一類脂肪酸類化合物、其制備方法及其用途與流程
本發明屬于藥物化學領域,涉及一類脂肪酸類化合物或其藥學上可接受的鹽及其制備方法,以及包含該類化合物的藥物組合物,該類化合物具有激活人源hepg2肝癌細胞中ampk和抑制c57bl/6j小鼠原代肝細胞葡萄糖輸出能力的活性,可應用于制備治療肥胖及糖尿病等疾病的藥物。
背景技術:
糖尿病(diabetesmellitus)是機體胰島功能減退、胰島素抵抗等因素導致的一系列代謝紊亂綜合癥,遺傳因素、免疫功能紊亂、精神因素等多種致病因子均會引發糖尿病。據世界衛生組織2011年的統計表明,全球的糖尿病患者已經達到3.66億人,糖尿病已成為除心腦血管疾病、惡性腫瘤外的第三大疾病。而我國已成為世界第一糖尿病大國,患病率為9.7%,已高于世界平均水平,患病總數已經達到9240萬,位居世界前列。
現在已經有許多用于治療ⅱ型糖尿病的藥物,其中包括二甲雙胍、磺酰脲類、dpp-4抑制劑、pparγ激動劑、α葡萄糖苷酶抑制劑、胰島素和glp-1類似物等。但是現有藥物存在療效不顯著,作用時間短的問題,并且有的藥物還存在如低血糖、體重增加、水腫、骨折、乳酸中毒和腸胃不適等副作用。
單磷酸腺苷激活蛋白激酶(ampk)在體內糖脂代謝中發揮主導作用,是反映細胞內能量狀態變化的能量計和代謝主開關。ampk激活能明顯改善2型糖尿病糖脂代謝紊亂,增強體內胰島素敏感性,已被確證成為治療2型糖尿病的新靶點。二甲雙胍、tzds類和黃連素等具有降糖降脂作用的臨床藥物被證明在細胞水平能激活ampk,為ampk作為抗糖尿病藥物新靶點奠定了基礎。
本發明人設計合成一類脂肪酸類化合物,此類化合物具有在細胞水平上顯著激活ampk的能力和具有抑制小鼠原代肝細胞葡萄糖輸出能力。其中部分化合物顯示了非常好的ampk激活活性,細胞水平可以顯著濃度梯度依賴刺激ampk、acc磷酸化;多數化合物具有明顯抑制糖輸出的作用,此類化合物作為糖尿病治療藥物具有很好的開發潛力。
技術實現要素:
發明人設計合成了一類脂肪酸類化合物,發現該類化合物具有激活apmk活性和抑制小鼠原代肝細胞葡萄糖輸出能力。通式ⅰ-ⅳ所示化合物具有激活ampk活性,可劑量依賴性地顯著促進hepg2細胞的ampk及acc的磷酸化和具有明顯抑制糖輸出的作用。
因此,本發明的目的在于提供一類新型具有激活ampk活性的脂肪酸類化合物或其藥學上可接受的鹽。
本發明的另一目的在于提供該類具有激活ampk活性的脂肪酸類化合物或其藥學上可接受的鹽的制備方法。
本發明的還一目的在于提供具激活ampk活性的脂肪酸類化合物藥物組合物,該組合物包含治療有效量的本發明所述的脂肪酸類化合物或其藥學上可接受的鹽作為活性成分及藥學上可接受的輔料。
本發明的再一目的在于提供該類脂肪酸類化合物或其藥學上可接受的鹽及其藥物組合物在制備作為ampk激活劑的藥物中的用途,特別是在制備用于治療肥胖或糖尿病的藥物中的用途。
本發明所述脂肪酸類化合物為如下通式i所示的化合物:
其中,r1、r2、r3、r4、r5、r6各自獨立地為h、c1~c4烷基、c1~c4烯基、c1~c4炔基、c3~c7環烷基、c3~c7環烯基、苯基或芐基,優選地,r1、r2、r3、r4、r5、r6各自獨立地為h、c1~c3烷基、c3~c6環烷基、c3~c6環烯基或苯基,
或者,r1和r2與和它們相鄰的碳原子一起形成c3~c7環烷基、或與和它們相鄰的碳原子一起形成c3~c7環烯基;
或r3和r4與和它們相鄰的碳原子一起形成c3~c7環烷基或與和它們相鄰的碳原子一起形成c3~c7環烯基;
或r5和r6與和它們相鄰的碳原子一起形成c3~c7環烷基或與和它們相鄰的碳原子一起形成c3~c7環烯基,
m、n各自獨立地為1、2、3、4、5或6,優選為2、3、4或5,
y1和y2各自獨立地為-oh、-cooh、-coor7、-so3h、-so2nhr、-po(oh)oet、-conhcn、
雙鍵為順式或反式。
在本發明中作如下定義,所述的烷基包括直鏈或支鏈的烷基,所述的鏈烯基包括直鏈或支鏈的鏈烯基。
在優選實施方式中,本發明所述脂肪酸類化合物為如下通式ii所示的化合物:
其中,r1、r2、r3、r4、r5、r6各自獨立地為h、c1~c4烷基、c1~c4烯基、c1~c4炔基、c3~c7環烷基、c3~c7環烯基、苯基或芐基;優選地,r1、r2、r3、r4、r5、r6各自獨立地為h、c1~c3烷基、c3~c6環烷基、c3~c6環烯基或苯基;更優選地,r1和r2為h,r3、r4、r5、r6各自獨立地為h、c1~c3烷基或苯基,
或者,r5和r6與其相鄰的碳原子一起形成c3~c7環烷基或與其相鄰的碳原子一起形成c3~c7環烯基,
或r3和r4與其相鄰的碳原子一起形成c3~c7環烷基或與其相鄰的碳原子一起形成c3~c7環烯基;
m、n各自獨立的為1、2、3、4、5或6,優選為2、3、4或5;
雙鍵是順式或者反式。
在另一優選實施方式中,本發明所述脂肪酸類化合物為如下通式iii所示的化合物:
