由乙醇酸或乙醇酸甲酯制備高分子量聚乙醇酸的方法與流程
本發明涉及一種聚乙醇酸的制備方法,尤其是涉及一種由乙醇酸或乙醇酸甲酯制備高分子量聚乙醇酸的方法。
背景技術:
傳統的高分子材料如pet、pe、pp、ps等雖給老百姓的生活帶來了很多便利,在社會經濟發展中承擔著重要作用,但由于回收利用不完善,且其本身在地球環境中不能降解或難以降解,造成嚴重的“白色污染”,隨著社會對環保的日益重視,各個國家和地區相繼頒發“限塑令”,發展以可降解的、對環境友好的高分子材料對傳統的不可降解的高分子材料的替代成為了重要的研究方向。
公開號為cn101302284a的中國專利發明報道了一種高分子量聚乳酸材料的制備方法,聚乳酸材料雖可生物降解,但其原料乳酸是以淀粉等生物質為原料制得的,成本高,且生產工藝苛刻,基于我國富煤少油的國情,亟需以煤化工副產物乙醇酸甲酯或乙醇酸為原料制備高分子量聚乙醇酸樹脂,該樹脂可生物降解,最終變為二氧化碳和水等對環境無害的物質,適合于制作包裝袋、一次性餐具等日用高分子材料,同時具有成本低、工藝簡單等優勢。
技術實現要素:
本發明的目的就是為了克服上述現有技術存在的缺陷而提供一種由乙醇酸或乙醇酸甲酯制備高分子量聚乙醇酸的方法。
本發明的目的可以通過以下技術方案來實現:
一種由乙醇酸或乙醇酸甲酯制備高分子量聚乙醇酸的方法,為一步法制取方法,主要步驟為:單體乙醇酸或乙醇酸甲酯在催化劑的作用下,進行預縮聚反應,制得特性粘度0.3-0.7dl/g的乙醇酸或乙醇酸甲酯預聚物;將乙醇酸或乙醇酸甲酯預聚物進行固相縮聚,制得特性粘度1.0dl/g以上的聚乙醇酸。
具體包括以下步驟:
(1)單體乙醇酸或乙醇酸甲酯在催化劑的作用下,進行預縮聚反應(也稱為熔融縮聚),制得特性粘度0.3-0.7dl/g的乙醇酸或乙醇酸甲酯預聚物;
(2)將步驟(1)得到的乙醇酸或乙醇酸甲酯預聚物用粉碎機粉碎至10-300目,優選為40-60目,得到乙醇酸或乙醇酸甲酯預聚物粉末;
(3)將步驟(2)得到的乙醇酸或乙醇酸甲酯預聚物粉末送至沸騰干燥機中,進行固相縮聚,制得特性粘度1.0dl/g以上的聚乙醇酸。
步驟(1)所述的催化劑由質量比為0.2:1-3:1的金屬氧化物催化劑和金屬鹽類催化劑復合組成;所述的金屬氧化物催化劑選自三氧化二銻、二氧化鍺、二氧化鈦、氧化錫或氧化鋅中的一種;所述的金屬鹽類催化劑選自醋酸鋅、辛酸亞錫、醋酸鈣、二水合醋酸鋅、氯化亞錫或二水合氯化亞錫中的一種。
步驟(1)中,催化劑中金屬氧化物催化劑用量為單體乙醇酸或乙醇酸甲酯質量的0.1%-2%。
本發明采用金屬氧化物催化劑和金屬鹽類催化劑復合組成,相較于普通單組份催化劑,可以更高效地分別作用于熔融縮聚和固相縮聚反應過程,有效提升單個過程的反應速率,且不會因催化劑濃度的提升而使得聚合物產生降解,從而提升全過程效率。
