一種(+)-丹參素及其類似物的制備方法與流程
本發明屬于醫藥技術領域,具體地說,涉及一種(+)-丹參素及其類似物的制備方法。
背景技術:
丹參素,化學名為r-(+)-β-3,4-二羥基苯基乳酸,其不僅是臨床上大規模使用的丹參注射液類制劑的重要質量標準成分,也是該類制劑發揮療效的主要活性成分。
藥理研究結果表明,丹參素可抑制血小板合成,提高心肌耐缺氧能力,保護心肌,增加冠狀動脈流量,具有抗氧化、抗心肌缺血、調血脂、抗血栓形成、神經細胞保護、防治肝纖維化和抗腫瘤等多種作用,為復方丹參滴丸、丹參注射液等的有效成分。由于直接從丹參中提取得到的物質為水溶性成分的混合物,不僅純度低,且丹參素含量較低,加之丹參資源有限,丹參素的提取純化工藝困難(參見貴州景峰cn104974033a,天津天士力cn102464581a,貴州拜特cn101633616a),影響了臨床用藥的質量和天然藥物的進一步開發和持續利用。因此,尋找有效的途徑來解決丹參素的資源問題一直以來備受人們關注。
人們首先合成了消旋體丹參素及其類似物[薛芬,戴華娟,丁麗蓉,上海第一醫學院學報,1983,10(2):133;鄧喜玲,陳學敏,江發壽等,中國醫藥工業雜志,2005,36(9):523;張彥文,馮淑華,天津醫科大學學報,2005,11(4):522;童元峰,郭曉赟,程永浩,吳松,中國藥物化學雜志,2007,17(2):92],所述合成一般是以3,4-二羥基苯甲醛為原料,與乙酰甘氨酸反應生成azactone,經水解再用鋅汞齊還原得到外消旋丹參素。由于丹參素結構中有兩個極易被氧化的酚羥基,因此,難以對外消旋丹參素進行手性拆分。之后, cn102924265a報道了一種(+)-丹參素的不對稱合成方法,但合成路線較長,且不對稱還原反應條件苛刻,所用不對稱還原試劑(-)-dip-cl的價格高昂,不適合產業化。此外,cn102863328a報道了以d-酪氨酸為起始原料,經重氮化反應及高價碘氧化作用得到丹參素的方法,但該方法成本高,且亞硝酸鹽及產生的重氮化物易導致環境污染。另外,cn103667371a公布了一種對羥基苯丙酮酸在對羥基苯乙酸間位羥基化酶的催化下合成3,4-二羥基苯丙酮酸,隨后在d-乳酸脫氫酶的催化下合成丹參素,但該方法生產周期較長,且后續純化困難。cn104926646a報道了一種化學合成丹參素以及相關類似物的方法,以肉桂酸酯類化合物為原料,不對稱環氧化后氫化反應或金屬催化的不對稱氫化反應;或者環氧化反應再經過重排反應得到β-苯基丙酮酸類化合物,然后經過金屬催化的不對稱氫化反應。但該方法所用不對稱環氧化以及不對稱還原反應試劑價格高昂,且反應步驟較長,產業化價值較低。因此,仍然需要開發新的(+)-丹參素及其類似物的制備工藝。
技術實現要素:
針對現有技術的上述不足之處,本發明的目的在于提供一種丹參素及其類似物的制備方法。與現有技術方法相比,應用該制備方法不僅可以避免使用高污染的鋅汞齊以及價格昂貴的不對稱硼烷及環氧試劑,而且實驗操作簡單,合成試劑價廉易得,且能以超過80%的總收率獲得單一光學純的(+)-丹參素及其類似物。
本發明的上述目的是通過下述的技術方案加以實現的:
一種由通式i表示的丹參素及其類似物的合成方法,
其特征在于,所述方法包括如下步驟:
(1)制備金屬有機試劑:將rx與金屬鎂試劑和/或金屬鋰試劑在溶劑中反應,得金屬化合物rmgx或rli,
