由四氟乙烯芯殼共聚物形成的致密制品及其制備方法與流程
本發明大體涉及四氟乙烯(tfe)芯殼共聚物,更具體而言,涉及由tfe芯殼共聚物形成的致密tfe共聚物膜。還提供包括致密tfe共聚物膜的致密制品以及使用tfe芯殼共聚物制造致密制品的方法。
背景技術:
阻隔膜用于各種各樣的技術,包括醫療和商業設備。例如,阻隔膜用于短期和長期可植入醫療裝置、密封件、墊圈、血液接觸表面、袋、容器和織物襯里。除了良好的阻隔性能之外,阻隔膜應具有良好的機械性能并且具有熱穩定性。已經構造了單層、多組分和多層阻隔膜作為阻隔材料,但是并未提供熱穩定性、強度和阻隔性能的組合。
聚四氟乙烯(ptfe)已被評估用作阻隔膜。使用ptfe的優點在于其可以在惡劣的化學環境和寬的溫度范圍內使用。例如,ptfe已在其他聚合物快速降解的惡劣化學環境中呈現了作為材料的實用性。ptfe還具有高達約260℃到低約-273℃的可用溫度范圍。然而,ptfe阻隔膜的機械性質差,例如抗張強度低,冷流阻抗或蠕變阻抗差,穿孔和磨蝕阻抗差,并且通常機械完整性差,使其不能用于許多材料工程應用中。
過去采用削磨工藝從較厚的預制品分割或刮削固體ptfe薄膜,制備了低孔隙率的ptfe制品。這些ptfe制品的特征是,強度低、冷流阻抗差以及膜長度方向和寬度方向上的承重能力差。ptfe細粉的例如柱塞式擠出等方法也已用于生產低孔隙率ptfe制品;然而,這中膜也具有相對較差的機械特性。還嘗試了通過在長度方向上拉伸來強化低孔隙率ptfe膜。然而,強度增益是最小的,并且由于該方法的特性而僅在一個方向上得到實現,從而大大降低了膜的實用性。
一種膨脹型聚四氟乙烯(eptfe)膜可根據授予戈爾公司(gore)的美國專利3,953,566所教導的方法進行制備。由該方法制備的多孔eptfe具有由原纖維互連的結點微結構,其強度比非膨脹型ptfe高,且保持非膨脹型ptfe的化學惰性和寬的可用溫度范圍。然而,這種膨脹型ptfe膜是多孔的,因而不能用作低表面張力流體的阻隔層,因為表面張力低于50達因-厘米的流體能通過膜的孔。
授予戈爾公司的美國專利3,953,566也教導了壓縮的eptfe制品,其中,采用平板印壓以加熱或不加熱的方式來致密化eptfe薄片。但是,壓機中發生冷流,導致非均勻部分,并且難以實現超過2.1g/cc的密度。因此,eptfe片作為阻隔膜的實用性受到限制。
用于形成tfe基阻隔膜的常規方法涉及膨脹、壓縮、以及隨后的變形或不變形的熱處理。另外,可不使用膨脹和壓縮工藝,通過在高于ptfe的結晶熔點的溫度下的干燥ptfe糊料的變形來生產高強度致密ptfe阻隔膜。盡管這種方法可產生高強度的致密氟聚合物膜,但是結晶度降低,滲透性未最優化,且該方法的規模受到限制。
因此,本領域中對于呈現出改進的阻隔性能(例如由耐甲烷滲透性證明)、改善的物理和機械性能(例如低蠕變)和高基質抗拉強度的tfe基阻隔膜存在需求,對制造tfe基阻隔膜的簡化方法也存在需求。
發明概述
本發明的一種實施方式涉及致密制品,該致密制品包括致密tfe共聚物膜,其具有在約50℃至約300℃之間的第一吸熱峰溫,在約320℃至約350℃之間的第二吸熱峰溫和在約350℃至約400℃之間的第三吸熱峰溫。基于所述致密tfe共聚物膜的總重量,tfe共聚物膜包括至少50%的tfe單體并含有至少3.0重量%的至少一種共聚單體的聚合單元。致密tfe共聚物膜的甲烷滲透率小于約20μg*微米/cm2/分鐘。致密制品具有小于約20%的空隙率。致密制品呈現改善的物理和機械性能,包括粘附性和阻隔性。致密tfe共聚物膜可直接從干燥預制件中產生,而不會像膨脹方法中通常所呈現的那樣增加干燥預制件的孔隙率。
