用于核酸遞送的陽離子脂質的制作方法
本發明涉及用于核酸遞送的陽離子脂質、包含其的脂質膜結構體及它們的用途。
背景技術:
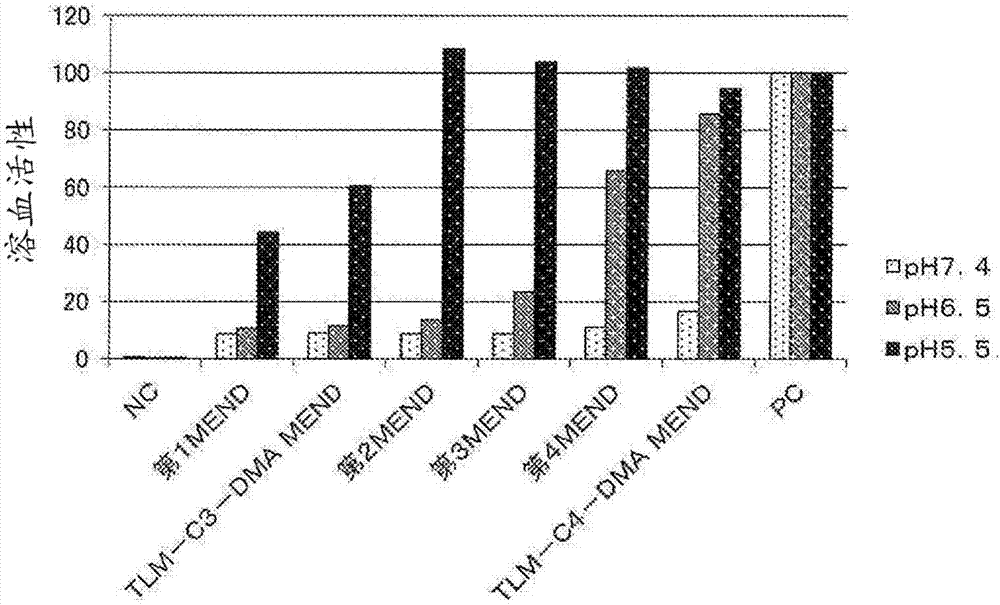
:核酸醫藥品將功能性核酸遞送至細胞質內而抑制致病蛋白質的表達,作為新一代的醫藥品而受到矚目,對其已進行了眾多研究。由于功能性核酸有在血中的穩定性較低、會迅速分解的缺點,因此,載體是必要的。作為遞送核酸的載體,可列舉病毒、聚合物膠束、脂質體等脂質膜結構體。雖然病毒載體因為表達效率高而最為常用,但具有致病性、抗原性,并且存在諸如難以賦予功能性的缺點。因此,需要安全性更高的非病毒載體的開發,聚合物膠束、脂質膜結構體等的研究正在開展。聚合物膠束為由聚乙二醇(以下稱為“peg”)、聚氨基酸等構成的載體,但臨床研究的例子仍較少。脂質膜結構體為由陽離子脂質、磷脂等構成的載體。其優點為易于對構成載體的成分的組成進行改變、利用結構修飾來賦予功能性。迄今為止已進行了多次臨床研究,是最常使用的非病毒載體。對將脂質膜結構體用作載體并有效地將功能性核酸遞送至細胞內而言,除了改善脂質膜結構體的血中穩定性、腫瘤積累性等藥物動力學以外,需要提高細胞的攝取、內質逃逸能力等細胞內動力學。對作為脂質膜結構體的構成成分之一的陽離子脂質而言,以賦予脂質膜結構體以ph響應性的目的使用。通過使用陽離子脂質,一方面使脂質膜結構體在血中等生理環境下穩定地存在。另一方面在細胞內那樣的酸性環境下使脂質膜結構體崩潰,從而使藥物能夠向細胞質內放出。陽離子脂質大致由疏水部分和親水部分構成,疏水部分為脂肪酸殘基、甾醇殘基等疏水性基團,親水部分為氨基或銨基等陽離子性基團。尤其是疏水部分含有2個疏水性基團,且親水部分含有1個氨基或銨基等陽離子性基團的結構(雙鏈型陽離子脂質)廣為人知。作為脂質膜結構體使用的陽離子脂質,可列舉作為公知化合物的1,2-二油酰基-3-二甲基氨基丙烷(以下,稱為“dodap”)、1,2-二亞油酰基-3-二甲基氨基丙烷(以下,稱為“dlindap”)等。對陽離子脂質中包含的氨基而言,通過伴隨周圍環境的ph的降低而質子化變化為陽離子性,從而賦予脂質膜結構體以ph響應性。給予至活體的脂質膜結構體被內體攝入。已知早期內體隨著向高爾基體附近進行移動,而向內部包含有許多內質網的晚期內體成熟,并與溶酶體結合。如果晚期內體與溶酶體結合,則功能性核酸會被溶酶體內的分解酶分解,因此,對于將功能性核酸有效地遞送至細胞內,需要在從早期內體起,到晚期內體與溶酶體結合為止的過程中,將功能性核酸向細胞質內放出。但是,非專利文獻1主張封裝了功能性核酸的脂質膜結構體僅在早期內體的階段向細胞質內放出功能性核酸,并且放出量為百分之幾。像這樣,盡管使用了傳統的陽離子脂質的脂質膜結構體是該領域中的技術進步,但利用其所實現的向細胞內的核酸遞送能力還不足夠滿意。現有技術文獻非專利文獻非專利文獻1:naturebiotechnology,31,638-646(1july2013)技術實現要素:發明要解決的問題本發明的課題在于提供一種陽離子脂質,其在作為用于遞送功能性核酸的載體的脂質膜結構體使用的情況下,可以實現比傳統的陽離子脂質更高的細胞內遞送效率。解決問題的方法已知具有較小的親水部分、較大的疏水部分的圓錐型結構的脂質,通常容易從層狀相向反向六角相轉變。與此相對,認為對傳統的雙鏈型陽離子脂質而言,由于氨基在活體內發生質子化而帶正電,氨基會彼此發生排斥導致親水部分擴張,因此脂質膜變得難以發生從層狀相向反向六角相的相轉變。因而,在包含傳統的雙鏈型陽離子脂質的脂質膜結構體中,脂質膜難以發生相轉變,脂質膜結構體與內體膜之間的膜融合能力不足,因此包含該脂質膜結構體的核酸導入劑的向細胞質內遞送功能性核酸的能力較低。本發明人等對上述課題進行了深入研究,結果發現:開發了為了抑制由靜電排斥導致的親水部分的擴張而具有1個或2個陽離子性基團,并且為了加大疏水部分而導入了1個至4個疏水性基團的陽離子脂質。包含本發明的陽離子脂質的脂質膜結構體,在活體內那樣的酸性條件下的膜融合能力較高,相比于使用了包含傳統的雙鏈型的陽離子脂質的脂質膜結構體的核酸導入劑,使用該脂質膜結構體的核酸導入劑向細胞質內遞送功能性核酸的能力顯著升高,從而完成了本發明。即,本發明包含以下內容。[1]由式(1)表示的陽離子脂質。[化學式1](式中,x1~x6中的任意4個各自獨立地表示由式(xa)表示的基團、由式(xb)表示的基團或羥基(其中,不包括該4個同時為羥基的情況),其余2個各自獨立地表示由式(xc)表示的基團或羥基(其中,不包括這2個同時為羥基的情況))[化學式2]-y1-r1(xa)(式中,r1表示碳原子數8~22個的脂肪族烴基或碳原子數8~22個的酰基;y1表示-o-或-nh-。)[化學式3](式中,r2表示甾醇殘基或脂溶性維生素殘基;z1表示碳原子數2或3個的亞烷基;y2表示-o-co-或-nh-co-。)[化學式4](式中,r3及r4獨立地表示碳原子數1~6個的烷基,r3和r4任選互相結合而形成環;z2表示碳原子數1~6個的亞烷基;y3表示-o-、-o-co-或-nh-co-;n表示0或1。)[2]根據[1]所述的陽離子脂質,其中,x1~x6中的任意4個各自獨立地為由式(xa)表示的基團或由式(xb)表示的基團,其余2個各自獨立地為由式(xc)表示的基團。[3]根據[1]所述的陽離子脂質,其中,x1~x6中的任意4個各自獨立地為由式(xa)表示的基團,其余2個各自獨立地為由式(xc)表示的基團。[4]根據[1]~[3]中任一項所述的陽離子脂質,其中,r1為碳原子數10~20個的脂肪族烴基或碳原子數10~20個的酰基。[5]根據[1]~[3]中任一項所述的陽離子脂質,其中,r1為含有不飽和鍵的碳原子數10~20個的脂肪族烴基、或碳原子數10~20個的酰基。[6]根據[1]~[5]中任一項所述的陽離子脂質,其中,y1為-o-。[7]根據[1]所述的陽離子脂質,其中,x1~x6中的任意4個各自獨立地為由式(xb)表示的基團,其余2個各自獨立地為由式(xc)表示的基團。[8]包含[1]~[7]中任一項所述的陽離子脂質的脂質膜結構體。[9]包含[8]所述的脂質膜結構體及核酸的核酸導入劑。發明的效果本發明涉及一種包括含有1個至4個疏水性基團的疏水部分,并含有1個或2個陽離子性基團的親水部分的陽離子脂質,及包含該陽離子脂質的脂質膜結構體、包含該脂質膜結構體及核酸的核酸導入劑。本發明的陽離子脂質可以形成穩定的脂質膜結構體,可以將作為脂質膜結構體的酸解離常數(以下,稱為“pka”)調整至中性附近。進一步,在利用使用本發明的陽離子脂質的核酸導入劑進行功能性核酸的導入時,可以僅在內體內那樣的弱酸性環境下顯示與內體膜的較高的膜融合能力,有效地將功能性核酸向細胞質內放出。即,通過利用使用本發明的陽離子脂質的核酸導入劑進行功能性核酸的導入,可以利用遞送至細胞質內的功能性核酸來實現有效的基因敲低(geneknockdown)。附圖說明[圖1]示出tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend、dlindapmend、dodapmend的溶血活性的圖。[圖2]示出第1~第4mend、tlm-c3-dmamend、tlm-c4-dmamend的溶血活性的圖。[圖3]示出由tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、dlindapmend、dodapmend導致的fvii敲低活性的圖。[圖4]示出由tlm-c3-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend導致的fvii敲低活性的圖。[圖5]示出由第1~第4mend、tlm-c3-dmamend、tlm-c4-dmamend導致的fvii敲低活性的圖。[圖6]示出由tlm-c3-dmammend、tdm-c3-dmammend導致的mrna表達活性的圖。具體實施方式以下,對本發明的實施方式進行說明。