吡啶胺基嘧啶衍生物甲磺酸鹽的結晶形式及其制備和應用的制作方法
本發明涉及吡啶胺基嘧啶衍生物甲磺酸鹽的結晶形式,具體的說,本發明涉及n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺甲磺酸鹽的結晶形式、其制備方法、包含該結晶形式的藥物組合物及該結晶形式在治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥中的應用。
背景技術:
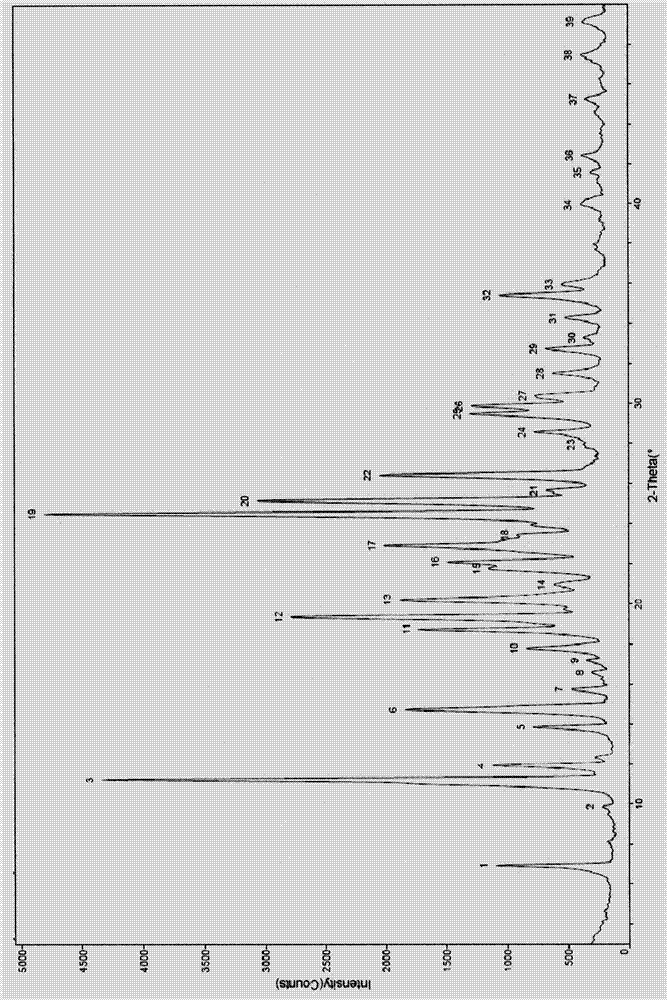
:表皮生長因子受體(egfr)被確認為是在細胞生長和增值過程中至關重要的驅動因素。表皮生長因子受體家族由egfr(erb-b1)、erb-b2(her-2/neu)、erb-b3和erb-b4組成。表皮生長因子受體與大部分癌癥的疾病進程有關,如肺癌、結腸癌、乳腺癌等。egfr的過度表達和突變已被明確證實是預后不好的乳腺癌的主要危險因素。目前,研究前沿的已經是不可逆的第三代egfr抑制劑。專利申請cn201410365911.4公開如下式(i)結構化合物,該化合物對egfr激活型突變(如19號外顯子缺失激活突變、或l858r激活突變)和t790m耐藥型突變的抑制活性顯著高于對野生型egfr(wtegfr)的抑制活性,有很好的選擇性,并且毒副作用較低,安全性好。晶型對化合物的物理性質有一定影響,對于具有多種晶型的藥用化合物,因其晶格結構不同,除可能導致具有不同的外觀如顏色、形狀外,也會引起某些物理性質如熔點、溶解性、密度、穩定性、吸濕性等不同,進而導致其在體內呈現出不同的溶出和吸收行為,在一定程度上會影響藥用化合物的臨床療效和安全性。特定的晶型可產生與無定型物質或者另一種晶型不同的熱力學行為。在實驗室中用諸如熔點儀、熱重分析(tga)或差示掃描量熱(dsc)技術測量熱性質可將某特定晶型與無定型或者另一晶型區別開來。而且特定的晶型可產生特定的光譜性質,如粉末x衍射圖譜數據和紅外光譜數據均可用于表征特定的晶型。技術實現要素:本發明所要解決的技術問題是提供式(i)化合物甲磺酸鹽的結晶形式、其制備方法,包含該結晶形式的藥物組合物及該結晶形式在治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥中的應用。本發明提供了式(i)所示n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的結晶形式i,本發明命名為晶型i,本發明提供的晶型i,其粉末x衍射圖在衍射角2θ值為4.58°±0.2°、14.08°±0.2°、15.00°±0.2°、16.40°±0.2°、17.84°±0.2°、18.30°±0.2°、20.26°±0.2°、21.10°±0.2°、21.88°±0.2°、22.66°±0.2°、25.58°±0.2°、26.78°±0.2°處具有特征峰。更進一步的,本發明提供的晶型i,其x射線粉末衍射圖(xrpd)如圖1所示。更進一步的,本發明提供的晶型i,在加熱至212.6℃開始出現吸熱峰,其差示掃描熱分析譜圖(dsc)如圖3所示。更進一步的,本發明提供的晶型i,在加熱至230℃時,重量損失約為1%,其熱重分析圖(tga)如圖5所示。本發明提供了化合物(i)的甲磺酸鹽的晶型i的制備方法,包括:a)將式(i)化合物懸浮于第一溶劑中,b)升溫至20~70℃,滴加事先溶于第二溶劑中的甲磺酸溶液,c)析晶、過濾得到晶型i。更進一步的,所述第一溶劑為水、酮、環醚或腈類溶劑或它們的混合溶劑;第二溶劑為水、酮、環醚或腈類溶劑或它們的混合溶劑。更進一步的所述第一溶劑為水與酮、環醚或腈類溶劑的混合溶劑;所述第二溶劑為酮、環醚或腈類溶劑,或水與酮、環醚或腈類溶劑的混合溶劑。更進一步的所述酮類溶劑包括但不限于丙酮,環醚類溶劑包括但不限于四氫呋哺或1,4-二氧六環,腈類溶劑包括但不限于乙腈。更進一步的,所述酮、環醚或腈類溶劑與水的混合溶劑,其中酮、環醚或腈類溶劑與水 的體積比是10∶1~25∶1,更進一步的酮、環醚或腈類溶劑與水的體積比是15∶1~19∶1。更進一步的,在b)步驟中升溫至35~55℃。本發明提供了化合物(i)的甲磺酸鹽的晶型i的制備方法,包括:a)將式(i)化合物懸浮于第一溶劑中,b)升溫至20~70℃,滴加事先溶于第二溶劑中的甲磺酸溶液,c)滴加第三溶劑d)析晶完過濾得到晶型i。更進一步的,所述第一溶劑為水、酮、環醚或腈類溶劑或它們的混合溶劑;第二溶劑為水、酮、環醚或腈類溶劑或它們的混合溶劑。更進一步的所述第一溶劑為水與酮、環醚或腈類溶劑的混合溶劑;所述第二溶劑為酮、環醚或腈類溶劑,或水與酮、環醚或腈類溶劑的混合溶劑。