本發明的第二實施方式涉及用于制備致密制品的方法,所述方法包括在不高于約335℃的溫度下拉伸tfe共聚物帶的干燥預制件,以形成致密tfe共聚物膜。該致密tfe共聚物膜具有在約50℃至約300℃之間的第一吸熱峰溫、在約320℃至約350℃之間的第二吸熱峰溫和在約350℃至約400℃之間的第三吸熱峰溫。可在不低于第一吸熱峰溫的溫度下拉伸干燥預制件。在另一實施方式中,在第一吸熱峰溫和第二吸熱峰溫之間的溫度下拉伸干燥預制件。在進一步的實施方式中,可在第二吸熱峰溫以內的溫度下拉伸干燥預制件。在進一步的實施方式中,可在約350℃至約400℃的溫度下拉伸干燥預制件。該方法可進一步包括形成干燥預制件,包括:潤滑tfe芯殼共聚物;在低于第二吸熱峰溫的溫度下對所述經潤滑的tfe共聚物施加壓力,以形成經潤滑的tfe共聚物帶;以及干燥所述經潤滑的tfe共聚物帶,以基本除去潤滑劑并形成干燥預制件。該致密tfe共聚物膜的甲烷滲透率小于約20μg*微米/cm2/分鐘。該致密制品具有小于約20%的空隙率。
附圖的簡要說明
采用附圖以幫助進一步理解本公開內容,其納入說明書中并構成說明書的一部分,附圖顯示了本公開內容的實施方式,與說明書一起用來解釋本公開內容的原理。
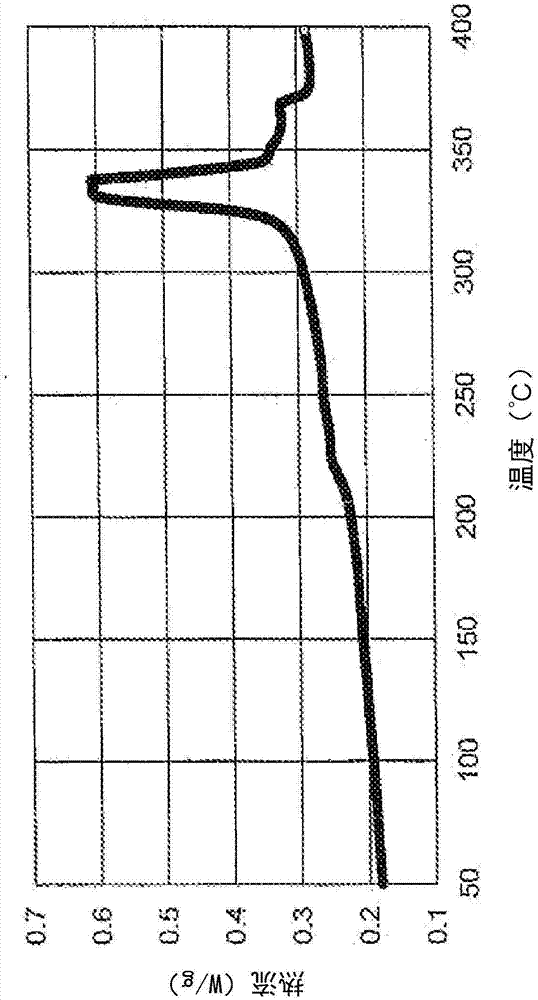
圖1是描繪根據本發明的至少一個實施方式的tfe-vdf致密制品的三個吸熱峰的差示掃描量熱掃描(dsc)圖;
圖2是描繪根據本發明的另一實施方式的tfe-ctfe致密制品的三個吸熱峰的差示掃描量熱掃描(dsc)圖;
圖3是描繪根據本發明的至少一個實施方式的tfe-vdf致密制品的三個吸熱峰的差示掃描量熱掃描(dsc)圖;以及
圖4是描繪根據本發明的至少一個實施方式的tfe-ctfe致密制品的三個吸熱峰的差示掃描量熱掃描(dsc)圖。
術語表
本文所用的術語“共聚單體”意在表示存在于芯殼四氟乙烯共聚物中的除四氟乙烯單體之外的任何共聚單體。
如本文所用,術語“基本上僅tfe單體”意在表示芯殼tfe共聚物中的殼部分含有(1)tfe單體或(2)tfe單體和不可量化的(痕量)共聚單體。
如本文所用,術語“共聚物”意在描述tfe單體和至少一種共聚單體的反應產物,其中共聚單體以基于tfe共聚物的總重量的至少3.0重量%的聚合單元的量存在于共聚物中。
如本文所用,術語“致密”用于描述空隙率低于約20%的致密制品。
如本文所用,術語“寬度”和“長度”分別類似于x方向和y方向。
本文所用的術語“潤滑劑”用于描述包括不可壓縮流體的加工助劑,在一些實施方式中用于描述由不可壓縮流體組成的加工助劑,所述加工助劑不是加工條件下聚合物的溶劑。流體-聚合物表面相互作用使得可能產生均勻混合物。
技術實現要素:
因注意,術語“tfe共聚物”、“tfe芯殼共聚物”和“芯殼tfe共聚物”在本文中可以互換使用。此外,術語“拉伸”、“經拉伸的”和“拉伸的”可分別與“變形”、“經變形的”和“變形的”互換使用。
本發明涉及致密四氟乙烯(tfe)共聚物膜和由tfe芯殼共聚物形成的致密制品。致密tfe共聚物膜可有利地直接從干燥預制件中產生,而不會像膨脹方法中通常所呈現的那樣增加干燥預制件的孔隙率。另外,本公開涉及致密制品的生產方法,該致密制品呈現改善的物理和機械性能,包括粘附性和阻隔性。致密制品包含致密tfe共聚物膜,其具有小于約20%的空隙率。tfe共聚物膜的甲烷滲透率小于約20μg*微米/cm2/分鐘。
通過使四氟乙烯單體與除了tfe以外的至少一種共聚單體進行共聚的方法形成具有芯殼結構的四氟乙烯(tfe)共聚物。如本文所用,術語“共聚單體”意在描述tfe共聚物中除了四氟乙烯之外的單體。共聚單體可以是能夠與tfe反應的烯鍵式不飽和單體,以便能夠與tfe單體聚合。例如,共聚單體可以是全氟烷基乙烯單體,例如全氟丁基乙烯(pfbe),全氟己基乙烯(pfhe),全氟辛基乙烯(pfoe),或者可以是全氟烷基乙烯基醚單體,例如全氟(甲基乙烯基醚)(pmve),全氟(乙基乙烯基醚)(peve)和全氟(丙基乙烯基醚)(ppve)。