步驟(1)所述的預縮聚反應條件為:單體乙醇酸或乙醇酸甲酯與催化劑加入反應釜中混合均勻,在反應釜絕對壓力200-500kpa下,反應溫度從150℃開始,以0.2-0.4℃/min升溫至200-220℃,脫除小分子的水或甲醇;然后以降壓速率1-6kpa/min,升溫速率0.35-0.65℃/min,使反應釜絕對壓力達到20pa-5kpa,溫度220-240℃,并在該條件下繼續反應0.5-4h,進一步脫除水或甲醇,最終制得特性粘度0.3-0.7dl/g的乙醇酸或乙醇酸甲酯預聚物。
步驟(3)中所述的固相縮聚工藝條件為:在溫度為110-220℃,載氣流量為10-500l/min下,乙醇酸或乙醇酸甲酯預聚物粉末在氣流的推動下處于沸騰狀態,在該條件下進行固相縮聚12-100h,最終制得特性粘度1.0dl/g以上的聚乙醇酸。固相縮聚工藝條件進一步優選為:在溫度為210℃,載氣流量為120l/min下,乙醇酸或乙醇酸甲酯預聚物粉末在氣流的推動下處于沸騰狀態,在該條件下進行固相縮聚72h。此優選工藝條件較其他工藝條件更具經濟性及操作性,具有工業化放大的潛在價值。
本發明中,預縮聚反應與固相縮聚均需嚴格控制反應的工藝參數及條件,否則均會導致最終聚乙醇酸的分子量達不到要求值。
在聚合反應過程中,需要控制反應過程的工藝參數及條件,要求能夠有效的提升反應速率,促進聚合物分子量的快速提升,而聚乙醇酸本身屬于易降解的聚合物,在提升聚合反應速率的同時,必須注意避免聚合物在反應過程中發生降解,在此基礎上再提升產品的分子量,并保證分子量的分布均勻,本發明的工藝條件正是針對聚乙醇酸預縮聚反應和固相縮聚反應過程的特點而提出,能夠促進聚合物分子量的快速提升,避免了聚合物在反應過程中發生降解,得到產品的分子量高,且分子量分布均勻。
步驟(3)中所述的固相縮聚所用載氣為氮氣、氦氣或二氧化碳中的任意一種。步驟(3)固相縮聚所用載氣經過純化后可循環利用,減少了投資成本。
預熱的載氣從沸騰干燥機底部從下而上通入吹掃物料(預聚物)使物料狀如“沸騰”進行反應,這樣可加快固相縮聚的傳熱和傳質,同時載氣迅速帶走反應副產物(水或甲醇)有利于平衡正向移動。
步驟(3)中固相縮聚無需再加催化劑。
本發明采用乙醇酸或乙醇酸甲酯先預聚制得低分子量預聚物,經粉碎后轉移至沸騰干燥器中進行固相縮聚制得高分子量聚乙醇酸。通過對反應工藝條件的控制,能夠促進聚合物分子量的快速提升,避免了聚合物在反應過程中發生降解,得到產品的分子量高,且分子量分布均勻;引入了更高效的沸騰床干燥機來作為固相縮聚反應設備,傳熱更加高效且均勻,傳質效率也更高,非常有利于小分子水或甲醇的脫除,提升反應速率,同時有效的避免了傳質、傳熱不均導致的聚合物品質下降,且產品分子量分布均一。相對于其他途徑制取高分子量聚乙醇酸具有流程短、能耗低、成本低及裝置投資成本和維護費用低等優勢。
常規高分子量聚乙醇酸的制備方法是采用乙醇酸或乙醇酸酯類先預聚成低聚物,低聚物再高溫裂解成乙交酯,乙交酯再開環聚合成高分子量聚乙醇酸。該工藝流程比較復雜,投資及能耗均較高。也有專利文獻報道不經乙交酯中間體而“一步法”制備聚乙醇酸的方法,但由于聚乙醇酸易發生降解,合成得到的聚乙醇酸分子量普遍不高。