更進一步的所述酮類溶劑包括但不限于丙酮,環醚類溶劑包括但不限于四氫呋喃或1,4-二氧六環,腈類溶劑包括但不限于乙腈。更進一步的,所述酮、環醚或腈類溶劑與水的混合溶劑,其中酮、環醚或腈類溶劑與水的體積比是10∶1~25∶1,更進一步的酮、環醚或腈類溶劑與水的體積比是15∶1~19∶1。更進一步的,在b)步驟中升溫至35~55℃。更進一步的,所述第三溶劑為c6-7烷烴、醚或酯類溶劑。更進一步的所述c6-7烷烴類溶劑包括但不限于正庚烷;醚類溶劑包括但不限于甲基叔丁基醚;酯類溶劑包括但不限于甲酸甲酯、乙酸乙酯、乙酸異丙酯、乙酸丙酯或乙酸丁酯。本發明提供了式(i)所示n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的結晶形式ii,本發明命名為晶型ii,本發明提供的晶型ii,其粉末x衍射圖在衍射角2θ值為6.94°±0.2°、11.24°±0.2°、11.94°±0.2°、14.72°±0.2°、18.74°±0.2°、19.38°±0.2°、20.22°±0.2°、22.10°±0.2°、22.92°±0.2°、24.48°±0.2°、25.14°±0.2°、26.42°±0.2°處具有特征峰。更進一步的,本發明提供的晶型ii,其x射線粉末衍射圖(xrpd)如圖2所示。更進一步的,本發明提供的晶型ii,在加熱至206.8℃開始出現吸熱峰,其差示掃描熱分 析譜圖(dsc)如圖4所示。更進一步的,本發明提供的晶型ii,在加熱至220℃時,重量損失約為0.95%,其熱重分析圖(tga)如圖6所示。本發明提供了化合物(ii)的甲磺酸鹽的晶型ii的制備方法,包括將式(i)化合物的甲磺酸鹽的晶型i在加熱條件下溶解于醇類溶劑中,冷卻析晶過濾得到晶型ii。更進一步的,所述的醇類溶劑包括但不限于甲醇或乙醇。本發明提供的式(i)化合物的甲磺酸鹽的結晶形式中,式(i)化合物與甲磺酸的摩爾比是1∶1。本發明提供了藥物組合物,包括式(i)化合物甲磺酸鹽的結晶形式i。本發明提供了藥物組合物,包括式(i)化合物甲磺酸鹽的結晶形式ii。本發明提供了藥物組合物,包括式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物。本發明進一步提供藥物組合物,包括式(i)化合物甲磺酸鹽的結晶形式i,以及藥學上可接受的載體、賦形劑或稀釋劑。本發明進一步提供藥物組合物,包括式(i)化合物甲磺酸鹽的結晶形式ii,以及藥學上可接受的載體、賦形劑或稀釋劑。本發明進一步提供藥物組合物,包括式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物,以及藥學上可接受的載體、賦形劑或稀釋劑。本發明還提供了藥物組合物在制備治療癌癥的藥物中的用途,所述藥物組合物包括式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物。本發明提供了用作抗腫瘤藥物的式(i)化合物甲磺酸鹽的結晶形式i。本發明提供了用作抗腫瘤藥物的式(i)化合物甲磺酸鹽的結晶形式ii。本發明提供了用作抗腫瘤藥物的式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物。本發明還提供式(i)化合物甲磺酸鹽的結晶形式i在制備治療癌癥的藥物中的用途。本發明還提供式(i)化合物甲磺酸鹽的結晶形式i在制備治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥的藥物中的用途。本發明還提供式(i)化合物甲磺酸鹽的結晶形式ii在制備治療癌癥的藥物中的用途。本發明還提供式(i)化合物甲磺酸鹽的結晶形式ii在制備治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥的藥物中的用途。本發明還提供式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物在制備治療癌 癥的藥物中的用途。本發明還提供式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物在制備治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥的藥物中的用途。本發明還提供式(i)化合物甲磺酸鹽的結晶形式i在治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥方面的應用。本發明還提供式(i)化合物甲磺酸鹽的結晶形式ii在治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥方面的應用。