作為替代或附加方式,共聚單體可以是烯烴,例如乙烯、丙烯或異丁烯,諸如氯代三氟乙烯(ctfe)、六氟丙烯(hfp)、氟乙烯(cfh=ch2)、偏二氟乙烯(vdf)、六氟異丁烯(hfib)和三氟乙烯(cf2=cfh)的氟代單體,以及具有以下通式的氟化間二氧雜環戊烯:
其中,r1和r2是f或包含至少一個氟原子的1-3碳烷基,x、y可以是f和/或h;
具有以下通式的氟化間二氧雜環戊烯:
其中,rf是包含1-5個碳原子的全氟烷基,r1、r2可以是f和/或cf3;或
具有以下通式的氟化間二氧雜環戊烷:
其中,r1、r2可以是f和/或包含1-5個碳原子的全氟烷基。
芯殼tfe共聚物通過聚合法制備,其包括將tfe單體和至少一種共聚單體置于加壓反應器中,用自由基引發劑引發聚合反應,在聚合反應期間將tfe單體和共聚單體供入到反應容器中,在聚合反應完成之前的一個時間點停止在聚合反應中添加共聚單體,并且通過僅將tfe單體供入到反應容器中來繼續聚合反應,直到反應完成。應理解,可以將多于一種的共聚單體供入到加壓反應器中,以產生多組分共聚物,例如三元聚合物(terpolymers)。
可以將初始加入的tfe單體和共聚單體作為預充料(precharge)引入反應器容器中。聚合反應開始后,可以依次加入共聚單體和tfe單體,例如,在tfe單體之前加入共聚單體。或者,tfe單體和共聚單體可以同時加入到反應容器中。在聚合反應期間,tfe單體和共聚單體可以遞增地或間歇地引入反應容器中。通過以更高濃度的水平將共聚單體加入到反應容器中,使得所產生的tfe共聚物中具有更高濃度的共聚單體。共聚單體可以以至少約1.0重量%、至少約2.0重量%、至少約3.0重量%、至少約3.5重量%、至少約4.0重量%、至少約4.5重量%或至少約5.0重量%的量加入至反應容器中。應注意,本文所述的向反應容器中加入tfe單體和/或共聚單體的重量百分數(%)基于供入反應器容器中的tfe單體和共聚單體的總重量。
在約15%至約90%、約20%至約70%、約20%至約60%或約30%至約60%的反應進度完成的時間點,停止向反應容器添加共聚單體。在至少一個實施方式中,在聚合反應的大致中間點停止共聚單體的添加,例如在約30%至約60%完成的時間點。然后通過僅添加tfe單體使聚合反應繼續進行,直至反應完成。可按需在反應完成之前從反應器中除去(例如排出)過量的共聚單體。
在聚合反應中,可使用基本非調聚(non-telogenic)分散劑。全氟辛酸銨(apfo或“c-8")是一種用于聚合反應的合適的分散劑的非限制性示例。可以使用程序化添加(預充料和泵送)將分散劑加入到反應容器中。應理解的是,需要成分純度以在本文所述的致密制品中實現所需的性能。除了可溶性有機雜質以外,可增大離子強度的離子性雜質能夠導致鏈轉移或鏈終止,因此要將其量最少化或者甚至消除。在至少一個實施方式中,使用超純水。
通過本文所述方法制備的tfe芯殼共聚物含有包括共聚單元的芯部分和基本僅包括tfe單體的殼部分。本文所用的“基本上僅tfe單體”意在表示所述殼含有(1)僅tfe單體或(2)tfe單體和痕量(例如不可量化)的共聚單體。tfe芯殼共聚物可含有至少約1.0重量%、至少約2.0重量%、至少約3.0重量%、至少約3.5重量%、至少約4.0重量%、至少約4.5重量%、至少約5.0重量%、至少約5.5重量%、至少約6重量%、至少約7重量%、至少約8重量%、至少約9重量%或至少約10重量%的共聚單體。因此,可存在于tfe共聚物中的四氟乙烯(例如,tfe單體)的量可以小于約99重量%、小于約98重量%、小于約97重量%、小于約96.5重量%、小于約96重量%、小于約95.5重量%或小于約95重量%。與tfe芯殼共聚物相關的重量百分比基于tfe共聚物的總重量。在一些實施方式中,tfe共聚物包括至少30重量%、至少40重量%、至少50重量%、至少約60重量%、至少約70重量%、至少約80重量%或至少90重量%的tfe單體。