其中,r3、r4、r5、r6各自獨立地為h、c1~c4烷基、c1~c4烯基、c1~c4炔基、c3~c7環烷基、c3~c7環烯基、苯基或芐基,優選地,r3、r4、r5、r6各自獨立地為h、c1~c3烷基、c3~c6環烷基、c3~c6環烯基或苯基,
或者r5和r6與其相鄰的c一起形成c3~c7環烷基或與和它們相鄰的碳原子一起形成c3~c7環烯基,
或r3和r4與其相鄰的c一起形成c3~c7環烷基或與和它們相鄰的碳原子一起形成c3~c7環烯基,
m、n各自獨立地為1、2、3、4、5或6,優選為2、3、4或5。
在另一優選實施方式中,本發明所述脂肪酸類化合物為如下通式iv所示的化合物:
其中,r3、r4、r5、r6各自獨立地為h、c1~c4烷基、c1~c4烯基、c1~c4炔基、c3~c7環烷基、c3~c7環烯基、苯基或芐基,優選地,r3、r4、r5、r6各自獨立地為h、c1~c3烷基、c3~c6環烷基、c3~c6環烯基或苯基;
或者r5和r6與其相鄰的碳原子一起形成c3~c7環烷基或與其相鄰的碳原子一起形成c3~c7環烯基,
或r3和r4與其相鄰的碳原子一起形成c3~c7環烷基或與其相鄰的碳原子一起形成c3~c7環烯基。
m、n各自獨立地為1、2、3、4、5或6,優選為2、3、4或5,
p為1、2或3,優選為2。
本發明所述的具有激活ampk活性的脂肪酸類化合物最優選為如下化合物:
本發明所述通式ⅰ-ⅳ所示化合物或其藥學上可接受的鹽可以通過常規有機化學反應制備。通式ⅰ-ⅲ所示的化合物可選擇如下路線來實現。
路線一:
(1)取代的乙酸乙酯a1與二溴烷烴a2在二異丙基氨基鋰(lda)和有機溶劑存在的條件下縮合,得到中間體a3;同樣的條件下,將a1替換為
(2)等當量的a3和a4與對甲基苯磺酰甲基異腈在nah、四丁基碘化銨(tbai)、二甲基亞砜(dmso)存在的條件下反應,得到化合物a5;
(3)a5用濃鹽酸脫去氫氰酸和對甲苯磺酸反應,得到a6;反應可以在有機溶劑下進行,有機溶劑優選為二氯甲烷(dcm);
(4)a6在堿性條件下脫去酯基,經過稀鹽酸酸化得到a7;所述堿性條件優選為koh;所述脫去酯基的反應優選在有機溶劑如乙醇的存在下進行;
(5)a7再用硼氫化鈉還原酸化得到a8;
(6)a8在催化劑對甲苯磺酸的甲苯溶液回流,得到化合物a9或a10。
路線二:
(1)乙炔鈉a11與溴代物a12在dmf中反應,得到中間體a13;
(2)a13炔基化合物與溴代物a2在正丁基鋰和六甲基磷酰三胺(hmpa)條件下縮合,得到中間體a14;
(3)中間體a14用溴化鋰在有機溶劑中回流,轉化為中間體a15;所述有機溶劑優選為丙酮;
(4)a15與a16或者a17在二異丙基氨基鋰(lda)條件下縮合,得到中間體a18;
(5)a18加氫得到中間體a19;
(6)a19在堿性條件下脫去酯基,經稀鹽酸酸化得到a20;所述堿性條件優選為koh;所述脫去酯基的反應優選在有機溶劑如乙醇的存在下進行。
路線二的中間體a15還可以采用路線三制備。
路線三:
(1)1-炔基-醇化合物a21在吡啶對甲苯磺酸鹽(ppts)和2-四氫吡喃(thp)存在下,得到中間體a23;1-羥基-溴化物a22在吡啶對甲苯磺酸鹽和2-四氫吡喃(thp)存在下,得到中間體a24;
(2)a23和a24在正丁基鋰和hmpa條件下縮合,得到中間體a25;
(3)a25在對甲苯磺酸(tsoh)和有機溶劑存在下,脫除保護基得到中間體a26;所述有機溶劑優選為甲醇;
(4)a26用四溴化碳和三苯基磷(pph3)轉化為中間體a15。
路線一-三中:反應式中r3、r4、r5、r6、n和m的定義與前述相同。
通式ⅱ所示的化合物由如下路線四來實現,
路線四:
(1)中間體a18與頻哪醇聯硼酸酯在三苯基磷鉑催化下生成中間體a26;
(2)a26與碘化物r1i或r2i偶聯,得到中間體a27;
(3)a27在堿性條件下脫去酯基,經稀鹽酸酸化得到通式ⅱ化合物。
其中,r3、r4、r5、r6、m和n的定義與前述相同。
通式ⅲ所示的化合物由如下路線五來制備。
路線五:
中間體a20加碘甲烷和乙基鋅試劑,得到中間體a28,a28在堿性條件下脫去酯基,經稀鹽酸酸化得到通式ⅲ化合物。
其中,r3、r4、r5、r6、m和n的定義與前述相同。
通式ⅳ所示的化合物由如下路線六來實現。
路線六:
(1)化合物a29首先轉化為溴代烯烴中間體a30;
(2)中間體a30在碘化鉀和碘化亞銅條件下轉化為中間體a31;
(3)a31再與硼試劑a32和a33偶聯,得到中間體a34;
(4)a34在堿性條件下脫去酯基,經稀鹽酸酸化得到通式ⅳ化合物;所述堿性條件優選為koh;所述脫去酯基的反應優選在有機溶劑如乙醇的存在下進行。
其中,r3、r4、r5、r6、m、n和p的定義與前述相同。
本發明提供的激活ampk的活性化合物,從細胞水平和整體動物水平改善糖脂代謝,可用于制備治療肥胖或糖尿病的藥物。
