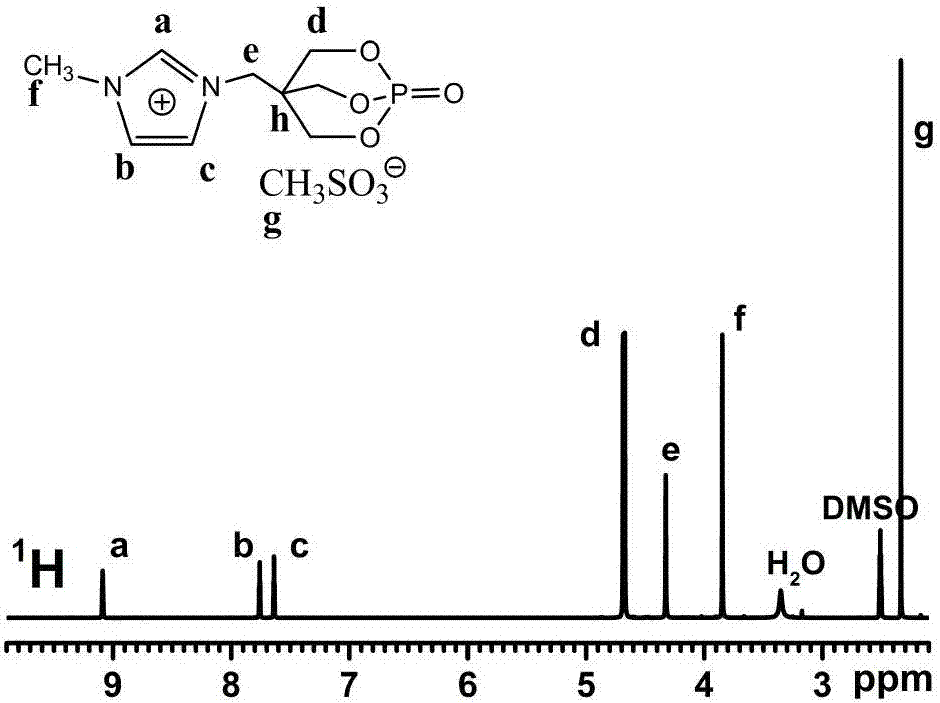
本發明的目的是針對現有技術存在的缺陷,首先提供一種含籠狀磷酸酯結構的咪唑離子液體型阻燃劑,該阻燃劑不含鹵素,具有優異的熱穩定性能。本發明的目的之二是提供上述含籠狀磷酸酯結構的咪唑離子液體型阻燃劑的制備方法。本發明提供的含籠狀磷酸酯結構的咪唑離子液體型阻燃劑,其特征在于其結構通式如下:式中X為C=1-20的烷基、苯基或磷酸酯基,其中磷酸酯基的結構如下:Y為C=1-20的烷基、苯基或對甲苯基,將該阻燃劑應用到聚碳酸酯中,在添加量為0.1~5wt%時,其極限氧指數為29.5~32.0%。以上阻燃劑中所述的Y優選C=1-8的烷基或對甲苯基。本發明提供的上述含籠狀磷酸酯結構的咪唑離子液體型阻燃劑的制備方法,其特征在于該方法的工藝步驟和條件如下:1)在N2保護下,將1-氧基磷雜-4-羥甲基-2,6,7-三氧雜雙環[2.2.2]辛烷(PEPA)和縛酸劑依次加入溶劑Ⅰ中,攪拌至大部分PEPA溶解混合均勻后再緩慢滴加取代磺酸氯,并在25-50℃下反應6-15h,反應結束后蒸餾除去溶劑Ⅰ,再經洗滌、干燥后得到中間體,其中縛酸劑與PEPA摩爾比為1-1.2:1;PEPA與取代磺酰氯的摩爾比為1:1-1.5,其反應過程如下:2)將所得中間體先完全溶解在溶劑II中,然后加入取代咪唑,并在N2保護下升溫至60-85℃反應48-72h,蒸餾除去溶劑II,經洗滌、干燥后即可,其中取代咪唑與中間體的摩爾比為1-1.2:1;其反應過程如下:上述方法中所用的溶劑Ⅰ為二氯甲烷、丙酮或四氫呋喃中的任一種。上述方法中所用的溶劑II為丙酮、乙腈、1-甲基咪唑、1-乙基咪唑或1-丙基咪唑中的任一種。上述方法中所用的縛酸劑為三乙胺或吡啶。上述方法中所用的取代磺酰氯的結構式為:式中Y為C=1-20的烷基、苯基或對甲苯基,優選C=1-8的烷基或對甲苯基。上述方法中所用的取代咪唑的結構式為:式中X為C=1-20的烷基、苯基或磷酸酯基,其中磷酸酯基的結構如下:本發明與現有技術相比,具有以下優點:1、由于本發明提供的含籠狀磷酸酯結構的咪唑離子液體型阻燃劑的結構中不含鹵素,因而該阻燃劑能符合現今環保可持續發展的要求,具有良好的應用前景;2、由于本發明提供的含籠狀磷酸酯結構的咪唑離子液體型阻燃劑的陰離子中含有磺酸根,因而在聚碳酸酯材料中只需要添加很少量的(0.1wt%)阻燃劑即可達到較好的阻燃效果,減少了添加型阻燃劑對材料力學性能帶來的負面影響。3、由于本發明中含籠狀磷酸酯結構的咪唑離子液體型阻燃劑具有優異的熱穩定性能,其分解溫度均在聚碳酸酯等大部分高分子材料的加工溫度以上,因而可有效避免其在加工過程中出現氧化分解,以致喪失優異的阻燃性。4、由于本發明提供的含籠狀磷酸酯結構的咪唑離子液體型阻燃劑在使用時可單獨使用,無需與其它阻燃成分進行復配就具有較好的阻燃性,因而既避免了尋找與之匹配的其它阻燃成分進行復配的麻煩,又可減少實際應用的限制。5、本發明提供的制備方法工藝操作簡單,成本低廉,且反應所得產物收率高,反應原料取代咪唑可回收再次利用,環保節約。附圖說明圖1是本發明實例1所制備的陰離子為甲基磺酸根的咪唑類離子液體型阻燃劑(簡記為[Pmim]CH3SO3)的1HNMR譜圖。