本發明還提供式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物在治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥方面的應用。本發明還提供一種治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥的方法,所述方法包括對患者施用式(i)化合物甲磺酸鹽的結晶形式i、或包括治療有效量的式(i)化合物甲磺酸鹽的結晶形式i和藥物可接受載體、賦形劑或稀釋劑的藥物組合物。本發明還提供一種治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥的方法,所述方法包括對患者施用式(i)化合物甲磺酸鹽的結晶形式ii、或包括治療有效量的式(i)化合物甲磺酸鹽的結晶形式ii和藥物可接受載體、賦形劑或稀釋劑的藥物組合物。本發明還提供一種治療哺乳動物尤其人類由egfr激活型或耐藥型突變體介導的疾病、特別是癌癥的方法,所述方法包括對患者施用式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物、或包括治療有效量的式(i)化合物甲磺酸鹽的結晶形式i和結晶形式ii的混合物及藥物可接受載體、賦形劑或稀釋劑的藥物組合物。本發明所提及癌癥包括但不限于,例如肺癌、卵巢癌、宮頸癌、乳腺癌、胃癌、結腸直腸癌、胰腺癌、膠質瘤、膠質母細胞瘤、黑色素瘤、前列腺癌、白血病、淋巴瘤、非霍奇金淋巴瘤、肝細胞癌、胃腸道基質瘤(gist)、甲狀腺癌、膽管癌、子宮內膜癌、腎癌、間變性大細胞淋巴瘤、急性髓細胞白血病(aml)、多發性骨髓瘤、間皮瘤,尤其對于表皮生長因子受體790位蘇氨酸突變為蛋氨酸(egfrt790m)的腫瘤類型有更好的應用。舉例來說,本發明式(i)化合物的甲磺酸鹽的結晶形式可作為和用于治療非小細胞癌(egfrt790m)的藥物。本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物可給藥于哺乳動物包括人,可以口服、直腸、腸胃外(靜脈內、肌肉內或皮下)、局部給藥(粉劑、軟膏劑或滴劑)、或瘤內給藥。本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物的給藥劑量可以大0.05~50mg/kg體重/天,例如0.1~45mg/kg體重/天,更例如0.5~35mg/kg體重/天本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物可以配制為用于口服給藥的固體劑型,包括,但不限于膠囊劑、片劑、丸劑、散劑和顆粒劑等。在這些固體劑型中,本發明式(i)化合物的甲磺酸鹽作為活性成分與至少一種常規惰性賦形劑(或載體)混合,例如與檸檬酸鈉或磷酸二鈣,或與下述成分混合:(1)填料或增容劑,例如,淀粉、乳糖、蔗糖、葡萄糖、甘露醇和硅酸等;(2)粘合劑,例如羥甲基纖維素、藻酸鹽、明膠、聚乙烯基吡咯烷酮、蔗糖和阿拉伯膠等;(3)保濕劑,例如,甘油等;(4)崩解劑,例如瓊脂、碳酸鈣、馬鈴薯淀粉或木薯淀粉、藻酸、某些復合硅酸鹽和碳酸鈉等;(5)緩溶劑,例如石蠟等;(6)吸收加速劑,例如,季銨化合物等;(7)潤濕劑,例如鯨蠟醇和單硬脂酸甘油酯等;(8)吸附劑,例如,高嶺土等;和(9)潤滑劑,例如,滑石、硬脂酸鈣、硬脂酸鎂、固體聚乙二醇、十二烷基硫酸鈉等,或其混合物。膠囊劑、片劑和丸劑中也可包含緩沖劑。所述固體劑型如片劑、糖丸、膠囊劑、丸劑和顆粒劑可采用包衣和殼材料例如腸溶衣和其他本領域公知的材料進行包衣或微囊化。它們可包含不透明劑,并且,這種組合物中活性成分的釋放可以延遲的方式在消化道內的某一部分中釋放。可采用的包埋組分的實例是聚合物質和蠟類物質。必要時,活性成分也可與上述賦形劑中的一種或多種形成微膠囊形式。本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物可以配制為用于口服給藥的液體劑型,包括,但不限于藥學上可接受的乳液、溶液、懸浮液、糖漿和酊劑等。除了作為活性成分的式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物外,液體劑型可包含本領域中常規采用的惰性稀釋劑,如水和其他溶劑,增溶劑和乳化劑,例如,乙醇、異丙醇、碳酸乙酯、乙酸乙酯、丙二醇、1,3-丁二醇、二甲基甲酰胺以及油類,特別是棉籽油、花生油、玉米胚油、橄欖油、蓖麻油和芝麻油等或這些物質的混合物等。