本發明提供一類治療肥胖或糖尿病等用途藥物組合物,該組合物包含治療有效量的通式i所示的化合物或其藥學上可接受的鹽中的一種或多種作為活性成分,以及藥學上可接受的輔料,例如分散劑、賦形劑、崩解劑、抗氧化劑、甜味劑、包衣劑等。該組合物可以按照藥學領域常規的制備方法來制備,并可制成各種常規的劑型,例如片劑、包衣片劑、膠囊、粉劑等。
附圖說明
圖1為化合物促進人源hepg2肝癌細胞ampk磷酸化的柱狀圖;
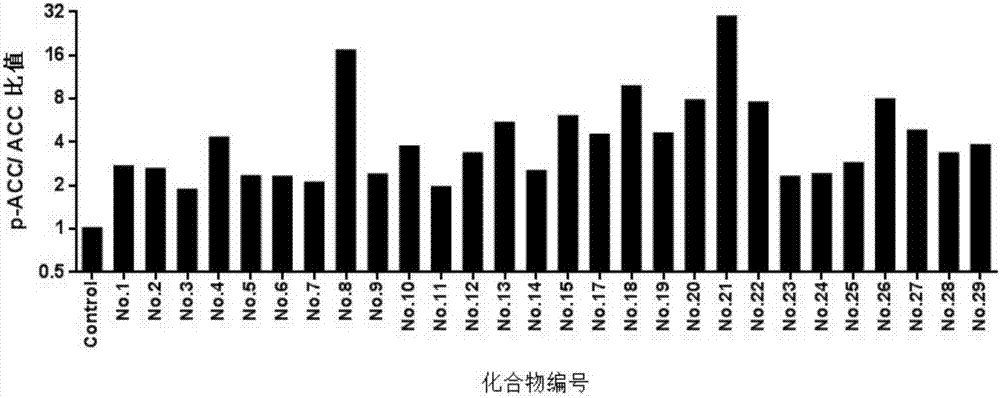
圖2為化合物促進人源hepg2肝癌細胞acc磷酸化的柱狀圖;
圖3為化合物抑制小鼠原代肝細胞糖輸出的柱狀圖。
具體實施方式
下面結合具體實施例對本發明作進一步闡述,但本發明不局限于這些實施例。
化合物制備實施例
在下述制備實施例中,nmr用varian生產的mercury-vx300m儀器測定,nmr定標:δh7.26ppm(cdcl3);質譜用agilent1200quadrupolelc/ms液質聯用儀;試劑主要由上海化學試劑公司提供;tlc薄層層析硅膠板由山東煙臺會友硅膠開發有限公司生產,型號hsgf254;化合物純化使用的正相柱層析硅膠為山東青島海洋化工廠分廠生產,型號zcx-11,200-300目。
制備實施例11-(5-(6-氧-7-雜螺[4.8]十三烷-8-基)戊基)環戊烷碳酸(化合物5)
(1)1-(5-溴戊基)環戊烷碳酸甲酯(化合物b3)
氬氣保護下,將1.28g(10mmol)環戊基甲酸乙酯與2.49g(11mmol)1,5-二溴戊烷加入反應瓶中,50ml四氫呋喃溶解,冷卻至0℃,緩慢滴加lda(0.467mmol)約1小時,加完室溫反應過夜,次日tlc監測反應完全。加入飽和氯化銨溶液猝滅反應,乙酸乙酯萃取3次,有機相用飽和食鹽水洗,無水na2so4干燥,減壓旋除溶劑,柱層析分離,pe:ea(體積比)=100:1,得化合物b3,淡黃色液體1.7g,收率62%。1hnmr(300mhz,cdcl3)δ3.63(s,3h),3.36(t,2h),2.05-2.09(m,2h),1.78-1.83(m,2h),1.54-1.60(m,6h),1.35-1.44(m,4h),1.18-1.21ppm(m,2h).
(2)6-羰基-十一烷烴-1,11-二環戊烷甲酸甲酯(化合物b4)
氬氣保護下,將化合物b3(1.7g,6.2mmol)、對甲基苯磺酰甲基異腈(0.64g,3.1mmol)和四丁基碘化銨(0.23g,0.62mmol)溶于10mldmso中,冷卻至0℃分批加入氫化鈉(60%)(0.3g,7.4mmol),加完室溫反應過夜,次日tlc監測反應完全。加入水猝滅反應,二氯甲烷萃取3次,有機相用飽和食鹽水洗,無水na2so4干燥,減壓旋蒸除去溶劑,得一粗產物。直接下步反應。粗產物加入20mldcm,加入4ml濃鹽酸,室溫攪拌1小時,加水稀釋,二氯甲烷萃取3次,有機相用飽和食鹽水洗,無水na2so4干燥,減壓旋蒸除去溶劑,柱層析分離,pe:ea(體積比)=30:1,得化合物b4,淡黃色液體850mg,收率65%。1hnmr(300mhz,cdcl3)δ3.63(s,6h),2.33(t,4h),2.04-2.09(m,4h),1.52-1.58(m,16h),1.40-1.52(m,4h),1.17-1.23ppm(m,8h).
(3)6-羰基-十一烷烴-1,11-二環戊烷甲酸(化合物b5)
將化合物b4(422mg,1mmol)溶于10ml乙醇中,加入koh(500mg,8.9mmol)的水溶液(2ml),加熱回流3小時,tlc監測反應完全。冷卻反應液,旋蒸除去大部分乙醇,加水稀釋,dcm萃取除去少量雜質。加入2nhcl調至酸性,二氯甲烷萃取3次,有機相用飽和食鹽水洗,無水na2so4干燥,減壓旋除溶劑,得一粗產物300mg。直接下步反應。
(4)6-羥基-十一烷烴-1,11二環戊烷甲酸(化合物b5)
化合物b5(366mg,1mmol)溶解于10ml甲醇中,冷卻至至0℃,加入硼氫化鈉(76mg,2mmol),攪拌0.5h,再分批加入304mg(8mmol)硼氫化鈉,加完室溫攪拌1h,tlc監測反應完全。加水稀釋,加入2nhcl調至酸性,二氯甲烷萃取3次,有機相用飽和食鹽水洗,無水na2so4干燥,減壓旋除溶劑,柱層析分離,dcm:meoh(體積比)=50:1至20:1,得化合物b6,320mg,收率87%.
1hnmr(300mhz,cdcl3)δ3.56-3.58(m,1h),2.10-2.15(m,4h),1.62-1.68(m,16h),1.45-1.52(m,8h),1.17-1.23ppm(m,8h).
(5)1-(5-(6-氧-7-雜螺[4.8]十三烷-8-基)戊基)環戊烷碳酸(化合物5)
化合物b6(40mg,0.22mmol)在80ml甲苯,加入30mg對甲苯磺酸,回流2天,tlc監測反應完全。減壓旋除溶劑,柱層析分離,dcm:meoh(體積比)=100:1得化合物b7,30mg,收率85%.