tfe共聚物以分散在水性介質中的細顆粒的形式產生。在一個實施方式中,tfe共聚物可以通過凝結或干混來與至少約5重量%、至少約5.5重量%、至少約6重量%、至少約6.5重量%、至少約7重量%、至少約7.5重量%、至少約8重量%、至少約8.5重量%、至少約9重量%、至少約9.5重量%或至少約10重量%的tfe均聚物、熱塑性聚合物、tfe共聚物及其組合來進行共混或組合。應注意,在混合另外的聚合物時使用的重量%基于聚合物共混物的總重量。合適的熱塑性聚合物的非限制性示例包括但不限于:氟化乙烯丙烯(fep),聚偏氟乙烯(pvdf),全氟(烷基乙烯基)醚(pave),全氟彈性體材料(ffkm),四氟乙烯、六氟丙烯和偏氟乙烯(thv)的聚合物,全氟烷氧基烷烴(pfa),乙烯和四氟乙烯的共聚物(etfe),以及聚氯三氟乙烯(pctfe)。可選擇所要共混的聚合物的類型和/或量,以提供所需的機械或末端功能性質。
tfe芯殼共聚物以分散在水性介質中的細顆粒的形式生產,并可加工成致密tfe共聚物膜,而不需在高于聚四氟乙烯(ptfe)的結晶熔化溫度下進行熱處理。致密tfe共聚物膜直接在約400℃以下和約300℃以上的變形溫度從干燥的擠出物中產生,而不會像膨脹方法中通常所呈現的那樣增加干燥預制件的孔隙率。
為了形成致密tfe共聚物膜,tfe共聚物可經受柱塞式擠出工藝,其中tfe共聚物與合適的潤滑劑(例如,
然后,干燥的預制件可在至少一個方向上在400℃以下的溫度下(例如,300℃至400℃)進行變形或拉伸,以形成致密tfe共聚物膜。如本文所用,術語“致密”用于描述空隙率低于約20%的tfe共聚物膜或制品。致密tfe共聚物膜的空隙率可小于約20%、小于約15%、小于約10%、小于約8%、小于約5%、小于約3%或小于約1%。
在示例性實施方式中,干燥預制件在縱向和橫向(例如,x和y方向)上同時變形以形成致密tfe共聚物膜。縱向(y方向)的變形可以以約10%/秒或更低、約1,000%/秒或更高或者約10%/秒至約1,000%/秒的拉伸速率來進行。橫向(x方向)的變形可以以約10%/秒或更低、約1,000%/秒或更高或者約10%/秒至約1,000%/秒的拉伸速率來進行。應理解,干燥預制件的變形可以在x或y方向上進行,或者在x和y方向上依次或同時地進行,其中使用縮放機,或在拉幅機或類似機器中連續進行。合適的拉伸比例可顯著變化,例如從1:1至1:1,000或從1:1至1:10,000或更高,并以變化的拉伸速率變化。
致密tfe共聚物膜具有至少三個獨立的吸熱峰溫。第一吸熱峰溫在低于約300℃的溫度下出現。在至少一個實施方式中,在約50℃至約300℃的溫度下出現第一吸熱峰溫。第二吸熱峰溫在約320℃至約350℃之間出現。第三吸熱峰溫在約350℃至約400℃之間出現。在示例性實施方式中,第三吸熱峰溫在約380℃時出現。應理解,本文所用的術語“第一”、“第二”和“第三”不代表任何類型的順序,而是用于區分tfe共聚物膜中存在的三個獨立和不同的吸熱峰溫。還應注意,tfe芯殼共聚物可以通過與另外的tfe均聚物、熱塑性聚合物和/或另一tfe共聚物(如本文所述)共混而制備共混物,這可以產生額外的吸熱峰溫。
此外,致密tfe共聚物膜薄,并且厚度可小于約250微米、小于約150微米、小于約100微米、小于約50微米、小于約30微米、小于約10微米、小于約5微米或小于約1微米。