1.本發明的陽離子脂質本發明提供由式(1)表示的陽離子脂質。[化學式5](式中,x1~x6中的任意4個獨立地表示由式(xa)表示的基團、由式(xb)表示的基團或羥基(其中,不包括該4個同時為羥基的情況),其余2個獨立地表示由式(xc)表示的基團或羥基(其中,不包括這2個同時為羥基的情況)。)[化學式6]-y1-r1(xa)(式中,r1表示碳原子數8~22個的脂肪族烴基或碳原子數8~22個的酰基;y1表示-o-或-nh-。)[化學式7](式中,r2表示甾醇殘基或脂溶性維生素殘基;z1表示碳原子數2或3個的亞烷基;y2表示-o-co-或-nh-co-。)[化學式8](式中,r3及r4獨立地表示碳原子數1~6個的烷基,r3和r4任選互相結合而形成環;z2表示碳原子數1~6個的亞烷基;y3表示-o-、-o-co-或-nh-co-;n表示0或1。)在式(1)中,x1~x6中的任意4個可以是獨立地表示由式(xa)表示的基團、由式(xb)表示的基團或羥基(其中,不包括該4個同時為羥基的情況),其余2個可以是獨立地表示由式(xc)表示的基團或羥基(其中,不包括這2個同時為羥基的情況),優選x1~x6中的任意4個獨立地表示由式(xa)表示的基團或由式(xb)表示的基團,其余2個獨立地表示由式(xc)表示的基團或羥基(其中,不包括這2個同時為羥基的情況),更優選x1~x6中的任意4個獨立地表示由式(xa)表示的基團或由式(xb)表示的基團,其余2個獨立地表示由式(xc)表示的基團。作為式(1)的一個形態,在x1~x6中的任意4個獨立地表示由式(xa)表示的基團或由式(xb)表示的基團,其余2個獨立地表示由式(xc)表示的基團的情況下,雖然該x1~x6中由式(xa)表示的基團、由式(xb)表示的基團及由式(xc)表示的基團的組合是任意的,但作為該組合的具體例,可列舉以下(1)~(8)的組合。(1)x1、x2、x5及x6獨立地表示由式(xa)表示的基團,并且x3及x4獨立地表示由式(xc)表示的基團。(2)x1、x3、x4及x6獨立地表示由式(xa)表示的基團,并且x2及x5獨立地表示由式(xc)表示的基團。(3)x2、x3、x4及x5獨立地表示由式(xa)表示的基團,并且x1及x6獨立地表示由式(xc)表示的基團。(4)x1、x2、x3及x4獨立地表示由式(xa)表示的基團,并且x5及x6獨立地表示由式(xc)表示的基團。(5)x1、x2、x5及x6獨立地表示由式(xb)表示的基團,并且x3及x4獨立地表示由式(xc)表示的基團。(6)x1、x3、x4及x6獨立地表示由式(xb)表示的基團,并且x2及x5獨立地表示由式(xc)表示的基團。(7)x2、x3、x4及x5獨立地表示由式(xb)表示的基團,并且x1及x6獨立地表示由式(xc)表示的基團。(8)x1、x2、x3及x4獨立地表示由式(xb)表示的基團,并且x5及x6獨立地表示由式(xc)表示的基團。其中,從原料采購、制造的容易程度的觀點考慮,優選(1)的組合,尤其優選其中,x1、x2、x5及x6的由式(xa)表示的基團全部相同,并且x3及x4的由式(xc)表示的基團相同的(1)的組合。另外,從原料采購、制造的容易程度的觀點考慮,優選(3)的組合,尤其優選其中,x2、x3、x4及x5的由式(xa)表示的基團全部相同,并且x1及x6的由式(xc)表示的基團相同的(3)的組合。作為式(1)的一個形態,在x1~x6中的任意4個獨立地表示由式(xa)表示的基團或由式(xb)表示的基團的情況下,例如,其余2個可以為一方為由式(xc)表示的基團且另一方為羥基,或者也可以為其余2個均為由式(xc)表示的基團。在式(1)中,表示羥基的x1~x6的數量優選為3以下,更優選為1以下,特別優選為0。以下對于式(xa)、式(xb)及式(xc)中的各基團的定義進行詳述。[式(xa)]式(xa)呈-y1-r1的結構。r1表示碳原子數8~22個的脂肪族烴基或碳原子數8~22個的酰基。該脂肪族烴基及酰基中包含的碳原子數優選為10~20個。該脂肪族烴基及酰基可以為直鏈狀,也可以具有支鏈或環,但優選為直鏈狀。該脂肪族烴基及酰基可以含有飽和也可以含有不飽和鍵,但優選為含有不飽和鍵。在該脂肪族烴基及酰基含有不飽和鍵的情況下,其中包含的不飽和鍵的數量通常為1~6個,優選為1~3個,更優選為1~2個。其中包含的不飽和鍵優選為碳-碳雙鍵。作為碳原子數8~22個的脂肪族烴基,可列舉例如:辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基、二十烷基、二十一烷基、二十二烷基、辛烯基、壬烯基、癸烯基、十一碳烯基、十二碳烯基、十三碳烯基、十四碳烯基、十五碳烯基、十六碳烯基、十七碳烯基、十八碳烯基、十九碳烯基、二十碳烯基、二十一碳烯基、二十二碳烯基、辛二烯基、壬二烯基、癸二烯基、十一碳二烯基、十二碳二烯基、十三碳二烯基,十四碳二烯基,十五碳二烯基,十六碳二烯基、十七碳二烯基、十八碳二烯基、十九碳二烯基、二十碳二烯基、二十一碳二烯基、二十二碳二烯基、十八碳三烯基、二十碳三烯基、二十碳四烯基、二十碳五烯基、二十二碳六烯基、異十八烷基、四甲基十六碳烯基(植烷基)等。優選為癸基、十二烷基、十四烷基、十六烷基、十八烷基、二十烷基、癸烯基、十二碳烯基、十四碳烯基、十六碳烯基、十八碳烯基、二十碳烯基、癸二烯基、十二碳二烯基、十四碳二烯基、十六碳二烯基、十八碳二烯基、二十碳二烯基等含有飽和或不飽和鍵的碳原子數10~20個的脂肪族烴基,更優選為癸烯基、十二烯基、十四碳二烯基、十六碳二烯基、十八碳二烯基等含有不飽和鍵的碳原子數10~20個的脂肪族烴基。作為碳原子數8~22的酰基,可列舉例如:辛酰基、壬酰基、癸酰基、十一碳酰基、十二碳酰基、十三碳酰基、十四碳酰基、十五碳酰基、十六碳酰基、十七碳酰基、十八碳酰基、十九碳酰基、二十碳酰基、二十一碳酰基、二十二碳酰基、辛烯酰基、壬烯酰基、癸烯酰基、十一碳烯酰基、十二碳烯酰基、十三碳烯酰基、十四碳烯酰基、十五碳烯酰基、十六碳烯酰基、十七碳烯酰基、十八碳烯酰基、十九碳烯酰基、二十碳烯酰基、二十一碳烯酰基、二十二碳烯酰基、辛二烯酰基、壬二烯酰基、癸二烯酰基、十一碳二烯酰基、十二碳二烯酰基、十三碳二烯酰基、十四碳二烯酰基、十五碳二烯酰基、十六碳二烯酰基、十七碳二烯酰基、十八碳二烯酰基、十九碳二烯酰基、二十碳二烯酰基、二十一碳二烯酰基、二十二碳二烯酰基、十八碳三烯酰基、二十碳三烯酰基、二十碳四烯酰基、二十碳五烯酰基、二十二碳六烯酰基、異硬脂酰基、四甲基十六碳酰基(植物酰基)、視黃酰基等,優選為癸酰基、十二碳酰基、十四碳酰基、十六碳酰基、十八碳酰基、二十碳酰基、癸烯酰基、十二碳烯酰基、十四碳烯酰基、十六碳烯酰基、十八碳烯酰基、二十碳烯酰基、癸二烯酰基、十二碳二烯酰基、十四碳二烯酰基、十六碳二烯酰基、十八碳二烯酰基、二十碳二烯酰基等含有飽和或不飽和鍵的碳原子數10~20個的酰基,更優選為癸烯酰基、十二碳烯酰基、十四碳二烯酰基、十六碳二烯酰基、十八碳二烯酰基等含有不飽和鍵的碳原子數10~20個的酰基。在式(1)包含2個以上由式(xa)表示的基團的情況下,各r1可以相同也可以不同,但優選為全部相同。y1為-o-或-nh-,優選為-o-。在式(1)包含2個以上由式(xa)表示的基團的情況下,各y1可以相同也可以不同,但優選為全部相同。作為優選的由式(xa)表示的基團,為由式(xa)表示的基團中,r1為含有不飽和鍵的碳原子數10~20的脂肪族烴基或酰基,y1為-o-的基團。在式(1)包含2個以上由式(xa)表示的基團的情況下,各個由式(xa)表示的基團可以相同也可以不同,但優選為全部相同。[式(xb)]式(xb)呈-y2-z1-co-r2的結構。r2表示甾醇殘基或脂溶性維生素殘基,優選為脂溶性維生素殘基。作為甾醇殘基,可列舉例如:膽甾烯基(cholesteryl)(膽甾醇殘基)、膽甾烷基(cholestaryl)(膽甾烷醇殘基)、β-谷甾烯基(β-谷甾醇殘基)、羊毛甾烯基(羊毛甾醇殘基)、麥角甾烯基(麥角甾醇殘基)等。