1hnmr(300mhz,cdcl3)δ5.33-5.34(m,1h),2.15-2.17(m,4h),1.97-2.09(m,4h),1.62-1.64(m,12h),1.40-1.49(m,6h),1.26-1.29ppm(m,10h).
同樣的方法,得到如下化合物:
用異丙酸乙酯替換反應物b1得到化合物1:
1hnmr(300mhz,cdcl3)δ5.34-5.36(m,2h),1.95-1.98(m,4h),1.50-1.55(m,4h),1.21-1.42(m,10h),1.18ppm(s,12h).
用異丙酸乙酯替換反應物b1,1,6-二溴己烷替換反應物b2,得到化合物2:1hnmr(300mhz,cdcl3)δ5.34-5.35(m,2h),1.96-1.98(m,4h),1.50-1.52(m,4h),1.21-1.42(m,14h),1.17ppm(s,12h).
用異丙酸乙酯替換反應物b1,1,4-二溴丁烷替換反應物b2,得到化合物3:1hnmr(300mhz,cdcl3)δ5.29-5.38(m,2h),1.92-1.95(m,4h),1.43-1.52(m,4h),1.21-1.42(m,6h),1.13ppm(s,12h).
用異丙酸乙酯替換反應物b1,1,7-二溴庚烷替換反應物b2,得到化合物4:1hnmr(300mhz,cdcl3)δ5.34-5.35(m,2h),1.94-1.95(m,4h),1.48-1.52(m,6h),1.21-1.42(m,16h),1.17ppm(s,12h).
用1,6-二溴己烷替換反應物b2,得到化合物6:1hnmr(300mhz,cdcl3)δ5.33-5.34(m,2h),2.12-2.16(m,4h),1.94-2.02(m,4h),1.62-1.64(m,12h),1.40-1.49(m,6h),1.26-1.29ppm(m,12h).
用1,4-二溴丁烷替換反應物b2,得到化合物7:1hnmr(300mhz,cdcl3)δ5.33-5.42(m,2h),2.14-2.18(m,4h),1.96-1.99(m,4h),1.62-1.64(m,10h),1.40-1.49(m,6h),1.26-1.29ppm(m,6h).
用1,7-二溴庚烷替換反應物b2,得到化合物8:1hnmr(300mhz,cdcl3)δ5.33-5.36(m,2h),2.12-2.16(m,4h),1.96-1.99(m,4h),1.62-1.64(m,12h),1.40-1.49(m,6h),1.26-1.29ppm(m,16h).
用2-苯基-丙酸乙酯替換反應物b1得到化合物9:1hnmr(300mhz,cdcl3)δ7.26-7.36(m,10h),5.34-5.36(m,2h),2.02-2.22(m,4h),1.86-2.03(m,4h),1.54(s,6h),1.22-1.42(m,10h).
用環己基甲酸乙酯替換反應物b1得到化合物10:1hnmr(300mhz,cdcl3)δ5.32-5.34(m,2h),2.06-2.09(m,4h),1.94-1.98(m,4h),1.45-1.65(m,10h),1.15-1.45(m,18h),0.88-0.96ppm(m,2h).
用環丁基甲酸乙酯替換反應物b1得到化合物10:1hnmr(300mhz,cdcl3)δ5.32-5.34(m,2h),2.43-2.45(m,4h),1.96-1.99(m,4h),1.81-1.92(m,10h),1.76-1.1.82(m,4h),1.25-1.36ppm(m,8h).
化合物12
1hnmr(300mhz,cdcl3)δ5.34-5.36(m,2h),2.36-2.43(t,4h),1.96-2.05(m,4h),1.53-1.65(m,4h),1.22-1.40(m,10h).
用環丁基甲酸乙酯替換反應物b1,1,6-二溴己烷替換反應物b2,得到化合物13:1hnmr(300mhz,cdcl3)δ5.33-5.35(m,2h),2.43-2.45(m,4h),1.96-1.99(m,4h),1.81-1.92(m,10h),1.75-1.1.82(m,4h),1.11-1.28ppm(m,12h).
用2-甲基丁酸乙酯替換反應物b1,得到化合物14:1hnmr(300mhz,cdcl3)δ5.33-5.35(m,2h),1.96-2.02(m,4h),1.60-1.78(m,4h),1.38-1.58(m,4h),1.22-1.40(m,8h),1.11(s,6h),0.85ppm(t,6h).
用環戊烯甲酸乙酯替換反應物b1,1,6-二溴己烷替換反應物b2,得到化合物15:
1hnmr(300mhz,cdcl3)δ5.60(s,4h),5.32-5.34(m,2h),2.88-2.94(d,j=16hz,4h),2.27-2.32(d,j=16hz,4h),1.94-1.96(m,4h),1.67-1.72(m,4h),1.19-1.38ppm(m,18h).
用環戊烯甲酸乙酯替換反應物b1,得到化合物16:
1hnmr(300mhz,cdcl3)δ5.60(s,4h),5.32-5.34(m,2h),2.88-2.93(d,j=16hz,4h),2.27-2.32(d,j=16hz,4h),1.96-1.99(m,4h),1.67-1.73(m,4h),1.25-1.38ppm(m,14h).
用2-苯基-丙酸乙酯替換反應物b1,1,6-二溴己烷替換反應物b2,得到化合物171hnmr(300mhz,cdcl3)δ7.26-7.40(m,10h),5.34-5.36(m,2h),2.02-2.22(m,4h),1.86-2.03(m,4h),1.55(s,6h),1.18-1.42(m,14h).
用2-苯基-丙酸乙酯替換反應物b1,1,7-二溴庚替換反應物b2得到化合物18:1hnmr(300mhz,cdcl3)δ7.23-7.38(m,10h),5.34-5.36(m,2h),2.02-2.22(m,4h),1.86-2.03(m,4h),1.55(s,6h),1.18-1.42(m,18h).
用2-甲基丁酸乙酯替換反應物b1,1,6-二溴己烷替換反應物b2,得到化合物19:1hnmr(300mhz,cdcl3)δ5.33-5.35(m,2h),1.96-2.02(m,4h),1.58-1.78(m,4h),1.38-1.58(m,4h),1.22-1.40(m,14h),1.11(s,6h),0.86ppm(t,6h).