(2)制備中間體2(r),3-環氧丙酸及其酯:將l-絲氨酸在-5至-15℃下溶解在水中,滴加亞硝酸鈉-氫溴酸溶液并攪拌;萃取后取有機層濃縮、干燥,溶于乙醇中,然后在冰浴下加入koh,攪拌反應8至15小時;反應完成后加入水,調節ph至3,析晶,抽濾,洗滌并干燥,得到中間體2(r),3-環氧丙酸;然后在常規酯化條件下與醇r1oh反應得到酯;
(3)不對稱加成及脫保護:然后將步驟(1)的金屬化合物與步驟(2)的2(r),3-環氧丙酸及其類似物溶液反應,得到加成產物;然后脫去保護基,得到通式i所示的化合物;
其中,r表示未取代的或者取代的選自下列的基團:苯基、吡啶基、噻吩基、呋喃基、吡咯基和咪唑基,所述取代的取代基選自羥基、硝基、氰基、甲酰基或者未取代的或取代的選自下列的基團:烷基、環烷基、環氧基、烷環氧基、烯基、炔基、芳基、芳氧基、芳烷基、芳烷氧基、雜環基、雜芳基、雜芳烷基、雜芳氧基、雜烷芳氧基、酰基、酰氧基、羥烷基、氨基、酰胺基、一烷基氨基、二烷基氨基、芳基氨基、芳烷基氨基、氨基烷基、烷氧羰基、芳氧羰基、芳烷氧羰基、烷氧基烷基、芳氧基烷基、芳烷氧基烷基、烷硫基、硫代烷基、烷氧羰基氨基、芳氧羰基氨基、芳烷氧羰基氨基、羧酸或其衍生物、磺酸或其衍生物;
r1表示氫或者未取代的或取代的選自下列的基團:烷基、環烷基、烯基、炔基、芳基、芳烷基、雜環基、雜芳基、雜芳烷基;
在r和r1中,所述烷基優選為c1-c10烷基,更優選為c1-c6烷基,更優選為c1-c3烷基;所述環烷基優選為c3-c10環烷基,更優選為c3-c8環烷基,更優選為c5-c7環烷基;所述雜環基優選為具有1-3個選自o、n、s的雜原子的c3-c10雜環烷基,更優選為c3-c8雜環烷基,更優選為c5-c7雜環烷基;所述烯基優選為c2-c10烯基,更優選為c2-c6烯基,更優選為c2-c3烯基;所述炔基優選為c2-c10炔基,更優選為c2-c6炔基,更優選 為c2-c3炔基;所述芳基優選為c6-c40芳基,更優選為c6-c20芳基,更優選為c6-c10芳基;所述雜芳基優選為具有1-3個選自o、n、s的雜原子的c6-c40雜芳基,更優選為c6-c20雜芳基,更優選為c6-c10雜芳基;
優選地,r表示未取代的或者取代的選自下列的基團:苯基、吡啶基、噻吩基、呋喃基、吡咯基和咪唑基,所述取代的取代基選自羥基、硝基、氰基、甲酰基;
r1表示氫或者c1-c6烷基;
更優選地,r表示未取代的或者取代的苯基,所述取代的取代基選自羥基、硝基、氰基、甲酰基;
r1表示氫或者c1-c3烷基;
x為cl、br或i,
其中,所述步驟(2)中的常規酯化條件為:在酸催化劑的作用下加熱至60-140℃,優選70-130℃,更優選80-120℃下反應,其中,所述酸催化劑為本領域公知的用于酯化反應的催化劑,例如,濃硫酸、對甲苯磺酸。
本發明具體相關合成路線如下:
丹參素及其類似物的合成路線:
所述制備方法中,優選地,步驟(1)所用金屬鎂試劑為如mg粉、甲基溴化鎂、乙基溴化鎂、異丙基氯化鎂,金屬鋰試劑為如正丁基鋰、異丁基鋰以及叔丁基鋰;
所述制備方法中,優選地,步驟(1)的反應溫度為0~-80℃;
所述制備方法中,優選地,步驟(1)所用的溶劑為乙醚、四氫呋喃、二氧六環、1,2-二甲氧基乙烷或甲基叔丁基醚,這些溶劑可以單獨使用或者以一定比例的混合物形式使用,或者以其與烴,如甲苯、庚烷、或環己烷的混合物形式使用;