本方法提出了“熔融縮聚+固相縮聚”的方法來制備高分子量聚乙醇酸:選擇了合適的聚合反應工藝條件,能夠促進聚合物分子量的快速提升,避免了聚合物在 反應過程中發生降解,得到產品的分子量高,且分子量分布均勻;引入更有利于固相縮聚的沸騰干燥機來進行固相縮聚反應,沸騰床干燥機的傳熱更加高效且均勻,傳質效率也更高,非常有利于小分子水或甲醇的脫除,提升反應速率,有效的避免了傳質、傳熱不均導致的聚合物品質下降,且產品分子量分布均一。因此,相較于常規制備方法而言,本發明具有流程短、能耗低、成本低及裝置投資成本和維護費用低等優勢,且產品分子量高,分子量分布更均一。
附圖說明
圖1為預縮聚反應的工藝示意圖;
圖2為固相縮聚工藝示意圖。
具體實施方式
下面結合附圖對本發明實施方式進行詳細說明。
一種由乙醇酸或乙醇酸甲酯制備高分子量聚乙醇酸的方法,為一步法制取方法,具體包括以下步驟:
(1)單體乙醇酸或乙醇酸甲酯在催化劑的作用下,進行預縮聚反應,預縮聚反應條件為:單體乙醇酸或乙醇酸甲酯與催化劑加入反應釜中混合均勻,在反應釜絕對壓力200-500kpa下,反應溫度從150℃開始,以0.2-0.4℃/min升溫至200-220℃,脫除小分子的水或甲醇;然后以降壓速率1-6kpa/min,升溫速率0.35-0.65℃/min,使反應釜絕對壓力達到20pa-5kpa,溫度220-240℃,并在該條件下繼續反應0.5-4h,進一步脫除水或甲醇,最終制得特性粘度0.3-0.7dl/g的乙醇酸或乙醇酸甲酯預聚物。
(2)將步驟(1)得到的乙醇酸或乙醇酸甲酯預聚物用粉碎機粉碎至10-300目,優選為40-60目,得到乙醇酸或乙醇酸甲酯預聚物粉末;
(3)將步驟(2)得到的乙醇酸或乙醇酸甲酯預聚物粉末送至沸騰干燥機中,進行固相縮聚,固相縮聚工藝條件為:在溫度為110-220℃,載氣流量為10-500l/min下,乙醇酸或乙醇酸甲酯預聚物粉末在氣流的推動下處于沸騰狀態,在該條件下進行固相縮聚12-100h,最終制得特性粘度1.0dl/g以上的聚乙醇酸。固相縮聚工藝條件進一步優選為:在溫度為210℃,載氣流量為120l/min下,乙醇酸或乙醇酸甲酯預聚物粉末(40~60目)在氣流的推動下處于沸騰狀態,在該條件下進行固相縮 聚72h。此優選工藝條件較其他工藝條件更具經濟性及操作性,具有工業化放大的潛在價值。
步驟(1)所述的催化劑由質量比為0.2:1-3:1的金屬氧化物催化劑和金屬鹽類催化劑復合組成;所述的金屬氧化物催化劑選自三氧化二銻、二氧化鍺、二氧化鈦、氧化錫或氧化鋅中的一種;所述的金屬鹽類催化劑選自醋酸鋅、辛酸亞錫、醋酸鈣、二水合醋酸鋅、氯化亞錫或二水合氯化亞錫中的一種。步驟(1)中,催化劑中金屬氧化物催化劑用量為單體乙醇酸或乙醇酸甲酯質量的0.1%-2%。
步驟(3)中所述的固相縮聚所用載氣為氮氣、氦氣或二氧化碳中的任意一種。步驟(3)固相縮聚所用載氣經過純化后可循環利用,減少了投資成本。