除了這些惰性稀釋劑外,本發明液體劑型也可包含常規助劑,如潤濕劑、乳化劑和懸浮劑、甜味劑、矯味劑和香料等。所述懸浮劑包括,例如,乙氧基化異十八烷醇、聚氧乙烯山梨醇和脫水山梨醇只、微晶纖維素、甲醇鋁和瓊脂等或這些物質的混合物。本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物可以配制為用于腸胃外注射的劑型,包括,但不限于生理上可接受的無菌含水或無水溶液、分散液、懸浮液或乳液,以及用于重新溶解成無菌的可注射溶液或分散液的無菌粉末。 適宜的載體、稀釋劑、溶劑或賦形劑包括水、乙醇、多元醇及其適宜的混合物。本發明化合物或其藥學上可接受的鹽也可以配制為用于局部給藥的劑型,包括如軟膏劑、散劑、栓劑、滴劑、噴射劑和吸入劑等。作為活性成分的本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物在無菌條件下和生理上可接受的載體及任選的防腐劑、緩沖劑,或必要時可能需要的推進劑一起混合。本發明還提供藥物組合物,它含有本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物,以及藥學上可接受載體、賦形劑或稀釋劑。在制備藥物組合物時,通常是將本發明式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物與藥學上可接受載體、賦形劑或稀釋劑混合。可以按常規制備方法將所述本發明組合物配制為常規藥物制劑。例如片劑、丸劑、膠囊劑、散劑、顆粒劑、乳液劑、混浮劑、分散液、溶液劑、糖漿劑、酏劑、軟膏劑、滴劑、栓劑、吸入劑、噴射劑等。本發明所述的式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii或結晶形式i與結晶形式ii的混合物可以單獨給藥,或者與其他藥學上可接受的治療劑聯合給藥,特別是與其他抗腫瘤藥物組合。所述治療劑包括但不限于:作用于dna化學結構的藥物抗腫瘤藥如順鉑,影響核苷酸合成的抗腫瘤藥物如甲氨蝶呤(mtx)、5-氟尿嘧啶(5fu)等,影響核酸轉錄的抗腫瘤藥物如阿霉素、表阿霉素、阿克拉霉素、光輝霉素等,作用于微管蛋白合成的抗腫瘤藥物如紫杉醇、長春瑞濱等,芳香化酶抑制劑如氨魯米特、蘭特隆、來曲唑、瑞寧德等,細胞信號通路抑制劑如表皮生長因子受體抑制劑伊馬替尼(imatinib)、吉非替尼(gefitinib)、埃羅替尼(erlotinib)等。待組合的各成分可同時或順序的給予,以單一制劑形式或以不用制劑的形式給予。所述組合不僅包括本發明化合物和一種其他活性劑的組合,而且也包括本發明化合物和兩種或更多種其他活性劑的組合。本發明式(i)化合物的甲磺酸鹽的結晶形式灌胃給藥的絕對生物利用度的測定方法如下:靜脈給藥:健康sd大鼠,隨機分組。測試物質按一定劑量d靜脈給藥,于給藥前及給藥后5min、15min、0.5h、1.0h、2.0h、4.0h、8.0h、12h和24h經眼球后靜脈叢取血,分離制備血漿,采用液相色譜/串聯質譜法測定血漿中藥物的濃度,得到藥物濃度-時間曲線。灌胃給藥:健康sd大鼠,隨機分組。測試物質按一定劑量d灌胃給藥,于給藥前和給藥后0.5、1.0、2.0、4.0、6.0、8.0、10、12和24h經大鼠眼球后靜脈叢取靜脈血,分離制備血漿,采用液相色譜/串聯質譜法測定血漿中藥物的濃度,得到藥物濃度-時間曲線。經劑量校正,按藥時曲線下面積(auc0-t)計算,得到絕對生物利用度f,計算公式f=(auc灌胃×d靜脈)/(auc靜脈×d灌胃)×100%。本發明式(i)化合物的甲磺酸鹽的結晶形式溶解度的測定方法如下:稱取各物質適量置于棕色量瓶中,加入不同溶劑,超聲20s,使其分散均勻后于25℃,200rpm震搖24h,取出,在12000rpm下離心10min,吸取上清液并用相應溶劑稀釋一定倍數后使用hplc測定濃度,并測定ph值。本發明的有益效果為:本發明提供的式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii經試驗證明在動物體內具有優異的生物利用度。本發明提供的式(i)化合物甲磺酸鹽的結晶形式i或結晶形式ii相對于式(i)化合物在不同ph值的溶劑中具有更好的溶解度。附圖說明附圖1是式(i)化合物甲磺酸鹽的晶型i的xrpd圖附圖2是式(i)化合物甲磺酸鹽的晶型ii的xrpd圖附圖3是式(i)化合物甲磺酸鹽的晶型i的dsc圖附圖4是式(i)化合物甲磺酸鹽的晶型ii的dsc圖附圖5是式(i)化合物甲磺酸鹽的晶型i的tga圖附圖6是式(i)化合物甲磺酸鹽的晶型ii的tga圖下面結合具體實施例,進一步闡述本發明。