甾醇殘基優選為膽甾烯基或膽甾烷基。作為脂溶性維生素殘基,可列舉例如:視黃醇殘基、視黃醛殘基、麥角甾醇殘基、7-羥基膽甾醇殘基、7-脫氫膽甾醇殘基、鈣化醇殘基、膽骨化醇殘基、二氫麥角鈣化醇殘基、二氫速固醇殘基、生育酚殘基、生育三烯酚殘基等。脂溶性維生素殘基優選為視黃醇殘基或生育酚殘基。在式(1)包含2個以上由式(xb)表示的基團的情況下,各r2可以相同也可以不同,但優選為全部相同。z1表示碳原子數2或3的亞烷基,該亞烷基可以為直鏈狀也可以具有支鏈,但優選為直鏈狀。作為碳原子數2或3的亞烷基,例如可列舉亞乙基、三亞甲基等,優選為三亞甲基。在式(1)包含2個以上由式(xb)表示的基團的情況下,各z1可以相同也可以不同,但優選為全部相同。y2為-o-co-或-nh-co-,優選為-o-co-。對y2的鍵的方向沒有限制,例如在y2為-o-co-的情況下,式(xb)優選呈-o-co-z1-co-r2的結構。另外,例如在y2為-nh-co-的情況下,式(xb)優選呈-nh-co-z1-co-r2的結構。在式(1)包含2個以上由式(xb)表示的基團的情況下,各y2可以相同也可以不同,但優選為全部相同。作為優選的由式(xb)表示的基團,為r2為甾醇殘基(優選為膽甾烯基或膽甾烷基)或脂溶性維生素殘基(優選為視黃醇殘基或生育酚殘基),z1為碳原子數2或3的亞烷基(優選為亞乙基或三亞甲基,更優選為三亞甲基),y2為-o-co-的由式(xb)表示的基團。在式(1)包含2個以上由式(xb)表示的基團的情況下,各由式(xb)表示的基團可以相同也可以不同,但優選為全部相同。[式(xc)]式(xc)呈-(y3-z2)n-nr3r4的結構。r3及r4獨立地表示碳原子數1~6的烷基。r3及r4可以為直鏈狀、支鏈狀及環狀中的任意,并且也可以r3與r4互相結合而形成環。該烷基的碳原子數優選為1~3。作為直鏈狀或支鏈狀的碳原子數1~6的烷基,可列舉例如:甲基、乙基、丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、戊基、異戊基、新戊基、叔戊基、1,2-二甲基丙基、2-甲基丁基、2-甲基戊基、3-甲基戊基、2,2-二甲基丁基、2,3-二甲基丁基、環己基等。作為在r3與r4互相結合并形成環的情況下的-nr3r4的例子,具體可列舉:氮丙啶基、氮雜環丁烷基、唑啉基(azolidyl)、哌啶基等。r3及r4優選為甲基、乙基、丙基或異丙基,更優選為甲基。r3和r4可以相同也可以不同,優選r3與r4相同。在式(1)包含2個由式(xc)表示的基團的情況下,各r3可以相同也可以不同,但優選為相同。在式(1)包含2個由式(xc)表示的基團的情況下,各r4可以相同也可以不同,但優選為相同。z2表示碳原子數1~6的亞烷基。z2可以為直鏈狀也可以具有支鏈,但優選為直鏈狀。作為碳原子數1~6的亞烷基,可列舉例如:亞甲基、亞乙基、三亞甲基、異亞丙基、四亞甲基、異亞丁基、五亞甲基、新亞戊基、六亞甲基等。z2優選為亞甲基、亞乙基、三亞甲基、異亞丙基、四亞甲基、六亞甲基,更優選為亞乙基、三亞甲基。在式(1)包含2個由式(xc)表示的基團的情況下,各z2可以相同也可以不同,但優選為相同。y3表示-o-、-o-co-或-nh-co-,優選為-o-co-。對y3的鍵的方向沒有限制,例如在y3為-o-co-的情況下,式(xc)優選呈-(o-co-z2)n-nr3r4的結構。另外,例如在y3為-o-的情況下,式(xc)優選為呈-(o-z2)n-nr3r4的結構,在y3為-nh-co-的情況下,式(xc)優選為呈-(nh-co-z2)n-nr3r4的結構。在式(1)包含2個由式(xc)表示的基團的情況下,各y3可以相同也可以不同,但優選為相同。n為0或1,優選為1。在n為1的情況下,式(xc)呈-y3-z2-nr3r4的結構,在n為0的情況下,式(xc)呈-nr3r4的結構。在式(1)包含2個由式(xc)表示的基團的情況下,各n可以相同也可以不同,但優選為相同。作為優選的由式(xc)表示的基團,為r3及r4獨立地表示碳原子數1~3的烷基(優選為甲基、乙基、丙基或異丙基,更優選為甲基),z2為碳原子數1~6的亞烷基(優選為亞甲基、亞乙基、三亞甲基、異亞丙基、四亞甲基、六亞甲基、更優選為亞乙基、三亞甲基),y3為-o-co-,n為1的由式(xc)表示的基團。在式(1)包含2個由式(xc)表示的基團的情況下,各由式(xc)表示的基團可以相同也可以不同,但優選為相同。作為本發明的陽離子脂質的具體例,可列舉表1所述的tlm-c2-dma(1,2,5,6-四亞油酰基-3,4-二(二甲基氨基乙酰基)-d-甘露醇)、tlm-c3-dma(1,2,5,6-四亞油酰基-3,4-二(3-二甲基氨基丙酰基)-d-甘露醇)、tlm-c4-dma(1,2,5,6-四亞油酰基-3,4-二(4-二甲基氨基丁酰基)-d-甘露醇),tdm-c3-dma(1,2,5,6-四(癸烯基)-3,4-二(3-二甲基氨基丙酰基)-d-甘露醇)、tlmes-c3-dma(四亞油酰基-二(3-二甲基氨基丙酰基)-d-甘露醇)等。[表1]下面對本發明的陽離子脂質的制造方法進行說明。作為本發明的陽離子脂質的制造方法,可列舉向具有6個羥基的由式(1’)表示的化合物中,導入由式(xa)表示的基團和/或由式(xb)表示的基團、以及由式(xc)表示的基團的方法,例如:(i)導入xa和/或xb之后導入xc的方法,(ii)導入xc之后導入xa和/或xb的方法,(iii)同時導入xa和/或xb和xc的方法,或基于這些的方法等。[化學式9]對本發明的陽離子脂質的制造方法沒有特別限制,優選為上述(i)方法。雖然以下舉出了該(i)方法的具體例,但本發明的陽離子脂質的制造方法并不特別限定于該方法。作為起始化合物,可列舉例如,在具有6個羥基的式(1’)表示的化合物中有2個羥基利用保護基保護的化合物,或在式(1’)表示的化合物中有4個羥基利用保護基保護的化合物等。需要說明的是,就這些起始化合物而言,可以容易地取得市售品,或者可以按照本身公知的方法或基于公知的方法的方法進行制造。作為向式(1’)導入的保護基,可列舉例如:異亞丙基、亞芐基、苯甲酰基、芐基、三苯甲基、4-甲氧基三苯甲基、4,4’-二甲氧基三苯甲基、三烷基硅烷基(例如,三甲基硅烷基、三乙基硅烷基、叔丁基二甲基硅烷基、叔丁基二苯基硅烷基等)等。對該保護基的導入方法沒有特別的限制,可以根據本身公知的方法或基于其的方法進行。例如,在制造作為本發明的陽離子脂質為式(1)中的x1、x2、x5及x6為由式(xa)表示的基團(r1:脂肪族烴基、y1:-o-,此外的符號如式(xc)所定義),x3及x4為由式(xc)表示的基團(y3:-co-o-,n:1)時的式(5)表示的化合物的情況下,對由該式(5)表示的化合物而言,可以使用由式(1a)表示的化合物作為起始化合物,經過以下的工序1(醚化)、工序2(脫保護)、工序3(酯化)、或者工序1(醚化)、工序2(脫保護)、工序4(酯化)、工序5(氨基化)來制造。[化學式10]式中,a表示保護基(例如異亞丙基、亞芐基、苯甲酰基、芐基、三苯甲基、4-甲氧基三苯甲基、4,4’-二甲氧基三苯甲基、三烷基甲硅烷基等),b表示脫離基(例如:碘原子、溴原子、氯原子、甲烷磺酰氧基、對甲苯磺酰氧基、三氟甲烷磺酰氧基等),d表示羥基或鹵原子(例如:碘原子、溴原子、氯原子等),e表示由式(e)表示的基團或乙烯基。[化學式11]b-z2-(e)(式中,z2表示碳原子數1~6的亞烷基,b表示脫離基(例如:碘原子、溴原子、氯原子、甲烷磺酰氧基、對甲苯磺酰氧基、三氟甲烷磺酰氧基等)。)工序1(醚化)通過將由式(1a)表示的化合物與由r1-b(式中的r1及b與上述同義)表示的化合物發生反應,得到由式(2)表示的化合物。在反應中可以使用氫氧化鉀、氫化鈉、叔丁醇鉀等堿催化劑,也可以無催化劑進行。優選將氫氧化鉀作為催化劑使用。作為使用的催化劑量,相對于由式(1a)表示的化合物通常為6~20摩爾當量,優選為8~12摩爾當量。