其中步驟(1)所述預縮聚反應的工藝如圖1所示,單體乙醇酸或乙醇酸甲酯與催化劑加入反應釜1中,反應釜1上方連接精餾塔2,用于收集副產物甲醇或水,反應釜1內設攪拌裝置,反應釜1還順序連接冷凝器3與真空系統4,以提供預縮聚反應的反應條件,預縮聚反應結束后從反應釜1下方出料。
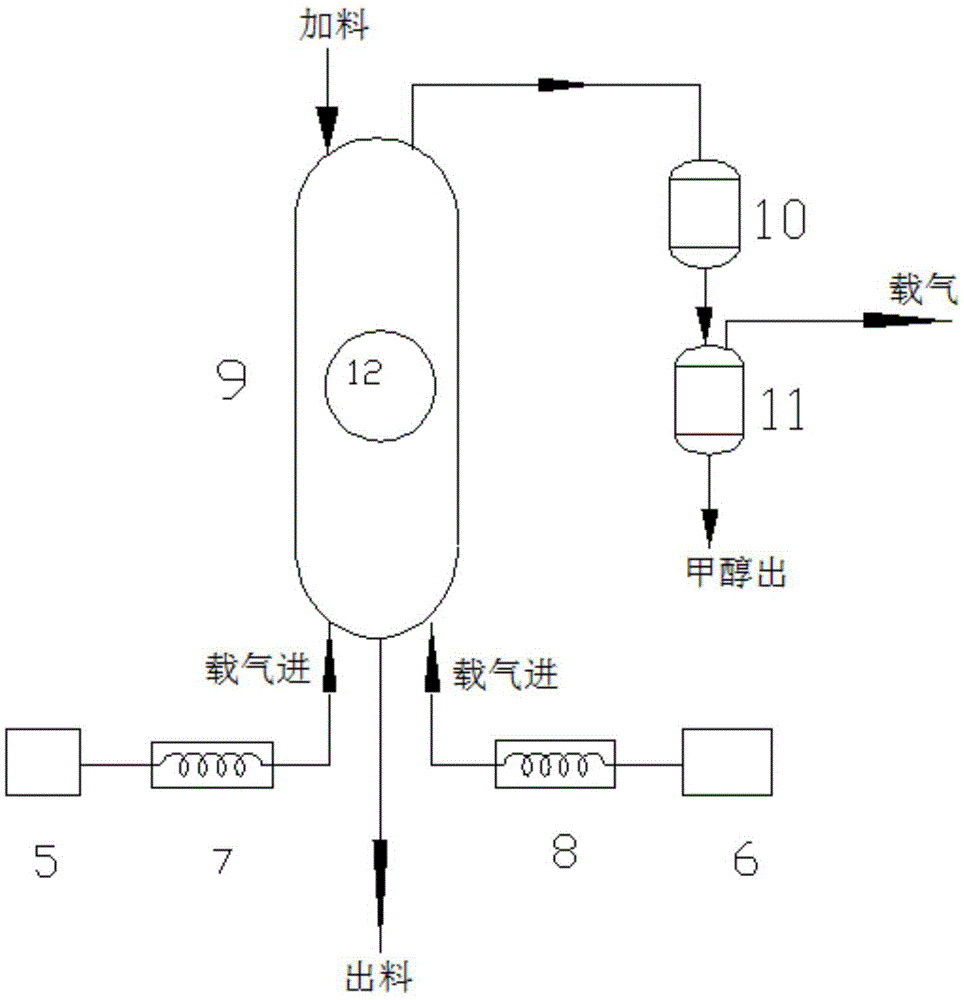
步驟(3)所述的固相縮聚工藝如圖2所示,乙醇酸或乙醇酸甲酯預聚物粉末加入到沸騰干燥機9中,沸騰干燥機9頂部為加料口,底端為出料口,設有兩個載氣源,分別為第一載氣源5與第二載氣源6,第一載氣源5的出氣管路經第一預熱器7后通入沸騰干燥機9的底部,第二載氣源6的出氣管路經第二預熱器8后通入沸騰干燥機9的底部。在沸騰干燥機9的頂部順序連接冷凝器10與氣液分離器11,同時在沸騰干燥機9的中部設置視窗12。
預熱的載氣從沸騰干燥機9底部從下而上通入吹掃物料(乙醇酸或乙醇酸甲酯預聚物粉末)使物料狀如“沸騰”進行反應,這樣可加快固相縮聚的傳熱和傳質,同時載氣迅速帶走反應副產物(水或甲醇)進入冷凝器10與氣液分離器11,甲醇或水從氣液分離器11底部流出,載氣從氣液分離器11頂部流出,經過純化后可循環利用。
以下所述實施例目的在于使本領域普通技術人員更易于理解本工藝方法,而不限制本發明。
實施例1
稱取乙醇酸甲酯3kg,加入3g二氧化鈦、15g辛酸亞錫,加入到5l的聚合反應釜中,混合均勻,控制反應釜內絕對壓力為500kpa,升溫至150℃開始反應,以升溫速率0.2℃/min升至200℃,副產物甲醇從精餾塔塔頂排出收集。然后以降壓 速率6kpa/min降至20pa,同時溫度以0.35℃/min升至240℃,在20pa、240℃的條件下,繼續反應0.5h,可得特性粘度0.50dl/g的乙醇酸甲酯低聚物。
實施例2
稱取乙醇酸甲酯3kg,加入60g的三氧化二銻、20g的二水合氯化亞錫,加入到5l的聚合反應釜中,混合均勻,控制反應釜內絕對壓力為200kpa,升溫至150℃開始反應,以升溫速率0.4℃/min升至220℃,副產物甲醇從精餾塔塔頂排出收集;然后以降壓速率1kpa/min降至5kpa,同時溫度以0.65℃/min升至240℃,在5kpa、240℃條件下,繼續反應4h,可得特性粘度0.30dl/g的乙醇酸甲酯低聚物。
實施例3
稱取乙醇酸甲酯3kg,加入27g的二氧化鍺、13.5g的二水合醋酸鋅,加入到5l的聚合反應釜中,混合均勻,控制反應釜內絕對壓力為350kpa,升溫至150℃開始反應,以升溫速率0.3℃/min升溫至210℃,副產物甲醇從精餾塔塔頂排出收集;然后以降壓速率3.5kpa/min降至1kpa,同時溫度以0.5℃/min升至230℃,在1kpa、230℃的條件下繼續反應2h,可得特性粘度0.70dl/g的乙醇酸甲酯低聚物。
實施例4