在一個示例性實施方式中,干燥預制件可如上所述在至少一個方向上在第一吸熱峰溫(即300℃)以上的溫度下進行拉伸,以形成致密tfe共聚物膜。在另一實施方式中,干燥預制件可如上所述在至少一個方向上在第一吸熱峰溫與第二吸熱峰溫之間的溫度(即,約300℃至約320℃)下變形。在進一步的實施方式中,干燥預制件可如上所述在至少一個方向上在第二吸熱峰溫以內(即約320℃至約350℃)的溫度下進行拉伸,以形成致密tfe共聚物膜。在進一步的另一實施方式中,干燥預制件可在約350℃至約400℃的溫度下進行拉伸。在至少另一實施方式中,干燥預制件可在至少一個方向上在約300℃至約335℃的溫度下變形。在另一實施方式中,干燥預制件可在約400℃以下的溫度下變形,以形成致密tfe共聚物膜。在進一步的實施方式中,干燥預制件可在約370℃以下、約335℃以下或約320℃以下的溫度下變形,以形成致密tfe共聚物膜。在示例性實施方式中,在300℃以上的溫度下變形。
可在干燥預制件拉伸或變形為致密tfe共聚物膜之前和/或之后任選地將干燥預制件加熱至變形溫度(例如,預加熱和/或后加熱)。于是,在一個實施方式中,在拉伸干燥預制件之前和/或之后將干燥預制件加熱至300℃以上的溫度。在另一實施方式中,在拉伸之前和/或之后,將干燥預制件加熱至第一吸熱峰溫和第二吸熱峰溫之間的溫度(即,約300℃至約320℃)。在另一實施方式中,在拉伸之前和/或之后,將干燥預制件加熱至第二吸熱峰溫以內的溫度(即,約320℃至約350℃)。另外,在拉伸之前和/或之后,將干燥預制件加熱至約350℃至約400℃的溫度。
致密tfe共聚物膜和致密制品可用作阻隔材料。致密tfe共聚物膜的甲烷滲透性小于約20μg*微米/cm2/分鐘、小于約15μg*微米/cm2/分鐘、小于約10μg*微米/cm2/分鐘、小于約5μg*微米/cm2/分鐘、小于約1.0μg*微米/cm2/分鐘、或小于約0.5μg*微米/cm2/分鐘。此外,致密tfe共聚物膜在至少一個方向上的基體拉伸強度大于或等于約5,000psi,大于或等于約25,000psi,大于或等于約50,000psi,大于或等于約75,000psi,或大于或等于約100,000psi,或更高。
此外,致密tfe共聚物膜和包含致密tfe共聚物膜的致密制品呈現常規聚四氟乙烯(ptfe)均聚物所不能實現的粘附特性。即,致密tfe共聚物膜和致密制品可以在比ptfe均聚物粘附到其自身或其它基材上所需的溫度和/或時間和/或壓力更低的溫度和/或更短的時間和/或更低壓力下粘附至其自身或其它材料上,所述其他基材例如有粘附促進基材、聚合物基材或金屬基材。這種粘附特性使得能夠形成阻隔材料,而不需要粘附其他基材,所述基材每單位體積具有更差的阻隔和機械性能。其結果是,致密tfe共聚物膜和致密制品的阻隔性質得到最大化。
可以將致密tfe共聚物膜和包括致密tfe共聚物膜的致密制品層疊、粘附或以其他方式結合(例如,熱、機械或化學結合)到基材上。合適的基材的非限制性示例包括但不限于:氟化乙烯丙烯(fep),全氟烷氧基烷烴(pfa),聚四氟乙烯(ptfe),四氟乙烯、六氟丙烯和偏氟乙烯(thv)的聚合物,聚氨酯,聚酰胺,乙烯乙烯基醇(evoh)以及聚氯乙烯(pvc)。該基材還可以是金屬片、無機片或壓敏粘合劑。這種層疊結構可以促進或增強與附加層(例如紡織品)的進一步結合。
測試方法
應理解,雖然下文描述了某些方法和設備,但也可選擇性地采用本領域普通技術人員確定適用的其它方法或設備。
差示掃描量熱法(dsc)
將ta儀器(tainstruments)q2000dsc和ta儀器標準鋁盤和蓋用于差示掃描量熱法(dsc),實施了該測試。使用sartoriusmc210p微量天平進行了重量測定。所有譜圖中以y軸正方向記錄吸熱峰。
通過使用由設備附帶的thermaladvantage軟件提供的calibrationwizard來進行q2000的校準。所有校準和所得的掃描數據在50ml/分鐘的恒定氮氣流下進行。