雖然反應可以使用溶劑,也可以無溶劑進行,但由于由式(1a)表示的化合物為高極性的固體,需要在反應體系中分散,因此優選使用溶劑。作為溶劑,可以使用不阻礙反應,由式(1a)表示的化合物可分散的溶劑,可列舉例如:己烷、甲苯、二甲基甲酰胺、二甲亞砜(以下,稱為“dmso”)等。這些中,優選甲苯。反應溫度通常為20~150℃,優選為40~100℃。反應時間通常為1~50小時,優選為10~30小時。得到的由式(2)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序2(脫保護)對由式(2)表示的化合物的保護基脫保護,得到包含2個游離的羥基的由式(3)表示的化合物。在反應中使用酸催化劑。作為酸催化劑,可列舉鹽酸、乙酸、硫酸、磷酸、對甲苯磺酸一水合物等,優選為鹽酸。作為使用的催化劑量,相對于由式(2)表示的化合物通常為1~50摩爾當量,優選為5~20摩爾當量。在反應中使用溶劑。作為溶劑,可列舉甲醇、乙醇、異丙醇、水等,優選為甲醇、乙醇。反應溫度通常為20~70℃,優選為40~60℃。反應時間通常為1~12小時,優選為4~8小時。得到的由式(3)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序3(酯化)可以通過將由式(3)表示的化合物與由式(p)(式中,r3、r4及z2與上述同義)表示的化合物發生反應,得到本發明的式(5)表示的化合物。在反應中,使用二環己基碳二亞胺(以下,稱為“dcc”)、二異丙基碳二亞胺(以下,稱為“dic”)、1-乙基-3-(二甲基氨基丙基)碳二亞胺鹽酸鹽(以下,稱為“edc”)等縮合劑。在反應中添加堿催化劑。作為堿催化劑,可列舉4-二甲基氨基吡啶(以下,稱為“dmap”)、吡啶、三乙胺等,優選為dmap。由式(p)表示的化合物的投入量,相對于由式(3)表示的化合物通常為2~10摩爾當量,優選為4~8摩爾當量。雖然反應可以使用溶劑,也可以無溶劑進行,但由于由式(p)表示的化合物為高極性的固體,需要在反應體系中溶解或分散,因此優選使用溶劑。作為可使式(p)表示的化合物溶解或分散的溶劑,可列舉例如,氯仿、二氯甲烷、甲苯、乙酸乙酯等,這些中,優選氯仿。反應溫度通常為10~60℃,優選為20~40℃。反應時間通常為1~20小時,優選為2~10小時。得到的由式(5)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序4(酯化)可以通過將由式(3)表示的化合物與由式(q)(式中的d、e與上述同義)表示的化合物發生反應,得到由式(4)表示的化合物。在由式(q)表示的化合物中的d為羥基的情況下,在反應中,使用dcc、dic、edc等縮合劑。可以使由式(3)表示的化合物與由式(q)表示的化合物直接反應,或也可以形成由式(q)表示的化合物的酸酐后再與式(3)表示的化合物反應。在由式(q)表示的化合物中的d為鹵原子的情況下,為了中和副產的鹵化氫而添加堿。作為堿,可列舉三乙胺、吡啶等。反應使用溶劑。作為溶劑,可列舉氯仿、二氯甲烷、甲苯、乙酸乙酯等,優選為甲苯。由式(q)表示的化合物的投入量,相對于由式(3)表示的化合物通常為2~10摩爾當量,優選為2~5摩爾當量。反應溫度通常為0~60℃,優選為10~40℃,反應時間通常為1~10小時,優選為1~5小時。得到的由式(4)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序5(氨基化)可以通過使含有r3及r4的仲胺與由式(4)表示的化合物發生反應,得到本發明的式(5)表示的化合物。在反應中可以使用碳酸鉀、碳酸鈉、叔丁醇鉀等堿催化劑,也可以無催化劑進行。反應可以使用溶劑,也可以無溶劑進行。對于溶劑,可以使用例如乙酸乙酯、二氯甲烷、氯仿、苯、甲苯、四氫呋喃(以下,稱為“thf”)等,這些中,優選甲苯。含有r3及r4的仲胺的投入量,相對于由式(4)表示的化合物通常為1~20摩爾當量,優選為5~10摩爾當量。反應溫度通常為10~100℃,優選為60~80℃。反應時間通常為1~10小時,優選為2~6小時。得到的由式(5)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。本領域技術人員可以通過適當選擇原料并基于本說明書的實施例的方法進行反應,來制造需要的本發明的陽離子脂質。另外,例如,在制造如下由式(9)表示的化合物的情況下,其中,在由式(9)表示的化合物中,式(1)中的x2、x3、x4及x5為由式(xa)表示的基團(r1:酰基、y1:-o-),x1及x6為由式(xc)表示的基團(n:1,y3:-co-o-,此外的符號如式(xc)所定義),對以該式(9)表示的化合物而言,可以使用由式(1b)表示的化合物作為起始化合物,經過以下的工序6(酯化)、工序7(脫保護)、工序8(酯化)、或者工序6(酯化)、工序7(脫保護)、工序9(酯化)、工序10(氨基化)來制造。[化學式12]式中,a表示保護基(例如異亞丙基、亞芐基、苯甲酰基、芐基、三苯甲基、4-甲氧基三苯甲基、4,4’-二甲氧基三苯甲基、三烷基硅烷基等),d表示羥基或鹵原子(例如:碘原子、溴原子、氯原子等),e表示由式(e)表示的基團或乙烯基。[化學式13]b-z2-(e)(式中,z2表示碳原子數1~6的亞烷基,b表示脫離基(例如:碘原子、溴原子、氯原子、甲烷磺酰氧基、對甲苯磺酰氧基、三氟甲烷磺酰氧基等)。)工序6(酯化)通過將由式(1b)表示的化合物與由r1-d(式中的r1及d與上述同義)表示的化合物發生反應,得到由式(6)表示的化合物。在r1-d的d為羥基的情況下,在反應中,使用dcc、dic、edc等縮合劑。對使用量而言,相對于式(1b)表示的化合物,通常為4~10摩爾當量,優選為5~8摩爾當量。在反應中添加堿催化劑。作為堿催化劑,可列舉dmap、吡啶、三乙胺等,優選為dmap。在反應中使用溶劑。作為溶劑,只要是不阻礙反應、基質可溶解的溶劑即可,沒有特別限定。具體而言,可列舉氯仿、二氯甲烷、甲苯、乙酸乙酯等,優選為氯仿。反應溫度通常為0~60℃,優選為10~40℃。反應時間通常為1~50小時,優選為10~30小時。得到的由式(6)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序7(脫保護)對由式(6)表示的化合物的保護基進行脫保護,得到包含2個游離的羥基的由式(7)表示的化合物。在a為三烷基甲硅烷基的情況下,在反應中使用氟化物。作為氟化物,可列舉:四丁基氟化銨、四丙基氟化銨、四乙基氟化銨、四甲基氟化銨、氫氟酸、氟化銫等,優選為四丁基氟化銨。作為氟化物的使用量,相對于式(6)表示的化合物通常為2~10摩爾當量,優選為4~8摩爾當量。在三烷基甲硅烷基的脫保護的過程中,生成為強堿的烷氧化物。由于如果存在烷氧化物,則會發生酯的分解、轉移,因此,需要迅速地中和。因而,優選向反應體系中添加作為質子源的酸。由于酸過強則會分解酯,因此優選為弱酸。具體而言,可列舉乙酸、乙二酸、檸檬酸、磷酸、苯甲酸、硼酸、三乙胺鹽酸鹽等,優選為乙酸。作為酸的使用量,相對于式(6)表示的化合物通常為2~10摩爾當量,優選為4~8當量。在反應中使用溶劑。作為溶劑,可列舉:四氫呋喃、氯仿、乙酸乙酯、乙醇、甲醇等,優選為四氫呋喃。反應溫度通常為0~70℃,優選為10~60℃。反應時間通常為1~12小時,優選為4~8小時。得到的由式(7)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序8(酯化)可以通過將由式(7)表示的化合物與由式(p)(式中,r3、r4及z2與上述同義)表示的化合物發生反應,得到本發明的式(9)表示的化合物。在反應中,使用dcc、dic、edc等縮合劑。反應時,可以使由式(7)表示的化合物與由式(p)表示的化合物直接反應,或也可以形成由式(p)表示的化合物的酸酐后再與由式(7)表示的化合物反應。在反應中添加堿催化劑。作為堿催化劑,可列舉dmap、吡啶、三乙胺等,優選為dmap。