應理解,這些實施例僅用于舉例說明本發明而不用于限制本發明的范圍。下列實施例中未注明具體條件的實驗方法,通常按照常規條件,或按照制造廠商所建議的條件。除非另外說明,否則份數和百分比分別為重量份和重量百分比。具體實施方式本發明所述的x射線粉末衍射圖在panalyticalempyreanx射線粉末衍射儀上采集。本發明所述的x射線粉末衍的方法參數如下:x射線反射參數:cu,kα1.540598;1.544426kα2/kα1強度比例:0.50電壓:45千伏特(kv)電流:40毫安培(ma)掃描范圍:自3.0至50.0度本發明所述的差示掃描量熱分析(dsc)圖在perkinelmerdsc8500上采集。本發明所述的差示掃描量熱分析的方法參數如下:溫度控制:起始溫度為50℃,50℃維持1min,以10℃/min的速度升溫至250℃保護氣體:氮氣本發明所述的熱重分析(tga)圖在netzschtg209f3上采集。本發明所述的熱重分析的方法參數如下:溫度控制:30℃維持5min,以10℃/min的速度升溫至400℃保護氣體:氮氣i.制備實施例實施例1:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺中間體1c:n2-甲基-n2-[2-(二甲胺基)乙基]-6-(2,2,2-三氟乙氧基)-3-硝基吡啶-2,5-二胺,制備方法引用專利申請cn201410365911.4實施例中間體2a:3-(2-氯嘧啶-4-基)-1-甲基-1h-吲哚,制備方法引用專利申請cn201410365911.4實施例化合物(ii):n2-甲基-n2-[2-(二甲胺基)乙基]-6-(2,2,2-三氟乙氧基)-n5-[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]-3-硝基吡啶-2,5-二胺的合成在圓底燒瓶中加入3-(2-氯嘧啶-4-基)-1-甲基-1h-吲哚(73mg,0.3mmol)、n2-甲基-n2-[2-(二甲胺基)乙基]-6-(2,2,2-三氟乙氧基)-3-硝基吡啶-2,5-二胺(100mg,0.3mmol)、三(二亞芐基丙酮)二鈀(14mg,0.015mmol),4,5-雙二苯基膦-9,9-二甲基氧雜蒽(14mg,0.03mmol),磷酸鉀(127mg,0.6mmol)和8ml二氧六環,氬氣保護下,95℃反應5h。過濾,濾液減壓蒸干,硅膠柱層析(二氯甲烷∶甲醇=20∶1),得到140mg產物。收率為86%。msm/z:545[m+1]。化合物(iii):n2-甲基-n2-[2-(二甲胺基)乙基]-6-(2,2,2-三氟乙氧基)-n5-[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]吡啶-2,3,5-三胺的合成在圓底燒瓶中加入n2-甲基-n2-[2-(二甲胺基)乙基]-6-(2,2,2-三氟乙氧基)-n5-[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]-3-硝基吡啶-2,5-二胺(150mg,0.27mmol)、二氧化鉑(60mg)和10ml甲醇,通入氫氣,室溫反應1h。過濾,制備板分離(二氯甲烷∶甲醇=10∶1),得到80mg目標化合物。收率為56%。msm/z:515[m+1]。化合物(i):n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的合成向圓底燒瓶中加入n2-甲基-n2-[2-(二甲胺基)乙基]-6-(2,2,2-三氟乙氧基)-n5-[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]吡啶-2,3,5-三胺(80mg,0.16mmol)和5ml二氯甲烷,冰水浴冷卻,加入0.5n丙烯酰氯的二氯甲烷溶液(0.5ml,0.25mmol)。冰水浴下反應1.5小時,反應液用50ml乙酸乙酯稀釋,飽和碳酸氫鈉溶液洗滌,有機相用無水硫酸鈉干燥,過濾,濾液減壓濃縮,制備板分離純化(二氯甲烷∶甲醇=10∶1),得到20mg目標產物。收率為23%。msm/z:569[m+1]。1hnmr(400mhz,dmso-d6)δ10.41(s,1h),10.27(s,1h),8.68(s,1h),8.44(s,1h),8.28(t,j=8.5hz,2h),8.18(s,1h),7.52(d,j=8.0hz,1h),7.29-7.14(m,3h),6.98(s,1h),6.28(d,j= 17.1hz,1h),5.76(d,j=10.4hz,1h),5.00(q,j=9.0hz,2h),3.89(s,3h),3.61(s,2h),3.28(s,2h),2.80(s,3h),2.73(s,6h)。