用2-甲基丁酸乙酯替換反應物b1,1,7-二溴庚烷替換反應物b2,得到化合物20:1hnmr(300mhz,cdcl3)δ5.33-5.35(m,2h),1.96-2.02(m,4h),1.58-1.70(m,4h),1.38-1.50(m,4h),1.22-1.40(m,18h),1.11(s,6h),0.86ppm(t,6h).
用環己基甲酸乙酯替換反應物b1,1,6-二溴己烷替換反應物b2,得到化合物21:1hnmr(300mhz,cdcl3)δ5.32-5.34(m,2h),2.04-2.05(m,4h),1.94-1.98(m,4h),1.45-1.65(m,12h),1.15-1.45(m,22h).
用環己基甲酸乙酯替換反應物b1,1,7-二溴庚烷替換反應物b2,得到化合物22:
1hnmr(300mhz,cdcl3)δ5.32-5.34(m,2h),2.04-2.08(m,4h),1.96-2.01(m,4h),1.45-1.65(m,12h),1.23-1.45(m,26h).
制備實施例2(z)-2,2,14,14-四甲基--7-烯基-十五碳二酸的制備(化合物24)
(1)2-(己-5-炔基氧基)四氫-2h-吡喃(b8)制備
化合物5-炔-1-己醇(9g,91mmol)和對甲苯磺酸吡啶鹽(25.1g,100mmol)溶解于100mldcm中,加入四氫吡喃(12.5ml,126mmol),室溫攪拌過夜。tlc監測反應完全。除去大部分dcm,約剩余30ml,加入約100ml乙醚攪拌0.5h,濾除固體,固體加乙醚洗一次,濃縮濾液,過柱,pe/ea(體積比)=50/1,得產物b8油狀物18g,收率>100%.
(2)2-(5-溴戊基)四氫-2h-吡喃(b9)制備
化合物5-溴-1-戊醇(15.1g,91mmol)和對甲苯磺酸吡啶鹽(25.1g,100mmol)溶解于100mldcm中,加入四氫吡喃(12.5ml,126mmol),室溫攪拌過夜。tlc監測反應完全。除去大部分dcm,約剩余30ml,加入約100ml乙醚攪拌0.5h,濾除固體,固體加乙醚洗一次,濃縮濾液,過柱,pe/ea(體積比)=50/1,得化合物b9,18g,收率79%。
(3)2,2'-(十一碳-5-炔-1,11-二雙(氧基))雙(四氫-2h-吡喃)(b10)制備
化合物b8(5.4g,30mmol)溶于40ml無水thf中,冷卻至-40℃,滴加正丁基鋰(19ml,30.4mmol)(約0.5h加完),再加入10mlhmpa,此溫度攪拌1h后,加入化合物b9(7.78g,31mmol)的10mlthf溶液。此溫度1h后,緩慢升至室溫攪拌過夜。加入飽和氯化銨溶液猝滅反應,分層,水相再用乙酸乙酯萃取1次,合并有機相,加水和飽和食鹽水洗滌。干燥濃縮過柱,pe/ea(體積比)=20/1→10/1→5/1,得產物b10,8.5g,收率80%。
(4)十一碳-5-炔-1,11-二醇(b11)制備
化合物b10(3g,8.5mmol)溶解于100ml甲醇和10ml中,加入對甲苯磺酸(300mg),室溫攪拌過夜。tlc監測反應完全。濃縮除去大部分的甲醇,加水稀釋,用乙醚萃取2次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮過柱,pe/ea(體積比)=2/1→1/1→1/2,得產物b11,1.1g,收率70%。
(5)1,11-二溴-十一碳-5-炔(b12)制備
化合物b11(1.1g,5.9mmol)和四溴化碳(5.95g,17.9mmol)溶于30ml無水dcm中,冷卻至0℃,滴加三苯基膦(4.7g,17.9mmol)的10mldcm溶液,室溫攪拌1h后,反應完全。濃縮至約15ml,加入50ml乙醚攪拌0.5h,濾除固體,濾液濃縮,過柱pe/ea=30/1,得溴代物b12,1.84g,收率100%。
(6)二乙基-2,2,14,14-四甲基-7-炔基-十五碳二酸(b13)制備
化合物b12(1.84g,5.9mmol)和異丁酸乙酯(2.73g,23.6mmol)溶于50ml無水thf中,冷卻至0℃,滴加lda(15.7ml,23.6mmol),(約0.5h加完),保持此溫度1h后,升至室溫攪拌過夜。tlc監測反應完全。加飽和氯化銨溶液猝滅反應,分層,水相加ea萃取2次,合并有機相,加水,飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1→20/1,得產物b13,1.8g,收率80%。
(7)二乙基-(z)-2,2,14,14-四甲基-7-烯基-十五碳二酸酯(b14)制備
一個大氣壓h2下,醋酸鎳(749mg,3mmol)懸浮于20ml無水乙醇,快速加入硼氫化鈉(114mg,3mmol),氫氣再置換2次,溶液變黑。室溫攪拌15分鐘后,加入乙二胺(0.45ml,6mmol)后加入化合物b13(1.8g,4.7mmol)的10ml無水乙醇溶液。室溫攪拌2h后,tlc監測反應完全。加入50ml乙醚稀釋,墊硅藻土過濾,濾液濃縮過柱,過柱pe/ea(體積比)=50/1,得產物b14,1.7g,收率95%。
(8)(z)-2,2,14,14-四甲基--7-烯基-十五碳二酸的制備(化合物24)制備
化合物b14(1.7g,4.45mmol)溶于50ml乙醇,加入koh(2g,35mmol)的10ml水溶液,加熱回流6h,tlc監測反應完全。冷卻后旋轉蒸發除去大部分乙醇,加水(40ml)稀釋,加乙醚萃取2次除雜質。水層加2nhcl調至酸性,加dcm萃取4次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱dcm/meoh(體積比)=100/1,得1.1g。
化合物24
1hnmr(600mhz,dmso)δ5.31-5.32(m,2h),1.96-1.98(m,4h),1.40-1.44(m,4h),1.22-1.30(m,4h),1.18-1.22(m,6h),1.06ppm(s,12h).