所述制備方法中,優選地,步驟(3)的脫保護基的試劑包括:
(1)lewis酸類:如alcl3/hsch2ch2sh、bbr3、bf3、bcl3、mgi2、hbr/bu4nbr、hbr/nai、hbr/acoh,或者
(2)氫化還原類:如pd/c、pd/caco3、pd/baso4、pt/c、rh/al2o3、raney鎳、pto2、ru/c;
所述制備方法中,優選地,步驟(3)在脫保護基時使用的溶劑為水、甲醇、乙醇、正丙醇、異丙醇、環己醇、乙酸、二氯甲烷、氯仿、乙醚、乙酸乙酯、甲苯、四氫呋喃、環己烷、二甲基甲酰胺中的一種或幾種;
所述制備方法中,優選地,步驟(3)中的所述lewis酸類試劑的用量為2(r),3-環氧丙酸摩爾量的2-5倍;或者
所述制備方法中,優選地,步驟(3)中的所述氫化還原類試劑的用量為2(r),3-環氧丙酸摩爾量的0.5-10%。
有益效果
根據本發明的方法,可以以更簡單的方法和過程制備光學純的丹參素及其類似物,所需的反應試劑低廉、易得,反應過程安全,不產生環境污染,反應總收率高,具有產業化價值。
附圖說明
圖1是根據本發明的實施例4和6所述的方法制備的(+)-丹參素,即光學純的r-(+)-β-3,4-二羥基苯基乳酸的hplc譜圖。
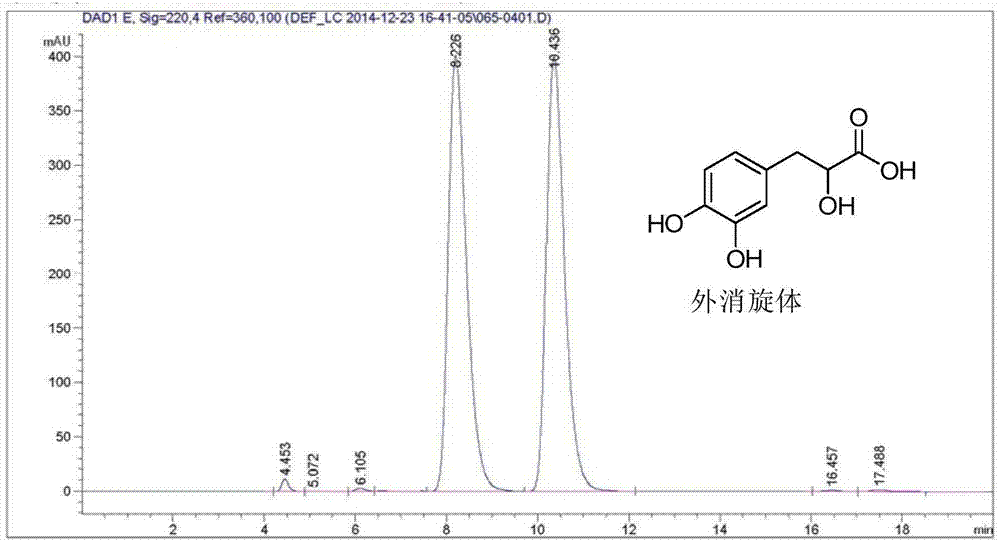
圖2是外消旋的β-3,4-二羥基苯基乳酸的hplc譜圖(按照鄧喜玲,陳學敏,江發壽等,中國醫藥工業雜志,2005,36(9):523記載的方法制造的消旋體丹參素)。
具體實施方式
以下將通過實施例詳細描述本發明,但本領域技術人員能夠理解,以下實施例僅用于說明本發明,而不是為了限制本發明的保護范圍。
在以下實施例中,原料l-絲氨酸購自(上海阿拉丁試劑公司),亞硝酸鈉、氫溴酸等試劑購自(國藥試劑),正丁基鋰、鎂試劑等(相關試劑購自百靈威試劑公司)采用如arkadykrasovskiy(angew.chem.int.ed.2004,43,3333–3336)所述的方法制備,產物采用分析儀器如nmr(bruker400mhz)、 hresims(agilentg6224atofspectrometer)、hplc(agilent1260,大賽璐od-h手性柱)進行相關結構及手性ee值測試。
實施例12(r),3-環氧丙酸(化合物1)
250ml三口燒瓶中加入l-絲氨酸10.0g(100mmol),水50ml,冷卻至-10℃,滴加亞硝酸鈉-氫溴酸溶液(20.7克亞硝酸鈉溶于3m氫溴酸溶液100ml),滴畢,繼續攪拌12小時;反應完成后,乙酸乙酯萃取(100ml×3),合并萃取液,減壓濃縮至干后溶于100ml乙醇中,然后于冰浴下加入11.2gkoh,攪拌反應12h。反應完成后加入500ml水,hcl調節ph至3,有大量晶體洗出,抽濾,冰水洗滌,干燥,得到白色晶體化合物1共78g,收率:88.6%。
實施例23,4-二芐氧基(+)-丹參素
3,4-二芐氧基溴苯(18.4g,50mmol)加入到500ml的乙醚溶液中,冷卻至-50℃,滴加1m的正丁基鋰溶液(50mmol),滴畢,繼續攪拌0.5小時;然后將上述反應液于-50℃條件下滴加到化合物1(50mmol)的乙醚溶液中,滴加完后室溫下反應0.5h。反應完成后,乙酸乙酯水萃取三次,合并萃取液并減壓濃縮至干,得到固體18.8g。收率:99%。
實施例33,4-二芐氧基(+)-丹參素(2)