由式(p)表示的化合物的投入量,相對于由式(7)表示的化合物通常為2~10摩爾當量,優選為4~8摩爾當量。雖然反應可以使用溶劑,也可以無溶劑進行,但由于由式(p)表示的化合物為高極性的固體,需要在反應體系中分散,因此優選使用溶劑。作為可使由式(p)表示的化合物分散的溶劑,可列舉例如,氯仿、二氯甲烷、甲苯、乙酸乙酯等,這些中,優選氯仿。反應溫度通常為10~60℃,優選為20~40℃。反應時間通常為1~20小時,優選為2~10小時。得到的由式(9)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序9(酯化)可以通過將由式(7)表示的化合物與由式(q)(式中的d、e與上述同義)表示的化合物發生反應,得到由式(8)表示的化合物。在由式(q)表示的化合物中的d為羥基的情況下,在反應中,使用dcc、dic、edc等縮合劑。可以使由式(7)表示的化合物與由式(q)表示的化合物直接反應,或也可以形成由式(q)表示的化合物的酸酐后再與由式(7)表示的化合物反應。在反應中添加堿催化劑。作為堿催化劑,可列舉dmap、吡啶、三乙胺等,優選為dmap。在由式(q)表示的化合物中的d為鹵原子的情況下,為了中和副產的鹵化氫而添加堿。作為堿,可列舉三乙胺、吡啶等。反應使用溶劑。作為溶劑,可列舉氯仿、二氯甲烷、甲苯、乙酸乙酯等,優選為甲苯。由式(q)表示的化合物的投入量,相對于由式(7)表示的化合物通常為2~10摩爾當量,優選為2~5摩爾當量。反應溫度通常為0~60℃,優選為10~40℃,反應時間通常為1~10小時,優選為1~5小時。得到的由式(8)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。工序10(氨基化)可以通過使含有r3及r4的仲胺與由式(8)表示的化合物發生反應,得到本發明的式(9)表示的化合物。在反應中可以使用碳酸鉀、碳酸鈉、叔丁醇鉀等堿催化劑,也可以無催化劑進行。反應可以使用溶劑,也可以無溶劑進行。對于溶劑,可以使用例如乙酸乙酯、二氯甲烷、氯仿、苯、甲苯、thf等,這些中,優選使用甲苯。含有r3及r4的仲胺的投入量,相對于由式(8)表示的化合物通常為1~20摩爾當量,優選為5~10摩爾當量。反應溫度通常為10~100℃,優選為60~80℃。反應時間通常為1~10小時,優選為2~6小時。得到的由式(9)表示的化合物可以通過提取、重結晶、吸附處理、再沉淀、柱色譜等方法進行適當純化。本領域技術人員可以通過適當選擇原料并基于本說明書的實施例的方法進行反應,來制造需要的本發明的陽離子脂質。2.本發明的脂質膜結構體下面對本發明的脂質膜結構體進行說明。本發明的脂質膜結構體,含有由上述式(1)表示的陽離子脂質(即,本發明的陽離子脂質)作為膜的構成脂質。這里,本發明中的“脂質膜結構體”是指,膜的構成脂質的親水基團朝向界面的水相側排列的脂質膜結構體。對本發明的脂質膜結構體的形態沒有特別限定,例如,作為本發明的陽離子脂質分散在水系溶劑中而成的形態,可列舉脂質體(例如:單層脂質體、多層脂質體等)、o/w型乳液、w/o/w型乳液、球狀膠束、帶狀膠束、或不特定的層狀結構物(unspecifiedlayerstructure)等。本發明的脂質膜結構體的形態優選為脂質體。本發明的脂質膜結構體除了本發明的陽離子脂質以外,也可以進一步含有這以外的其它構成成分。作為該其它構成成分,可列舉例如:脂質(例如:磷脂(例如:磷脂酰乙醇胺、磷脂酰肌醇、磷脂酰絲氨酸、磷脂酸、磷脂酰甘油、磷脂酰膽堿等)、糖脂、肽脂、膽甾醇、除了本發明的陽離子脂質以外的陽離子脂質、peg脂質等)、表面活性劑(例如:chaps、膽酸鈉鹽、辛基糖苷、n-d-葡萄糖-n-甲基烷酰胺類、poloxamer類、聚氧乙烯山梨糖醇酐脂肪酸酯類等)、peg、蛋白質等。本發明的脂質膜結構體中的該其它構成成分的含量,通常為5~90重量%,優選為10~30重量%。本發明的脂質膜結構體中包含的本發明的陽離子脂質可以僅為1種,也可以組合2種以上使用。通過組合2種以上本發明的陽離子脂質使用,可以將本發明的脂質膜結構體的pka在4~7的范圍中任意調整。通過將本發明的脂質膜結構體的pka調整為適于目的的值,能夠將功能性核酸高效率地遞送至細胞內。對本發明的脂質膜結構體中包含的本發明的陽離子脂質的含量沒有特別限定,例如,在將本發明的脂質膜結構體使用于后述的核酸導入劑中的情況下,本發明的脂質膜結構體,優選包含為了導入核酸的充分量的本發明的陽離子脂質。本發明的脂質膜結構體中包含的本發明的陽離子脂質的含量,通常為本發明的脂質膜結構體中包含的總脂質量(totallipidamount)的5~100mol%,優選為30~90mol%,更優選為50~70mol%。本發明的脂質膜結構體,可以通過將本發明的陽離子脂質及其它構成成分(脂質等)溶解或分散于適當的溶劑或分散介質,例如:水性溶劑、醇性溶劑中,并根據需要進行誘導組織化的操作進行制備。作為“誘導組織化的操作”,可列舉例如:乙醇稀釋法、簡單水和法、超聲波處理、加熱、渦旋震蕩(vortex)、醚注入法、法壓法(frenchpressmethod)、膽酸法、ca2+融合法、凍融法、反相蒸發法等本身公知的方法。3.本發明的核酸導入劑通過向包含本發明的陽離子脂質的脂質膜結構體導入核酸,并使其與活體內和/或活體外的細胞接觸,可以向該細胞內導入核酸。因此,本發明也提供核酸導入劑(以下,稱為“本發明的試劑”)。本發明的試劑的主要的特征為含有上述包含本發明的陽離子脂質的脂質膜結構體及核酸。作為一種形態,本發明的試劑可以含有本發明的脂質膜結構體及核酸,在這種情況下,優選該核酸已被導入至本發明的脂質膜結構體。這里,將核酸“導入”至本發明的脂質膜結構體是指將核酸封裝到由脂質雙重膜形成的空間中。對可導入本發明的脂質膜結構體的核酸沒有特別的限制,可以使用任意核酸。作為核酸的種類,可列舉例如:dna、rna、dna和rna的嵌合核酸、dna/rna的雜合等,但不限于這些。另外,對核酸而言,可使用1~3條鏈中的任意,優選為1條鏈或2條鏈。核酸可以為例如:作為嘌呤或嘧啶堿的n-糖苷的其它類型的核苷酸,或者具有非核苷酸骨架的其它低聚物(例如:市售的肽核酸(pna)等)或含有特殊的鍵的其它低聚物(其中,該低聚物含有具有在dna、rna中發現的那樣的堿基配對、允許堿基的附著的構造的核苷酸)等。并且該核酸可以為例如:添加了公知的修飾的核酸、具有該領域已知的標記的核酸、帶帽的核酸、經甲基化的核酸、將1個以上的天然的核苷酸用類似物進行置換而成的核酸、分子內核苷酸修飾后的核酸、具有非電荷鍵的核酸(例如,甲基膦酸酯、磷酸三酯、氨基磷酸酯、氨基甲酸酯等)、具有帶電荷的鍵或含硫鍵(例如:硫代磷酸酯、二硫代磷酸酯等)的核酸、例如蛋白質(例如:核酸酶、核酸酶抑制劑、毒素、抗體、信號肽、聚-l-賴氨酸等)、糖(例如:單糖等)等具有側鏈基團的核酸、具有插層化合物(intercalatingcompound)(例如:吖啶、補骨脂素等)的核酸,含有螯合化合物(例如:金屬、具有放射活性的金屬、硼、氧化性的金屬等)的核酸、含有烷基化劑的核酸、具有經修飾的鍵的核酸(例如:α異頭物型核酸等)等。對在本發明中可以使用dna的種類沒有特別的限制,可以根據使用的目的適當選擇,可列舉例如,質粒dna、cdna、反義dna、染色體dna、pac、bac等,優選為質粒dna、cdna、反義dna,更優選為質粒dna。質粒dna等環狀dna也可以適當利用限制酶等進行消化,而以線性dna形式使用。在本發明中可以使用的rna的種類沒有特別的限制,可以根據使用的目的適當選擇,可列舉例如sirna、mirna、shrna、反義rna、信使rna(mrna)、單鏈rna基因組、雙鏈rna基因組、rna復制子、轉移rna、核糖體rna等,優選為sirna、mirna、shrna、mrna、反義rna、rna復制子。在本發明中使用的核酸優選通過本領域技術人員通常使用的方法進行純化。