化合物23
1hnmr(300mhz,cd3cl)δ5.31-5.32(m,2h),1.96-1.98(m,4h),1.40-1.44(m,4h),1.18-1.22(m,8h),1.06ppm(s,12h)
化合物25
1hnmr(300mhz,cd3cl)δ5.30-5.32(m,2h),1.96-1.98(m,4h),1.40-1.45(m,4h),1.18-1.22(m,12h),1.06ppm(s,12h)
制備實施例3(e)-2,2,14,14-四甲基--7-烯基-十五碳二酸的制備(化合物30)
(1)(e)-2-(6-碘己烯-5-烯基氧基)四氫-2h-吡喃(b16)制備
化合物b8(460mg,2.5mmol)溶于20ml無水thf中,加入schwanz試劑(氯化雙環戊二烯基氫鋯)(1.63g,6.3mmol),室溫攪拌2h,冷卻至0℃,加入飽和i2/dcm(30ml)溶液,0℃攪拌10min后,加入飽和na2s2o3溶液20ml淬滅反應,dcm萃取2次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1→20/1,得產物b16,450mg,收率58%.
(2)(e)-乙基-2,2-二甲基-13-(四氫-2h-吡喃-2-氧)十三碳-8-烯酸甲酯(b18)制備
氬氣保護下,化合物b15(60mg,0.35mmol)中加入2ml0.5m的9-bbnthf溶液,室溫攪拌2小時后,加入化合物b16的2mldmf溶液,再依次加入cs2co3(156mg),asph3(24mg)和1ml水,氬氣置換5min后,加入pd(dppf)cl2(20mg),室溫攪拌過夜。加入30ml乙醚稀釋,加水和飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1→20/1,得產物b18,54mg,收率90%.
(3)(e)-乙基-13-羥基-2,2-二甲基十三碳-8-烯酸甲酯(b19制備)
化合物b19(350mg,0.95mmol)溶解于10ml甲醇和11ml中,加入對甲苯磺酸(25mg),室溫攪拌過夜。tlc監測反應完全。濃縮除去大部分的甲醇,加水稀釋,用乙醚萃取2次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮過柱,pe/ea(體積比)=4/1,得產物b19,200g,收率78%。
(4)(e)-乙基-13-溴-2,2-二甲基十三碳-8-烯酸甲酯(b20)制備
化合物b19(200mg,0.7mmol)和四溴化碳(302mg,0.91mmol)溶于10ml無水dcm中,冷卻至0℃,滴加三苯基膦(256g,0.98mmol)的2mldcm溶液,室溫攪拌1h后,反應完全。濃縮過柱,pe/ea(體積比)=20/1,得溴代物b20,206mg,收率87%。
(5)二乙基-(e)-2,2,14,14-四甲基-7-烯基-十五碳二酸酯(b21)制備
化合物b20(200mg,0.57mmol)和異丁酸乙酯(100mg,0.86mmol)溶于10ml無水thf中,冷卻至0℃,滴加lda(0.6ml,0.9mmol),(約0.5h加完),保持此溫度1h后,升至室溫攪拌過夜。tlc監測反應完全。加飽和氯化銨溶液猝滅反應,分層,水相加ea萃取2次,合并有機相,加水,飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1,得產物b21,177mg,收率80%.
(6)(e)-2,2,14,14-四甲基-7-烯基-十五碳二酸的制備(化合物30)制備
化合物b21(107mg,0.28mmol)溶于10ml乙醇,加入koh(200mg,3.5mmol)的2ml水溶液,加熱回流6h,tlc監測反應完全。冷卻后旋轉蒸發除去大部分乙醇,加水(10ml)稀釋,加乙醚萃取2次除雜質。水層加2nhcl調至酸性,加dcm萃取4次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱dcm/meoh(體積比)=100/1,得70mg。
化合物30
1hnmr(600mhz,dmso)δ5.35-5.36(m,2h),1.92-1.94(m,4h),1.40-1.43(m,4h),1.27-1.29(m,4h),1.17-1.19(m,6h),1.18ppm(s,12h).
制備實施例4(z)-2,2,7,8,14,14-六甲基十五碳-7-丁烯二酸(化合物28)
(1)(z)-二乙基-2,2,14,14-四甲基-7,8-雙(4,4,5,5-四甲基-1,3,2-二氧硼雜環戊烷-2-基)十五碳-7-烯二酸(b22)制備
頻哪醇聯硼酸酯(483mg,1.9mmol)和四三苯基磷鉑(70mg)置于25ml圓底燒瓶中,氬氣置換3次后,滴加b13(660mg,1.74mmol)的10mldmf溶液中,80℃反應過夜。加水稀釋,乙醚萃取,合并有機相,加水,飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=10/1,得產物b22,1g,收率90%.
1hnmr(300mhz,cdcl3)δ4.08-4.11(m,4h),2.13-2.16(m,4h),1.40-1.43(m,4h),1.25-1.33(m,10h),1.27(m,24h),1.17-1.19(t,6h),1.13ppm(s,12h)
(2)(z)-二乙基-2,2,7,8,14,14-六甲基十五碳-7-丁烯二酸酯(b23)制備
化合物b22(150mg,0.23mmol),碘甲烷(0.1ml)和三苯基膦(10mg)溶于1ml二氧六環中,氬氣置換3次后,加入醋酸鈀(20mg),koh(40mg)的0.2ml水溶液,90℃反應12h,加水和乙酸乙酯稀釋,乙酸乙酯萃取3次,合并有機相,加水,飽和食鹽水洗滌,干燥濃縮。過柱pe/ea=50/1,得產物b23,30mg,收率31%.
(3)(z)-2,2,7,8,14,14-六甲基十五碳-7-丁烯二酸(化合物28)制備
化合物b23(30mg,0.07mmol)溶于5ml乙醇,加入koh(50mg,0.89mmol)的1ml水溶液,加熱回流6h,tlc監測反應完全。冷卻后旋轉蒸發除去大部分乙醇,加水(4ml)稀釋,加乙醚萃取2次除雜質。水層加2nhcl調至酸性,加dcm萃取4次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱dcm/meoh(體積比)=100/1,得15mg.