稱取乙醇酸3kg,加入3g二氧化鈦、15g辛酸亞錫,加入到5l的聚合反應釜中,混合均勻,控制反應釜內絕對壓力為500kpa,升溫至150℃開始反應,以升溫速率0.2℃/min升至200℃,副產物水從精餾塔塔頂排出收集。然后以降壓速率6kpa/min降至20pa,同時溫度以0.35℃/min升至240℃,在20pa、240℃的條件下,繼續反應0.5h,可得特性粘度0.62dl/g的乙醇酸低聚物。
實施例5
稱取乙醇酸3kg,加入27g的二氧化鍺、13.5g的二水合醋酸鋅,加入到5l的聚合反應釜中,混合均勻,控制反應釜內絕對壓力為350kpa,升溫至150℃開始反應,以升溫速率0.3℃/min升溫至210℃,副產物水從精餾塔塔頂排出收集;然后以降壓速率3.5kpa/min降至1kpa,同時溫度以0.5℃/min升至230℃,在1kpa、230℃的條件下繼續反應2h,可得特性粘度0.48dl/g的乙醇酸低聚物。
實施例6
將實施例2所得低聚物粉碎篩分至200—300目,稱取500g,送至沸騰干燥機中進行固相縮聚反應,設定固相縮聚溫度為110℃,二氧化碳流量500l/min,在氣流的作用下,乙醇酸甲酯預聚物粉末處于“沸騰”狀態,反應100h后測得物料的 特性粘度為1.09dl/g。
實施例7
將實施例3所得低聚物粉碎篩分至10—20目,稱取500g,送至沸騰干燥機中進行固相縮聚反應,設定固相縮聚溫度為220℃,氦氣流量10l/min,在氣流的作用下,乙醇酸甲酯預聚物粉末處于“沸騰”狀態,反應12h后測得產物的特性粘度為1.12dl/g。
實施例8
將實施例1所得低聚物粉碎篩分至40—60目,稱取500g,送至沸騰干燥機中進行固相縮聚反應,設定固相縮聚溫度為210℃,氮氣流量120l/min,在氣流的作用下,乙醇酸甲酯預聚物粉末處于“沸騰”狀態,反應72h后測得產物的特性粘度為1.08dl/g。
實施例9
將實施例4所得低聚物粉碎篩分至200—300目,稱取500g,送至沸騰干燥機中進行固相縮聚反應,設定固相縮聚溫度為110℃,二氧化碳流量500l/min,在氣流的作用下,乙醇酸預聚物粉末處于“沸騰”狀態,反應100h后測得物料的特性粘度為1.11dl/g。
實施例10
將實施例5所得低聚物粉碎篩分至40—60目,稱取500g,送至沸騰干燥機中進行固相縮聚反應,設定固相縮聚溫度為210℃,氮氣流量120l/min,在氣流的作用下,乙醇酸預聚物粉末處于“沸騰”狀態,反應72h后測得產物的特性粘度為1.05dl/g。
上述的對實施例的描述是為便于該技術領域的普通技術人員能理解和使用發明。熟悉本領域技術的人員顯然可以容易地對這些實施例做出各種修改,并把在此說明的一般原理應用到其他實施例中而不必經過創造性的勞動。因此,本發明不限于上述實施例,本領域技術人員根據本發明的揭示,不脫離本發明范疇所做出的改進和修改都應該在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!