對本發明的試劑而言,例如,可以出于疾病的預防和/或治療的目的進行活體內(invivo)給予。因此,在本發明中使用的核酸優選相對于某給定的疾病具有預防和/或治療活性(預防、治療用核酸)。作為這樣的核酸,可列舉例如用于所謂基因治療的核酸等。對向本發明的脂質膜結構體導入核酸的方法沒有特別限制,例如,可以通過在形成本發明的脂質膜結構體時,使本發明的脂質膜結構體的構成成分與期望的核酸共存,來向本發明的脂質結構體導入核酸。例如,在利用乙醇稀釋法形成本發明的脂質膜結構體時,通過將核酸的水溶液與本發明的脂質膜結構體的構成成分(脂質等)的乙醇溶液利用渦旋震蕩等劇烈混合后,將混合物利用適當的緩沖液進行稀釋,從而得到導入了核酸的本發明的脂質膜結構體的懸浮液。利用單純水合法形成本發明的脂質膜結構體時,通過將本發明的脂質膜結構體的構成成分(脂質等)溶解于適當的有機溶劑,并將該溶液放入玻璃容器,通過減壓干燥而蒸餾除去溶劑,得到脂質薄膜,然后,這里添加核酸的水溶液,使其水合后,利用超聲波發生器進行超聲波處理,從而得到導入了核酸的本發明的脂質膜結構體的懸浮液。作為本發明的試劑的一種形態,可列舉例如,多功能性包絡型納米結構體(multifunctionalenvelope-typenanodevice,以下,稱為“mend”)。該mend例如可以通過將核酸與聚陽離子(例如,魚精蛋白等)之間的靜電復合體導入本發明的脂質膜結構體等來制備(kogureketal.,multifunctionalenvelope-typenanodevice(mend)asanon-viralgenedeliverysystem.adv.drugdeliv.rev.,60,559-571(1march2008)。該結構體(mend)可以用作將核酸等選擇地遞送至特定的細胞內的藥物遞送系統,對例如基于向樹突細胞導入抗原基因的dna疫苗、腫瘤的基因治療等有用。導入了核酸的本發明的脂質膜結構體的表面電荷(zeta電位)優選為-10~+10mv,進一步優選為-10~+5mv。在以往的基因導入中,主要使用了表面電荷帶正電的粒子。其理由在于促進與具有負電荷的細胞表面的硫酸肝素之間的靜電相互作用,雖然作為用于促進細胞的攝取的方法有用,但就正的表面電荷而言,存在產生在細胞內由于與導入基因之間的相互作用而導致轉錄抑制、由于與mrna之間的相互作用而導致翻譯抑制的可能性。通過在上述的范圍內調整表面電荷,可解決該問題。表面電荷的測定,可以用metasizernano(malverninstrumentsltd.)進行。通過將本發明的脂質膜結構體的構成成分的組成在不破壞本發明的目的范圍內適宜調整,可以將表面電荷調整為期望的值。通過使本發明的試劑與細胞接觸,可以將本發明的試劑中包含的核酸導入到細胞內。對該“細胞”的種類沒有特別限定,可使用原核生物及真核生物的細胞,優選為真核生物的細胞。對該真核生物的種類也沒有特別限定,可列舉例如:包括人的哺乳類(例如:人、猴、小鼠、大鼠、倉鼠、牛等)、鳥類(例如:雞、鴕鳥等)、兩棲類(例如:青蛙等)、魚類(例如:斑馬魚、鳉魚(稻田魚)等)等脊椎動物;昆蟲(例如:蠶、蛾、果蠅等)等非脊椎動物;植物;微生物(例如:酵母等)等。更優選的是,作為本發明的對象的細胞,優選為動物細胞(例如:脊椎動物細胞等)或者植物細胞,進一步優選為哺乳動物細胞。該細胞可以是包括癌細胞的培養細胞株,也可以是從個體、組織分離而成的細胞,或者組織或者組織片的細胞。另外,該細胞可以為貼壁細胞,也可以為非貼壁細胞。以下對在活體外(invitro)使本發明的試劑與細胞接觸的方法進行具體說明。對細胞而言,在與本發明的試劑接觸的數天前懸浮于適當的培養基中,以適當的條件進行培養。在與本發明的試劑接觸的時候,細胞可以處于增殖期,也可以不是。該接觸時的培養液可以是含血清培養基,也可以是不含血清培養基,優選培養基中的血清濃度為30重量%以下,更優選為20重量%以下。如果培養基中含有過量的血清等蛋白質,則存在阻礙本發明的試劑與細胞之間的接觸的可能性。對該接觸時的細胞密度沒有特別限定,可以考慮細胞的種類等進行適當設定,通常為1×104~1×107細胞/ml的范圍。向這樣制備的細胞,例如,添加上述的導入了核酸的本發明的脂質膜結構體的懸浮液。對該懸浮液的添加量沒有特別限定,可以考慮細胞數等進行適當設定。對與細胞接觸時該懸浮液中的本發明的脂質膜結構體的濃度而言,只要能夠實現目標核酸向細胞內的導入即可,沒有特別限定,作為脂質濃度,通常為1~100nmol/ml,優選為10~50nmol/ml,作為核酸的濃度,通常為0.01~100μg/ml,優選為0.1~10μg/ml。將上述的懸浮液添加至細胞后,培養該細胞。對培養時的溫度、濕度、co2濃度等而言,可以考慮細胞的種類進行適當設定。在細胞為源自哺乳動物的細胞時,通常溫度為約37℃,濕度為約95%,co2濃度為約5%。另外,雖然培養時間也可以考慮使用的細胞的種類等條件進行適當設定,但通常為0.1~24小時的范圍,優選為0.25~4小時的范圍,更優選為0.5~2小時的范圍。如果上述培養時間過短,則核酸不能充分地導入細胞內,如果培養時間過長,則細胞可能變虛弱。通過上述培養將核酸導入細胞內,優選為將培養基與新鮮的培養基交換,或向培養基中添加新鮮的培養基并進一步繼續培養。在細胞為源自哺乳動物的細胞時,優選新鮮的培養基含有血清或營養因子。另外,如上所述,通過使用本發明的試劑,不僅在活體外(invitro),在活體內(invivo)中能夠也將核酸導入細胞內。即,通過將本發明的試劑給予對象,例如,使導入了核酸的本發明的脂質膜結構體到達并與目標細胞接觸,在活體內將已導入該脂質膜結構體中的核酸導入細胞內。對可以給予本發明的試劑的對象沒有特別限定,可以舉出例如:包括人的哺乳類(例:人、猴、小鼠、大鼠、倉鼠、牛等)、鳥類(例:雞、鴕鳥等)、兩棲類(例:青蛙等)、魚類(例、斑馬魚、鳉魚等)等脊椎動物,昆蟲(例、蠶、蛾、果蠅等)等無脊椎動物、植物等。本發明的試劑的給予對象優選為人或其它哺乳動物。對目標細胞的種類沒有特別限定,可以通過使用本發明的試劑,向各種組織(例如,肝、腎、胰腺、肺、脾、心臟、血液、肌肉、骨、腦,胃、小腸、大腸、皮膚、脂肪組織等)中的細胞導入核酸。另外,本發明的試劑中包含的脂質膜結構體,也可以導入有除了核酸以外的化合物。在本發明的試劑含有導入了除了核酸以外的化合物的脂質膜結構體的情況下,就給予本發明的試劑的對象(例如:脊椎動物、無脊椎動物等)的方法而言,只要是能夠使該脂質膜結構體到達并與目標細胞接觸,并將已導入至該脂質膜結構體的化合物導入細胞內的方法即可,沒有特別限定,可以考慮導入化合物的種類、目標細胞的種類、部位等,適當選擇本身公知的給予方法(例如:口服給予、非口服給予(例如:靜脈內給予、肌肉內給予、局部給予、經皮給予、皮下給予、腹腔內給予、噴霧等)等)。對本發明的試劑的給予量而言,只要是能夠實現化合物向細胞內的導入的范圍即可,沒有特別限定,可以考慮給予對象的種類、給予方法、導入化合物的種類、目標細胞的種類、部位等進行適當選擇。對將化合物導入至本發明的脂質膜結構體的方法沒有特別的限制,例如,可以通過與上述的向本發明的脂質膜結構體導入核酸的方法同樣的方法,或基于其的方法制備。對本發明的試劑的劑型沒有特別限制,可列舉例如:注射劑(例如:皮下注射劑、靜脈內注射劑、肌肉內注射劑、腹腔內注射劑、點滴注射劑等)等。本發明的試劑可以通過將本發明的脂質膜結構體按照相應于用途(例如研究用試劑、醫藥等)的常規方法進行制劑化而制造。在將本發明的試劑作為研究用試劑提供的情況下,對該本發明的試劑而言,可以將本發明的脂質膜結構體直接使用,或者使用其與例如水、或者除了水以外的生理上允許的液體(例如,水溶性溶劑(例如,蘋果酸緩沖液等)、有機溶劑(例如:甲醇、乙醇、dmso等)或者水溶性溶劑和有機溶劑的混合液等)而成的無菌性溶液或者懸浮液進行提供。本發明的試劑可以適當含有本身公知的生理上允許的添加劑(例如:賦形劑、媒介物、防腐劑、穩定劑、粘合劑等)。