將樣品放置到盤中,記錄重量精確至0.01mg,樣品范圍為5.00mg至10.00mg。這些數值被輸入到用于q2000的thermaladvantage控制軟件中。將蓋放置在盤上并使用標準壓機進行卷曲。除了試樣制品以外,準備了用于參照的類似的盤,其重量也被輸入到軟件中。將裝有試樣制品的盤裝載到q2000中的樣品傳感器上,將空盤裝載到參照傳感器上。然后在-50℃下將樣品平衡并以10℃/分鐘的速度升至410℃。使用ta儀器的通用分析儀2000(universalanalysis2000)對數據進行了分析。
甲烷滲透性
標準程序:
用于測量甲烷滲透率的裝置包括不銹鋼測試室,其具有上半部分、下半部分、甲烷氣體入口和零空氣入口。術語“零空氣”是指通過催化劑床以除去空氣中的任何烴的壓縮空氣,使得甲烷是fid檢測器測量的唯一烴。首先用零空氣吹掃測試室的下半部分。測試膜夾在兩個半部之間并密封。緊密的密封由兩個o形圈形成。
然后通過入口將甲烷氣體和零空氣引入測試樣品。使用針閥和質量流量控制器(型號brooks5850e)分別控制甲烷氣體和零空氣的流量。甲烷氣體從底部入口進入,通過底部排氣出口排出,確保測試樣品沒有背壓。
滲透穿過測試樣品的甲烷氣體由零空氣擔載,并且被供入fid檢測器(型號8800b,baseline-mocon公司)。fid檢測器連續測量滲透穿過測試樣品的甲烷氣體的濃度。檢測器連接到數據采集系統以獲取電壓信號,然后使用已知的三點校準曲線將其轉換為甲烷濃度(c甲烷)值。
測試持續時間至少持續到甲烷濃度達到穩定狀態為止。測試時間通常為約15分鐘至約40分鐘。報告了在測試持續時間的最后兩分鐘內收集的數據(c甲烷)的平均值。
通過以下等式計算甲烷通量(以g/cm2/分鐘為單位):
甲烷通量=0.000654*c甲烷*r/a;
其中,c甲烷是以ppm為單位的平均甲烷濃度,r是以cm3/分鐘為單位的零空氣流速,a是以cm2為單位的測試試樣的面積。甲烷滲透率測量重復兩次,并報告了基于兩個試樣的甲烷通量的平均值。
累積程序:
在該程序中,使用了上述標準程序的以下變化形式。關閉零空氣入口和端口,同時將甲烷氣體引入測試樣品。沒有零空氣流入測試室的上半部分,滲透穿過測試樣品的甲烷氣體累積在測試室的上半部分中。在固定的甲烷氣體累積時間(通常約30分鐘至約60分鐘)之后,打開零空氣入口和端口,然后通過零空氣將累積的甲烷氣體輸送至fid檢測器,該檢測器測量在測試期間積聚在測試室中的甲烷氣體的濃度(c濃度)。使用上述等式來計算甲烷通量。
空隙百分比(%)
通過試樣的體積密度(ρ體積)和骨架密度(ρ骨架)之間的差異來估算樣品的空隙(%)。將測試樣品模切成直徑約20.3mm的圓形的試片(coupon)形狀。從測試樣品的不同位置切下四個試片。使用mitutoyolitematicvl-50a接觸計,在每個試片的四個不同位置測量了厚度。計算了每個試片的厚度平均值。基于四個試片的平均值報告測試樣品的厚度。使用微量天平(mettlertoledomodelat20)測量每個試片的重量。基于四個試片的平均值報告測試樣品的重量。
然后通過用測試樣品的重量除以測試樣品面積與測試樣品厚度的乘積來計算體積密度(ρ體積)。體積密度測量的變異系數(cv)通常小于5%,平均cv為約3.6%。
使用標準氦比重瓶(accupyc1340型,體積為1cm3的樣品杯)測量測試樣品的骨架密度(ρ骨架)或真密度。測試樣品質量保持在高于0.2g的水平,這是達到骨架密度值的99%以內所需的最小重量。首先使用已知體積為0.05656cm3的鋼球校準儀器。使用以下測試條件:吹掃循環=20,吹掃填充壓力和循環填充壓力=19.5psig。報告了同一測試樣品的20次測量的平均值。20次重復的變異系數(cv)小于0.2%。