化合物28
1hnmr(300mhz,cdcl3)δ1.94-1.96(m,4h),1.58(s,6h),1.48-1.50(m,4h),1.25-1.33(m,12h),1.13ppm(s,12h)
制備實施例57-(2-(5-羧基-5-甲基己基)環丙基)-2,2-二甲基庚酸的制備(化合物29)
(1)乙基-7-(2-(6-乙氧基-5,5-二甲基-6-氧代己基)環丙基)-2,2-二甲基庚酸酯(b24)制備
氮氣保護下,20ml反應瓶中加入干燥的dcm1ml,加入二乙基鋅試劑(1ml,1mmol),冷卻至-40℃,5min后,滴加二碘甲烷(0.54g,2mmol)的0.5mldcm溶液,-40℃反應1h后,加入三氯乙酸(16mg,0.1mmol)和dme(45mg,0.5mmol),加完升至-15℃反應1h,加入b14(193mg,0.5mmol)的0.5mldcm溶液,加完,室溫攪拌過夜。加飽和氯化銨溶液淬滅反應,乙酸乙酯萃取3次,合并有機相,加水,飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1,得產物b24,120mg,收率60%.
(2)7-(2-(5-羧基-5-甲基己基)環丙基)-2,2-二甲基庚酸(化合物29)制備
化合物b24(100mg,0.25mmol)溶于5ml乙醇,加入koh(10mg,0.18mmol)的1ml水溶液,加熱回流6h,tlc監測反應完全。冷卻后旋轉蒸發除去大部分乙醇,加水(4ml)稀釋,加乙醚萃取2次除雜質。水層加2nhcl調至酸性,加dcm萃取4次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱dcm/meoh(體積比)=100/1,得65mg.
化合物29
1hnmr(300mhz,cdcl3)δ1.1.45-1.52(m,4h),1.21-1.43(m,16h),1.18(s,12h),0.65-0.70ppm(m,2h)
制備實施例6(z)-2,2,17,17-四甲基-十八碳-9-丁烯二酸(化合物26)
(1)8-氯-辛-1-炔(b26)制備
化合物b25(1.6g,8mmol)于8ml無水dmf中,冷卻至0℃,加入乙炔鈉的二甲苯溶液(18%溶液,12mmol),加完后,室溫攪拌過夜。加乙醚和水分層,有機相加水,飽和食鹽水洗滌,干燥濃縮,減壓蒸餾,得1.2gb26.
(2)1,14-二氯-十四碳-7-炔(b27)制備
化合物b26(1.2g,8mmol)溶于15ml無水thf中,冷卻至-40℃,滴加正丁基鋰(5.2ml,1.6m,8.3mmol),加完后加入hmpa(4ml),攪拌1h后,加入b25(1.6g,8.2mmol)的5mlthf溶液,室溫攪拌過夜。加入飽和氯化銨溶液淬滅反應,分層,水相再用乙酸乙酯萃取1次,合并有機相,加水和飽和食鹽水洗滌。干燥濃縮過柱,pe/ea=100/1,得產物b27,1.6g,收率80%.
(3)1,14-二溴-十四碳-7-炔(b28)制備
化合物b27(100mg,0.4mmol)和溴化鋰(688mg,8mmol)溶于3-戊酮(7ml),油浴120℃反應6h,減壓除去戊酮,加乙酸乙酯和水分層,加飽和食鹽水洗滌,干燥濃縮得120mg,直接下步反應.
(4)二乙基-2,2,17,17-四甲基-7-炔基-十八碳-二酸酯(b29)制備
化合物b28(120mg,0.34mmol)和異丁酸乙酯(238g,2mmol)溶于5ml無水thf中,冷卻至0℃,滴加lda(1.3ml,2mmol),(約0.5h加完),保持此溫度1h后,升至室溫攪拌過夜。tlc監測反應完全。加飽和氯化銨溶液猝滅反應,分層,水相加ea萃取2次,合并有機相,加水,飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1,得產物b29,80g,收率56%。
(5)(z)-二乙基-2,2,17,17-四甲基-7-烯基-十八碳-二酸酯(b30)制備
一個大氣壓h2下,醋酸鎳(25mg,0.1mmol)懸浮于1ml無水乙醇,快速加入硼氫化鈉(7mg,0.18mmol),氫氣再置換2次,溶液變黑。室溫攪拌15分鐘后,加入乙二胺(15l),后加入化合物b29(75mg,0.17mmol)的1ml無水乙醇溶液。室溫攪拌2h后,tlc監測反應完全。加入10ml乙醚稀釋,墊硅藻土過濾,濾液濃縮過柱,過柱pe/ea(體積比)=50/1,得產物b30,45mg,收率60%。
(6)(z)-2,2,17,17-四甲基-7-烯基-十八碳-二酸制備(化合物26)制備
化合物b30(40mg,0.01mmol)溶于5ml乙醇,加入koh(100mg,1.7mmol)的1ml水溶液,加熱回流6h,tlc監測反應完全。冷卻后旋轉蒸發除去大部分乙醇,加水(4ml)稀釋,加乙醚萃取2次除雜質。水層加2nhcl調至酸性,加dcm萃取4次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱dcm/meoh(體積比)=100/1,得20mg。
化合物26
1hnmr(300mhz,cd3cl)δ5.30-5.32(m,2h),1.98-2.02(m,4h),1.48-1.53(m,4h),1.18-1.22(m,16h),1.06ppm(s,12h)
制備實施例76,6'-(環己烯-1,2-二基)雙(2,2-二甲基己酸)(化合物27)
(1)1,2-二溴-1-氯環己烷
五氯化磷(4.37g,21mmol)懸浮于10ml氯仿中,冷卻至0℃,滴加化合物b31(1.96g,20mmol)的10ml氯仿溶液,加完后,室溫攪拌2h,再回流2h,倒入50g冰中,加入固體nahco3,分出有機相,dcm萃取1次,合并有機相,再分別用碳酸氫鈉溶液,飽和食鹽水洗滌。無水硫酸鈉干燥濃縮后,加入5mldcm,冷卻至-5℃,緩慢滴加液溴(2.08g,13mmol),滴完-5℃攪拌10min后,再分別加入10%硫代硫酸鈉溶液,飽和食鹽水洗滌,干燥濃縮,直接用石油醚過柱,得2.2化合物b32。
(2)1,2-二溴環己-1-烯
向回流的koh(1g,18mmol)甲醇(8ml)溶液中,加入化合物b32(2.2g)的8ml甲醇溶液,回流3h后,冷卻加入6nhcl調至酸性,加水,dcm萃取2次,合并有機相,加飽和食鹽水洗滌,干燥濃縮后,直接用石油醚過柱,得化合物b33,1.1g,收率兩步,23%。
(3)1,2-二碘環己-1-烯
化合物b33(700mg,2.9mmol)、碘化鉀(4.9g,30mmol)和碘化亞銅(2.85g,15mmol)置于8mlhmpa中,油浴120℃反應15h,冷卻至室溫,加入20ml2nhcl和20ml苯,分層,再加苯萃取一次,合并有機相,分別加10%硫代硫酸鈉溶液,飽和食鹽水洗滌,干燥濃縮,直接用石油醚過柱,得828mg化合物b34,收率85%。
(4)二乙基-6,6'-(環己烯-1,2-二基)雙(2,2-二甲基己)
氬氣保護下,化合物b1535(100mg,0.58mmol)中加入3.5ml0.5m的9-bbnthf溶液,室溫攪拌2小時后,加入化合物b34(50mg,0.15mmol)的2mldmf溶液,再依次加入cs2co(156mg),asph(24mg)和1ml水,氬氣鼓泡置換5min后,加入pd(dppf)cl2(30mg),室溫攪拌過夜。加入30ml乙醚稀釋,加水和飽和食鹽水洗滌,干燥濃縮。過柱pe/ea(體積比)=50/1,得產物b37,70mg,收率70%.