另外,在將本發明的試劑作為醫藥提供的情況下,對該本發明的試劑而言,可以將本發明的脂質膜結構體直接使用,或者與醫藥上允許的公知的添加劑(例如:載體、香味劑、賦形劑、媒介物、防腐劑、穩定劑、粘合劑等)一起使用,通過以通常認定的制劑實施所要求的單位用量形態進行混合,以口服劑(例如:片劑、膠囊劑等)或者非口服劑(例如注射劑、噴霧劑等)的形式,優選為非口服劑(更優選注射劑)的形式進行制造。本發明的試劑除了成人用以外,也可以作為兒童用的制劑。本發明的試劑也能夠以試劑盒的形態進行提供。該試劑盒除了本發明的脂質膜結構體及核酸此外,可以包含向細胞導入核酸時使用的試劑。在一種形態中,本發明的試劑(或試劑盒)可以進一步包含聚陽離子(例如:魚精蛋白等)。本發明的試劑(或試劑盒),可以通過進一步包含聚陽離子(例如:魚精蛋白等),容易地將核酸與聚陽離子(例如:魚精蛋白等)而成的靜電復合體導入至本發明的脂質膜結構體而制備mend。該mend可用于核酸的細胞內導入。實施例以下,舉出實施例對本發明進行詳細說明,但本發明不限于該實施例。在實施例的說明中使用的簡稱的含義,分別如下所述。lin-ms:甲磺酸亞油酰酯mim:3,4-o-異亞丙基-d-甘露醇tlmim:1,2,5,6-四亞油酰基-3,4-o-異亞丙基-d-甘露醇tlm:1,2,5,6-四亞油酰基-d-甘露醇tlm-c2-br:1,2,5,6-四亞油酰基-3,4-二(溴乙酰基)-d-甘露醇dmap:4-二甲基氨基吡啶chol:膽甾醇peg2000-dmg:1,2-二肉豆蔻酰-sn-丙三醇,甲氧基聚乙二醇(peg分子量:2000)decenyl-ms:甲基磺酸癸烯基酯tdmim:1,2,5,6-四(癸烯基)-3,4-o-異亞丙基-d-甘露醇tdm:1,2,5,6-四(癸烯基)-d-甘露醇tbdps-cl:叔丁基二苯基氯硅烷dipea:二異丙基乙基胺dtbdps-m:1,6-二-(叔丁基二苯基甲硅烷基)-d-甘露醇dtbdps-tlmes:1,6-二-(叔丁基二苯基甲硅烷基)-2,3,4,5-四亞油酰基-d-甘露醇tlmes:四亞油酰基-d-甘露醇tbaf:四丁基氟化銨在表2及表3中,示出了以下的實施例及比較例中制造的陽離子脂質的名稱和結構。[表2][表3][制造例1]<甲硅烷基保護>dtbdps-m的合成向d-甘露醇(東京化成工業株式會社制)3.0g(16.5mmol)中添加n,n-二甲基甲酰胺48ml及dipea(關東化學株式會社制)6.4g(49.4mmol),一邊攪拌一邊冷卻至0~10℃。向其中滴加添加將tbdps-cl(東京化成工業株式會社制)13.6g(49.4mmol)溶解于n,n-二甲基甲酰胺16ml而成的溶液并使溫度不超過10℃。在滴加結束后,升溫至20℃,攪拌2.5小時。通過tlc分析(展開溶劑:氯仿/甲醇=9/1(v/v),以過錳酸鉀顯色)確認了d-甘露醇及單甲硅烷基體消失,結束反應。向反應溶液加入離子交換水120ml及甲苯60ml,攪拌10分鐘,之后靜置10分鐘使其分層。用離子交換水40ml再次水洗得到的甲苯層之后,濃縮了甲苯層。將得到的濃縮物質溶解于乙腈170ml之后,利用己烷170ml進行5次提取純化。蒸餾除去得到的乙腈層的溶劑,得到了9.2g的dtbdps-m。通過1h-nmr(600mhz,cdcl3)分析得到的dtbdps-m,確認為目的物質。δ1.03ppm(s,18h,(ch3-)3c-),δ3.79-3.89ppm(m,8h,-o-ch2-ch(-oh)-ch(-oh)-),δ7.25-7.72ppm(m,20h,tbu-si(-ph)2-)[實施例1](tlm-c2-dma的合成)<甲磺酰化>lin-ms的合成向脫水甲苯500g加入亞油酸醇100g(日油株式會社制,純度≥99%)(0.38mol)、三乙胺(關東化學株式會社制)46g(0.45mol)并溶解,在氮氣體氛圍中一邊攪拌一邊冷卻至10℃。以溫度為30℃以下的方式,經2小時滴加了甲磺酰氯(關東化學株式會社制)47g(0.41mol)。滴加結束后,通過tlc分析(展開溶劑:氯仿,磷酸硫酸銅顯色)確認了亞油酸醇的點已消失。添加乙醇5.2g(0.11mol),通過濾紙濾去不溶物。利用離子交換水150g洗滌濾液,并廢棄水層。再次進行水洗之后,向得到的有機層添加無水硫酸鎂20g進行脫水處理。通過濾紙濾去不溶物,用蒸發儀蒸餾除去濾液的溶劑,得到了120g的lin-ms。通過1h-nmr(600mhz,cdcl3)分析得到的lin-ms,確認為目的物質。δ0.89ppm(t,3h,ch3-ch2-),δ1.41-1.26ppm(m,16h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.75ppm(quint,2h,-ch2-ch2-o-),δ2.05ppm(q,4h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.77ppm(t,2h,-ch=ch-ch2-ch=ch-),δ3.00ppm(s,3h,-so2-ch3),δ4.22ppm(t,2h,-ch2-o-),δ5.41-5.31ppm(m,4h,-ch=ch-ch2-ch=ch-)<醚化>tlmim的合成向甲苯40g添加mim(東京化成工業株式會社制)2.0g(9.00mmol),進一步添加氫氧化鉀(關東化學株式會社制)4.2g(74.50mmol)、lin-ms18.6g(54.00mmol),在25℃攪拌5分鐘。隨后升溫至80℃,攪拌了14小時。根據1h-nmr分析,確認源自反應產物的峰出現,源自lin-ms峰的積分值的減少停止,并終止反應。向反應溶液添加甲苯60ml和離子交換水100ml,在20℃攪拌10分鐘,之后靜置10分鐘進行分層。除去水層之后,再次進行水洗。接著添加25wt%的食鹽水100ml,攪拌10分鐘,之后靜置10分鐘進行分層,除去水層。向得到的有機層添加無水硫酸鎂2.0g,進行脫水處理。通過濾紙濾去不溶成分,用蒸發儀蒸餾除去濾液的溶劑,得到了褐色液體11.5g。將得到的褐色液體10g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=100/0~98/2(v/v)),得到了tlmim3.0g。通過1hnmr(600mhz,cdcl3)分析得到的tlmim,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.38-1.29ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.38ppm(s,6h,-o-c(ch3)2-o-),δ1.56ppm(m,8h,-ch2-ch2-o-),δ2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),δ3.54-3.41ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-c(ch3)2-),δ3.67ppm(m,4h,-o-ch2-ch-ch-o-c(ch3)2-),δ4.06ppm(d,2h,-o-ch2-ch-ch-o-c(ch3)2-),δ5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-)<脫保護>tlm的合成向tlmim6.7g(5.51mmol)添加乙醇67ml、離子交換水4.0g(220.40mmol)、鹽酸(4.0m二噁烷溶液)(東京化成工業株式會社制)13.8ml(以鹽酸計為55.10mmol),在60℃攪拌6h。通過tlc分析(展開溶劑:氯仿/甲醇=99.5/0.5(v/v),磷酸硫酸銅顯色),確認tlmim的點已消失,結束反應。一邊將反應液冷卻至5℃一邊靜置13h進行分層,回收了有機層。回收的有機層利用氮氣鼓泡而蒸餾除去溶劑,得到了微褐色液體5.1g。將得到的微褐色液體4.6g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=98/2~9/1(v/v)),得到了tlm3.5g。