圖中各氫原子的化學位移具體如下:(δH,(CD3)2SO):9.1(1H,s,NCHN),7.7(1H,s,CCHNCH3),7.6(1H,s,CCHNCH2),4.7(6H,d,OCH2),4.3(2H,s,NCH2),3.8(3H,s,NCH3),2.3(3H,s,SCH3),從中可以看出該目標產物的結構與預期結構一致。圖2是本發明實施例1所制備的陰離子為甲基磺酸根的咪唑類離子液體型阻燃劑的13CNMR譜圖。圖中各碳原子的化學位移具體如下:(δc,(CD3)2SO):138.5(N-CH2-N),124.4(CH3-N-CH2),123.7(N-CH2-CH2),75.9(O-CH2),45.9(N-CH2-C4),40.3(CH3SO3),37.3and36.9(C4diffierentconformation),36.4(N-CH3),從中可以看出該目標產物的結構與預期結構一致。圖3是本發明實施例1所制備的陰離子為甲基磺酸根的咪唑類離子液體型阻燃劑的熱重分析曲線。從圖中可以看出,[Pmim]CH3SO3的初始分解溫度為339℃,最大失重溫度Tmax為368℃,熱失重速率峰值為11.6%/min,,在700℃時的殘炭高達36.6%,說明其自身成炭性非常好。具體實施方式下面給出實施例以對本發明作進一步說明。有必要在此指出的是,以下實施例不能理解為對本發明保護范圍的限制,如果該領域的技術熟練人員根據上述本
技術實現要素:
對本發明作出一些非本質的改進和調整,仍屬于本發明保護范圍。值得說明的是:以下實施例所得產品的極限氧指數是將其制成120×6.5×3.2mm3的標準樣條,按照ASTMD2863-97標準,在HC-2氧指數儀上測定的。實施例11)在N2保護下,將0.5molPEPA和0.5mol縛酸劑三乙胺依次加入到二氯甲烷中,室溫25℃下攪拌10min,再滴加0.5mol的甲基磺酰氯,滴加時間控制1.5h,滴加完后在25℃下反應6h,反應結束后蒸餾除去二氯甲烷,再經洗滌、干燥后得到中間體,收率85%,其結構如下:2)將所得0.5mol中間體溶解在溶劑1-甲基咪唑中,然后加入0.5mol的1-甲基咪唑,并在N2保護下升溫至85℃攪拌反應48h,蒸餾除溶劑1-甲基咪唑,殘留物用丙酮洗滌3-4次,干燥得到陰離子為甲基磺酸根的咪唑類離子液體型阻燃劑,收率為76%。其結構式如下:3)將所得阻燃劑以0.5wt%的添加量與聚碳酸酯(PC)在轉矩流變儀中進行共混得到的阻燃PC材料,其極限氧指數為30.0%,與純PC的氧指數25.5%對比提高了4.5個單位。實施例21)在N2保護下,將0.5molPEPA和0.6mol縛酸劑三乙胺依次加入丙酮中,室溫25℃下攪拌20min,再滴加0.75mol的對甲苯磺酰氯,滴加時間控制3h,滴加完后在25℃下反應8h,反應結束后蒸餾除去丙酮,再經洗滌、干燥后得到中間體,收率88%,其結構如下:2)將所得0.5mol中間體溶解在溶劑1-甲基咪唑中,然后加入0.6mol的1-甲基咪唑,并在N2保護下升溫至83℃攪拌反應58h,蒸餾除溶劑1-甲基咪唑,殘留物用丙酮洗滌3-4次,干燥得到陰離子為對...
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1