(5)6,6'-(環己烯-1,2-二基)雙(2,2-二甲基己酸(化合物27)制備
化合物b30(40mg,0.01mmol)溶于5ml乙醇,加入koh(100mg,1.7mmol)的1ml水溶液,加熱回流6h,tlc監測反應完全。冷卻后旋轉蒸發除去大部分乙醇,加水(4ml)稀釋,加乙醚萃取2次除雜質。水層加2nhcl調至酸性,加dcm萃取4次,合并有機相,加水和飽和食鹽水洗滌,干燥濃縮。過柱dcm/meoh(體積比)=100/1,得20mg.
化合物27
1hnmr(300mhz,cd3cl)δ1.89-1.91(m,8h),1.52-1.54(m,8h),1.19-1.22(m,8h),1.06ppm(s,12h)
實驗實施例
1.評價化合物對人源hepg2肝癌細胞中ampk信號通路的影響
(1)檢測目的:在人源肝癌細胞hepg2上檢測此類脂肪酸類化合物是否具有促進ampkα亞基t172位點磷酸化、ampk經典下游acc1s79位點磷酸化的小分子化合物。
(2)實驗原理:在細胞上平,小分子化合物激活ampk,可促進其α亞基t172位點及下游底物乙酰輔酶a羧化酶(acc)ser-79位的磷酸化。
(3)檢測試劑:totalampk、phospho-ampkα(t172)、phospho-acc(s79)、totalacc抗體均購自cellsignalingtechnology。
(4)實驗方法:
1)、人源肝癌細胞hepg2細胞以30萬每毫升的密度接種到24孔板,貼壁12h后,換為無血清高糖培養基饑餓12小時,分別加入本發明制備的化合物1~30處理(1‰dmso,50μm)6小時后(1‰dmso作為陰性對照),待收樣細胞用預冷的pbs洗一次,加入1×sds凝膠加樣緩沖液(50mmtris-hcl(ph6.8),100mmdtt,2%sds,10%甘油,0.1%溴酚藍)冰上裂解細胞10min(24孔板每孔加100μl)。
2)、樣品收入1.5mlep管中,100℃加熱10分鐘,然后12000g離心10分鐘后取上清液進行sds-page電泳,條件為濃縮膠:80伏,分離膠:120伏。
3)、電泳結束后,用biorad濕轉電轉儀系統將蛋白轉移至硝酸纖維素膜(恒壓100v,100min)。將目的條帶剪下后,將條帶置于封閉液(含5%bsa的tbst)中室溫封阻1小時。隨后將條帶置于一抗溶液(1:1000,溶于tbst)中4℃孵育過夜。
4)、第二天將目的條帶置于tbst中,室溫洗滌3×10分鐘。隨后將條帶置于二抗溶液(羊抗兔與羊抗鼠均為1:5000,溶于tbst)中室溫孵育1小時,接著tbst洗膜3×10分鐘后,用ecl試劑曝光。
(5)實驗結果:
如圖1、2所示,在我們獲得的此類脂肪酸類化合物中,多數化合物(50μm)能夠明顯促進ampk、acc的磷酸化。其中,化合物4,5,8,18,19,20等促進ampk磷酸化的作用超過陰性對照4倍;化合物4,8,18,20,21,22等促進acc磷酸化的作用超過陰性對照4倍。
2.考察化合物對c57bl/6j小鼠原代肝細胞葡萄糖輸出能力的影響。
(1)檢測目的:c57bl/6j小鼠原代肝細胞上檢測此類脂肪酸類化合物能否抑制肝細胞葡萄糖輸出的作用。
(2)實驗原理:小分子化合物作用于小鼠原代肝細胞,抑制糖異生,進而影響細胞葡萄糖輸出到培養基中,通過檢測不同化合物處理細胞后,培養基中的葡萄糖濃度,以評價化合物對糖異生的影響。
(3)檢測試劑:葡萄糖檢測試劑盒購自上海名典生物工程有限公司。
(4)實驗方法:
1)、分離c57bl/6j小鼠原代肝細胞以30萬每毫升的密度接種到24孔板,貼壁6h后,換為無血清低糖培養基饑餓12小時,分別加入本發明制備的化合物1~30處理(1‰dmso,50μm)6小時后(1‰dmso作為陰性對照),輕輕混勻培養基,每孔取50μl上清,葡萄糖氧化酶法檢測試劑盒測定葡萄糖濃度。
2)、棄培養基,用pbs洗細胞1次,甩干后,加入200μl250mm氫氧化鈉水溶液,裂解細胞30分鐘后,取裂解液,考馬斯亮藍法測定蛋白濃度。
3)、用蛋白濃度校正細胞葡萄糖輸出的量,并比較化合物調節肝細胞糖輸出的作用。
(5)實驗結果:
用我們獲得的化合物處理原代肝細胞后,未發現肝細胞的形態存在明顯的異常以及蛋白濃度的降低,說明化合物在次劑量和處理時間下,沒有明顯的毒性作用。如圖3所示此類脂肪酸類化合物,多數具有明顯抑制糖輸出的作用,其中,化合物1、2、6、13、15、20等的糖異生抑制作用超過陰性對照50%(如圖3)。
- 還沒有人留言評論。精彩留言會獲得點贊!