其中化合物(ii)、(iii)、(i)的制備方法參考專利申請cn201410365911.4實施例1。實施例2:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(451.5g,0.77mol)加至10l反應釜中,加入丙酮水溶液(5.42l,二者體積比15∶1),攪拌,氮氣置換,升溫至35-40℃。滴加甲磺酸(74.2g,0.76mol)的丙酮溶液(1.35l)。滴加完畢,控溫35-40℃攪拌15-18小時。滴加乙酸乙酯(3.39l),滴加完畢,緩慢降溫至20-25℃,過濾,乙酸乙酯(0.45l)洗濾餅。50℃真空干燥40-48小時,得晶型i(409.9g),收率80.09%。1hnmr(400mhz,dmso-d6)δ9.80(s,1h),9.23(s,1h),8.53(s,1h),8.42(s,1h),8.30(d,j=5.4hz,2h),8.23(s,1h),7.52(d,j=8.2hz,1h),7.25(t,j=7.2hz,1h),7.22(d,j=8.0hz,1h),7.15(t,j=7.4hz,1h),6.70(dd,j=17.0,10.2hz,1h),6.34(dd,j=17.0,1.7hz,1h),5.83(dd,j=10.3,1.6hz,1h),5.02(q,j=9.1hz,2h),3.88(s,3h),3.65(t,j=6.0hz,2h),3.33(t,j=6.0hz,2h),2.86(s,6h),2.81(s,3h),2.44(s,3h)。經檢測,本實施例得到晶型i粉末x衍射圖在衍射角2θ值為4.58°±0.2°、14.08°±0.2°、15.00°±0.2°、16.40°±0.2°、17.84°±0.2°、18.30°±0.2°、20.26°±0.2°、21.10°±0.2°、21.88°±0.2°、22.66°±0.2°、25.58°±0.2°、26.78°±0.2°處具有特征峰;其xrpd圖譜如圖1,dsc圖如圖3,tga圖如圖5。實施例3:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入四氫呋喃水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的四氫呋喃水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃攪拌15-18小時。緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(3.4g),收率57.95%。實施例4:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入乙腈水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的乙腈水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃攪拌15-18小時。滴加乙酸乙酯(25ml),滴加完畢,緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(4.3g),收率73.29%。實施例5:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入四氫呋喃水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的四氫呋喃水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃攪拌15-18小時。滴加乙酸乙酯(25ml),滴加完畢,緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,晶型i(4.75g),收率80.96%。實施例6:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入丙酮水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的四氫呋喃水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃,滴加乙酸乙酯(25ml),滴加完畢,緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(5.1g),收率86.93%。