然后使用以下等式計算空隙百分比(%):
空隙%=(ρ骨架-ρ體積)/ρ骨架*100
拉伸斷裂負荷測量和基體拉伸強度(mts)
使用配有平面夾具(flat-facedgrip)和0.445千牛壓力傳感器的
mts=(最大負荷/橫截面積)×(ptfe的固有密度)/試樣的固有密度);
其中,本發明的共聚物膜的體積密度相當于ptfe的固有密度,為2.2g/cc。
實施例
實施例1:
向裝有3槳葉攪拌器的50升的水平聚合反應器中添加1.5千克蠟、28千克去離子(di)水、18克全氟辛酸銨(apfo)和溶解于約50克di水的5克琥珀酸。反應器和內容物被加熱至高于蠟的熔點的溫度。將反應器反復抽真空并用tfe加壓(至約1atm或更低),直至氧水平降低至20ppm或更低。在抽真空和吹掃循環之間以60rpm的速度簡單攪拌內容物,以確保水脫氧。
將反應器加熱至83℃并以60rpm的速度攪拌。隨后,加入2.0mpa的vdf,然后加入tfe,直至壓力達到2.8mpa。此時,以80ml/分鐘的速度注入kmno4在去離子(di)水中的溶液(0.2g/l),直至添加了約1kg的tfe。以60ml/分鐘的速度添加kmno4,直至繼續加入2kg的tfe。然后以80ml/分鐘的速度添加kmno4,直到再消耗4kg的tfe。kmno4溶液的添加總量為4.61kg。
以40ml的增量加入約320g的20%的apfo溶液,在約1kg的tfe加入之后加入第一增量,然后每加入0.5kg的tfe之后加入后續的增量,以加入額外的2kg的tfe,然后再加入額外的3kg的tfe,從而在8kg的tfe反應之后加入了最終增量。
在首先加入的1kg的tfe耗盡之后,添加vdf和tfe。然后依次添加vdf和tfe,使得每消耗0.5kg的tfe后消耗0.5kg的vdf,直到消耗2kg的tfe和2kg的vdf。通過僅供入tfe來繼續進行聚合,直至聚合結束。
在反應器中加入了14kg的tfe之后停止聚合反應。制得的分散體的重量為48.90kg,含有33.21%的固體。用硝酸使分散體凝結并在130℃下干燥。
聚合物顆粒的原始分散體粒度(rdps)為0.321微米。共聚物中的vdf濃度通過質子核磁共振(nmr)質譜儀測定為27.9摩爾%(19.9重量%)。
使用上述細粉末樹脂按以下方式制備了包含tfe與vdf的芯殼共聚物的致密制品。將樹脂與isopar
使用縮放機,在300℃下將帶加熱480秒,然后以縱向方向和橫向方向均為約2.8:1的比例同時沿縱向和橫向拉伸,同時保持溫度為約300℃。平均工程應變率計算為約700%/秒。
對所得的致密制品進行表征,結果示于表1。上面在“甲烷滲透率”標題下描述的標準程序用于測量甲烷滲透率。圖1描繪了差示掃描量熱(dsc)掃描圖,顯示了致密制品的熔融轉變溫度峰(吸熱峰),清楚指示了三個峰,177.32℃處的第一峰,343.83℃處的第二峰,365.59℃處的第三峰。
實施例2:
向裝有3槳葉攪拌器的50升的水平聚合反應器中添加1.5千克蠟、28千克去離子(di)水、18克全氟辛酸銨(apfo)和溶解于約50克di水的5克琥珀酸。反應器和內容物被加熱至高于蠟的熔點的溫度。將反應器反復抽真空并用tfe加壓(至約1atm或更低),直至氧水平降低至20ppm或更低。在抽真空和吹掃循環之間以60rpm的速度簡單攪拌內容物,以確保水脫氧。將反應器加熱至83℃并以60rpm的速度攪拌。然后,添加2.8mpa的tfe,隨后以3.75ml/分鐘的速度注入kmno4在去離子(di)水中的溶液(0.6g/l),直至添加了約1kg的tfe。