3,4-二芐氧基溴苯(18.4g,50mmol)加入到500ml的乙醚溶液中,于冰浴下加入金屬鎂(15g,75mmol),然后緩慢升溫至回流1h,過濾得到濾液;然后將濾液在冰浴攪拌的條件下,滴加到化合物1(50mmol)的乙醚溶液,滴加完后室溫下反應0.5h。反應完成后,乙酸乙酯水萃取三次,合并萃取液并減壓濃縮至干,得到固體18.4g。收率:96%。
實施例4(+)-丹參素
3,4-二芐氧基(+)-丹參素(15.1g,40mmol)溶于200ml乙酸乙酯中,加入5%的pd/c(1g),在氫氣氛圍下,于室溫下繼續攪拌反應20h。反應完成后,硅藻土過濾,減壓除去溶劑,得粗產物為淺黃色油狀物,經乙醇重結晶,過濾,得到固體丹參素6.73g,產率85%。ee>99%(hplc,od-h,hexane/ipa=2:8,0.5ml/min,589nm,t=10.4min.[α]d20=+16°,(c0.1g/100ml,meoh).1hnmr(500mhz,dmso):δ2.77(d,1h),3.84(d,1h),4.20(d,1h),6.43(d,1h),6.59(d,2h);13cnmr(125mhz,dmso):δ40.63,72.12,114.75,117.24,122.27,133.06,143.23,143.82,173.33。hrmscalcdforc9h10o5na(m+na+)221.0420,221.0425。
實施例53,4-二甲氧氧基(+)-丹參素
3,4-二甲氧氧基(+)-丹參素甲酯(13.1g,51mmol)溶于甲醇水(4:1)中,加入naoh(4g,100mmol),于室溫下攪拌反應4h。反應完成后,調節ph至3,乙酸乙酯萃取3次,合并3次萃取液并減壓濃縮至干,得到3,4-二甲氧氧基(+)-丹參素11g,產率96%。
實施例6(+)-丹參素
3,4-二甲氧氧基(+)-丹參素(15.1g,40mmol)溶于200ml二氯甲烷中,加入bbr3(160mmol,1m),于室溫下攪拌反應20h。反應完成后,加水猝滅,乙酸乙酯萃取3次,合并3次萃取液并減壓濃縮至干,再經乙醇重結晶,過濾,得到固體丹參素6.0g,產率76%。ee>99%(hplc,od-h,hexane/ipa=2:8,0.5ml/min,589nm,t=10.4min.[α]d20=+16o,(c0.1g/100ml,meoh).1hnmr(500mhz,dmso):δ2.77(d,1h),3.84(d,1h),4.20(d,1h),6.43(d,1h),6.59(d,2h);13cnmr(125mhz,dmso):δ40.63,72.12,114.75,117.24,122.27,133.06,143.23,143.82,173.33。hrmscalcdforc9h10o5na(m+na+)221.0420,221.0425。
將實施例4和實施例6中制得的產物經過hplc檢測,得到如圖1所示的結果。
作為對比,圖2是外消旋的β-3,4-二羥基苯基乳酸的hplc譜圖(按照鄧喜玲,陳學敏,江發壽等,中國醫藥工業雜志,2005,36(9):523記載的方法制造的消旋體丹參素),由圖1和圖2的結果可以看出,本申請的實施例4和實施例6中制備的是光學純的(+)-丹參素,即r-(+)-β-3,4-二羥基苯基乳酸。
- 還沒有人留言評論。精彩留言會獲得點贊!