實施例7:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加 入四氫呋喃水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的四氫呋喃水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃攪拌15-18小時。滴加乙酸異丙酯(37.5ml),滴加完畢,緩慢降溫至20-25℃,過濾,乙酸異丙酯(5ml)洗濾餅。緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(4.4g),收率75.00%。實施例8:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入丙酮水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的四氫呋喃水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃,滴加甲酸乙酯(25ml),滴加完畢,緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(5.1g),收率86.93%。實施例9:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入丙酮水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的四氫呋喃水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃,滴加正庚烷(25ml),滴加完畢,緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(4.8g),收率81.82%。實施例10:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入乙腈水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的乙腈水溶液(7.5ml,二者體積比19∶1)。滴加完畢,控溫40-45℃攪拌15-18小時。緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(4.0g),收 率68.18%。實施例11:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型i的制備將n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺(5g,8.8mmol)加至100ml反應瓶中,加入乙腈水溶液(42.5ml,二者體積比19∶1),攪拌,氮氣置換,升溫至40-45℃。滴加甲磺酸(0.84g,8.7mmol)的乙腈溶液(7.5ml)。滴加完畢,控溫40-45℃,滴加甲酸甲酯(355ml),滴加完畢,緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型i(4.5g),收率76.70%。實施例12:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型ii的制備將按照實施例2所得的晶型i(5g,7.5mmol)加至100ml反應瓶中,加入甲醇(50.0ml),攪拌,升溫至50-55℃。控溫50-55℃攪拌15-18小時。緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型ii(3.1g),收率62.0%。1hnmr(400mhz,dmso-d6)δ9.80(s,1h),9.23(s,1h),8.53(s,1h),8.42(s,1h),8.30(d,j=5.4hz,2h),8.23(s,1h),7.52(d,j=8.2hz,1h),7.25(t,j=7.2hz,1h),7.22(d,j=8.0hz,1h),7.15(t,j=7.4hz,1h),6.70(dd,j=17.0,10.2hz,1h),6.34(dd,j=17.0,1.7hz,1h),5.83(dd,j=10.3,1.6hz,1h),5.02(q,j=9.1hz,2h),3.88(s,3h),3.65(t,j=6.0hz,2h),3.33(t,j=6.0hz,2h),2.86(s,6h),2.81(s,3h),2.44(s,3h)。