以4.6ml/分鐘的速度添加kmno4溶液,以額外加入1kg的tfe。隨后以50ml/分鐘的速度添加kmno4溶液,直到再次消耗1kg的tfe。然后以30ml/分鐘的速度添加kmno4溶液,直到再次消耗1kg的tfe。之后以40ml/分鐘的速度添加kmno4溶液,直到再次消耗1kg的tfe。隨后以50ml/分鐘的速度添加kmno4溶液,直到再次消耗1kg的tfe。隨后以25ml/分鐘的速度添加kmno4溶液,直到再次消耗1kg的tfe。隨后以2ml/分鐘的速度添加kmno4溶液,直到再次消耗1kg的tfe。kmno4溶液的添加總量為5.725kg。
以40ml的增量加入約320g的20%的apfo溶液,在消耗了1kg的tfe之后加入第一增量,然后每加入1kg的tfe之后加入增量,直到加入額外的6kg的tfe,從而在7kg的tfe反應之后加入了最終增量。
使用注射泵,借由液體進料將ctfe泵入反應器。在首先加入的1kg的tfe耗盡之后,添加ctfe和tfe。然后連續加入ctfe和tfe,從而每消耗1kg的tfe的情況下,有0.8l的ctfe被消耗。持續加入,直至總共消耗了3kg的tfe和1.6l的ctfe。通過僅供入tfe來繼續進行聚合,直至聚合結束。
在反應器中加入了14.1kg的tfe之后停止聚合反應。制得的分散體的重量為51.78kg,含有35.61%的固體。用硝酸使分散體凝結并在130℃下干燥。
聚合物顆粒的原始分散體粒度(rdps)為0.266微米。共聚物中的ctfe濃度通過質子核磁共振(nmr)質譜儀測定為13摩爾%(15重量%)。
使用上述tfe與ctfe的芯殼共聚物按以下方式制備了致密制品。將樹脂與isopar
使用縮放機,在300℃下將帶加熱480秒,然后以縱向方向和橫向方向均為約4.7:1的比例同時沿縱向和橫向拉伸,同時保持溫度為約300℃。平均工程應變率計算為約700%/秒。
對所得的致密制品進行表征,結果示于表1。上面在“甲烷滲透率”標題下描述的累積程序用于測量甲烷滲透率。
圖2描繪了差示掃描量熱(dsc)掃描圖,顯示了致密制品的熔融轉變溫度峰(吸熱峰),清楚指示了三個峰,223.3℃處的第一峰,332.52℃處的第二峰,368.57℃處的第三峰。
實施例3
使用實施例1的細粉末樹脂按以下方式制備了致密制品。將樹脂與isopar
使用縮放機,在370℃下將帶加熱480秒,然后以縱向方向和橫向方向均為3.3:1的比例同時沿縱向和橫向拉伸,同時保持溫度為約370℃。平均工程應變率計算為約700%/秒。
對所得的致密制品進行表征,結果示于表1。上面在“甲烷滲透率”標題下描述的標準程序用于測量甲烷滲透率。
圖3描繪了差示掃描量熱(dsc)掃描圖,顯示了致密制品的熔融轉變溫度峰(吸熱峰),清楚指示了三個峰,180.77℃處的第一峰,333.09℃處的第二峰,370.96℃處的第三峰。
實施例4
使用實施例2的細粉末樹脂按以下方式制備了致密制品。將樹脂與isopar
使用縮放機,在370℃下將帶加熱10秒,然后以縱向方向和橫向方向均為4.6:1的比例同時沿縱向和橫向拉伸,同時保持溫度為約370℃。平均工程應變率計算為約700%/秒。
對所得的致密制品進行表征,結果示于表1。上面在“甲烷滲透率”標題下描述的標準程序用于測量甲烷滲透。
圖4描繪了差示掃描量熱(dsc)掃描圖,顯示了致密制品的熔融轉變溫度峰(吸熱峰),清楚指示了三個峰,224.81℃處的第一峰,334.83℃處的第二峰,368.2℃處的第三峰。
表1
上文已經中概括性地并且結合具體實施方式描述本申請的發明。對本領域的技術人員來說顯而易見的是,可以在不偏離本發明的精神和范圍的情況下,對本文所述的實施方式進行各種修改和變動。因此,實施方式旨在覆蓋對本發明的這些修改和變動,只要這些修改和變動在所附權利要求及其等同方案的范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!