經檢測,本實施例得到晶型ii粉末x衍射圖在衍射角2θ值為6.94°±0.2°、11.24°±0.2°、11.94°±0.2°、14.72°±0.2°、18.74°±0.2°、19.38°±0.2°、20.22°±0.2°、22.10°±0.2°、22.92°±0.2°、24.48°±0.2°、25.14°±0.2°、26.42°±0.2°處具有特征峰;其xrpd圖譜如圖2,dsc圖如圖4,tga圖如圖6。實施例13:n-{2-{[2-(二甲胺基)乙基](甲基)胺基}-6-(2,2,2-三氟乙氧基)-5-{[4-(1-甲基-1h-吲哚-3-基)嘧啶-2-基]胺基}吡啶-3-基}丙烯酰胺的甲磺酸鹽的晶型ii的制備將按照實施例2所得的晶型i(5g,7.5mmol)加至100ml反應瓶中,加入乙醇(50.0ml),攪拌,升溫至50-55℃。控溫50-55℃攪拌15-18小時。緩慢降溫至20-25℃,過濾。50℃真空干燥40-48小時,得晶型ii(3.1g),收率62.0%。ii.活性測試實施例測試實施例1:sd大鼠(spraguedawley大鼠)藥物吸收實驗靜脈給藥:健康sd大鼠,雌雄各半共20只,體重200~280g,由上海西普爾-必凱實驗動物有限公司提供,隨機分成五組。分別按下表所列劑量靜脈給予式(i)化合物甲磺酸鹽晶型i、晶型ii、實施例1的式(i)化合物、對照例1和對照例2,于給藥前及給藥后5min、15min、0.5h、1.0h、2.0h、4.0h、8.0h、12h和24h經大鼠眼球后靜脈叢取靜脈血0.2ml,分離制備血漿,采用液相色譜/串聯質譜法測定血漿中藥物的濃度,得到藥物濃度-時間曲線。其主要的藥物動力學參數如下表1所示:參數晶型i晶型ii實施例1對照例1對照例2劑量d(mg/kg)2.52.52.53.04.0cmax(ng/ml)104.7125.481.3327.5630.2auc0-t(ng·h/ml)307.9362.0307.3437.8810.7t1/2(h)4.734.833.962.721.71表1表1中,對照例1物質結構如下,按專利申請cn201410365911.4實施例2方法制備得到,對照例2物質結構如下,按專利申請cn201410365911.4實施例16方法制備得到,t1/2:消除半衰期;cmax:血漿中藥物的最高濃度;auc0-t:藥時曲線下面積灌胃給藥:健康sd大鼠,雌雄各半共20只,體重200~280g,由上海西普爾-必凱實驗動物有限公司提供,隨機分成五組。分別按下表所列劑量灌胃給予式(i)化合物甲磺酸鹽晶型i、晶型ii、實施例1的式(i)化合物、對照例1和對照例2,于給藥前和給藥后0.5、1.0、2.0、4.0、6.0、8.0、10、12和24h經大鼠眼球后靜脈叢取靜脈血0.2ml,分離制備血漿,采用液相色譜/串聯質譜法測定血漿中藥物的濃度,得到藥物濃度-時間曲線。其主要的藥物動力學參數如下表2所示:參數晶型i晶型ii實施例1對照例1對照例2劑量d(mg/kg)101010610cmax(ng/ml)45.336.717.612.6328.3auc0-t(ng·h/ml)613.2566.3231.777.66172.2t1/2(h)7.985.794.148.605.76.f(%)49.839.118.858.98.5表2,其中對照例1、2的結構與制備方法同表1經劑量校正后,按auc0-t計算,得到絕對生物利用度f,計算公式f=(auc灌胃×d靜脈)/(auc靜脈×d灌胃)×100%,得到的絕對生物利用度f見上表2。結論:式(i)化合物的甲磺酸鹽的晶型i、晶型ii灌胃給藥的絕對生物利用度明顯優于實施例1式(i)化合物、對照例1和對照例2灌胃給藥的絕對生物利用度。測試實施例2:溶解度試驗考察實施例1的式(i)化合物、式(i)化合物的甲磺酸鹽的晶型i、式(i)化合物的甲磺酸鹽的晶型ii在不同ph緩沖液下的溶解度。測定方法:稱取各物質適量置于棕色量瓶中,加入不同溶劑,超聲20s,使其分散均勻后于25℃,200rpm震搖24h,取出,在12000rpm下離心10min,吸取上清液并用相應溶劑稀釋一定倍數后使用hplc測定濃度,并測定ph值。結果見表。結論:式(i)化合物的甲磺酸鹽的晶型i及晶型ii在溶劑ph值為1.0、4.5、6.8時的溶解度均明顯優于實施例1的式(i)化合物。在本文中提及的所有文獻均通過引用被并入本申請中。此外還應指明的是,在閱讀了本申請的上述公開內容之后,本領域技術人員可以無需背離本發明的精神和范圍,對本發明作出各種修飾、改動或修改,但這些變化形式同樣都應落于本申請所附權利要求書所記載的范圍。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1