通過1hnmr(600mhz,cdcl3)分析得到的tlm,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.38-1.28ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.56ppm(m,8h,-ch2-ch2-o-),2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),3.30ppm(d,2h,-oh),3.51-3.42ppm(m,6h,-o-ch2-ch-ch-oh,-o-ch2-ch-ch-oh),3.68-3.52ppm(m,8h,-ch2-ch2-o-),3.82ppm(d,2h,-o-ch2-ch-ch-oh),5.39-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-)<酯化>tlm-c2-br的合成將tlm1.0g(0.85mmol)和溴乙酸(東京化成工業株式會社制)354.5mg(2.55mmol)溶解于氯仿10ml后,添加dmap(廣榮化學工業株式會社制)51.9mg(0.43mmol)、dic(東京化成工業株式會社制)321.8mg(2.55mmol),在25℃攪拌1小時。通過tlc分析(展開溶劑:僅氯仿,磷酸硫酸銅顯色),確認tlm的點已消失。隨后,利用離子交換水10ml、25wt%食鹽水10ml洗滌了反應液,并回收有機層。向回收的有機層添加無水硫酸鎂1.0g進行脫水處理,通過濾紙過濾分離不溶成分。用蒸發儀蒸餾除去濾液的溶劑,得到了微褐色液體1.6g。將得到的微褐色液體利用硅膠色譜進行純化(洗脫液:己烷/乙酸乙酯=100/0~97/3(v/v)),得到了tlm-c2-br980mg。通過1hnmr(600mhz,cdcl3)分析得到的tlm-c2-br,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),1.38-1.27ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),1.52ppm(m,8h,-ch2-ch2-o-),2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),3.51-3.37ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-co-),3.60ppm(m,4h,-o-ch2-ch-ch-o-co-),3.84ppm(s,4h,-o-co-ch2-),5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-),5.51ppm(d,2h,-o-ch2-ch-ch-o-co-)<氨基化>tlm-c2-dma的合成將tlm-c2-br300mg(0.21mmol)溶解于thf2.2ml之后,添加二甲基胺(2.0mthf溶液)(東京化成工業株式會社制)846μl(以二甲基胺計1.68mmol),在25℃攪拌4小時。通過tlc分析(展開溶劑:氯仿/甲醇=96/4(v/v),磷酸硫酸銅顯色),確認了反應產物的點出現,以及作為原料的tlm-c2-br的點消失。向反應溶液添加氯仿8ml、離子交換水10ml,攪拌10分鐘后,靜置10分鐘進行分層,除去水層。隨后,進行利用離子交換水10ml的水洗3次,利用25wt%食鹽水10ml的水洗一次,回收有機層。向回收的有機層添加無水硫酸鎂0.5g,進行脫水處理。通過濾紙濾去不溶成分,用蒸發儀蒸餾除去濾液的溶劑,得到了tlm-c2-dma260mg。通過1hnmr(600mhz,cdcl3)分析得到的tlm-c2-dma,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),1.38-1.27ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),1.52ppm(m,8h,-ch2-ch2-o-),2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),2.35ppm(s,12h,-n-(ch3)2),2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),3.16ppm(q,4h,-o-co-ch2-),3.49-3.35ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-co-),3.55ppm(m,4h,-o-ch2-ch-ch-o-co-),5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-),5.47ppm(d,2h,-o-ch2-ch-ch-o-co-)[實施例2](tlm-c3-dma的合成)<酯化>將tlm1.0g(0.85mmol)和二甲基氨基丙酸鹽酸鹽(東京化成工業株式會社制)783.8mg(5.10mmol)溶解于氯仿10ml之后,添加dmap51.9mg(0.43mmol)、dcc(tama化學工業株式會社制)1.1g(5.10mmol)、在25±5℃攪拌1小時。通過tlc分析(展開溶劑:氯仿/甲醇=85/15(v/v),磷酸硫酸銅顯色),確認tlm的點已消失。通過濾紙濾去反應液中的不溶物,向得到的濾液添加氯仿10ml、離子交換水20ml、甲醇30ml洗滌,回收有機層。進一步向有機層添加離子交換水20ml、甲醇40ml洗滌,回收有機層。向回收的有機層添加無水硫酸鎂1.0g,進行脫水處理。通過濾紙濾去不溶成分,用蒸發儀蒸餾除去濾液的溶劑,得到了微褐色液體1.6g。將得到的微褐色液體利用硅膠色譜進行純化(洗脫液:氯仿/甲醇=98/2~96/4(v/v)),得到了tlm-c3-dma102mg。通過1hnmr(600mhz,cdcl3)分析得到的tlm-c3-dma,確認為目的物質。δ0.89(t,12h,ch3-ch2-),δ1.38-1.27(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.52(m,8h,-ch2-ch2-o-),δ2.05(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.23(s,12h,-n-(ch3)2),δ2.49(t,4h,-o-co-ch2-ch2-),δ2.60(m,4h,-o-co-ch2-ch2-),δ2.77(t,8h,-ch=ch-ch2-ch=ch-),δ3.43(m,8h,-ch2-ch2-o-),δ3.53(m,6h,-o-ch2-ch-ch-o-co-,-o-ch2-ch-ch-o-co-),δ5.39-5.30(m,18h,-ch=ch-ch2-ch=ch-,-o-ch2-ch-ch-o-co-)[實施例3](tlm-c4-dma的合成)<酯化>將tlm0.5g(0.43mmol)和二甲基氨基丁酸鹽酸鹽(acrosorganics;thermofisherscientific公司制)427.6mg(2.55mmol)溶解于氯仿5ml之后,添加51.9mg的dmap(0.43mmol)、321.8mg的dic(tama2.55mmol),在25℃攪拌4小時。通過tlc分析(展開溶劑:氯仿/甲醇=85/15(v/v),磷酸硫酸銅顯色),確認tlm的點已消失。隨后,向反應液添加離子交換水5ml、乙醇5ml洗滌,回收有機層。再次進行相同的洗滌,通過向回收的有機層添加硫酸鎂0.5g進行脫水處理。通過濾紙濾去不溶成分,用蒸發儀蒸餾除去濾液的溶劑,得到了微褐色液體515.8mg。將得到的微褐色液體中的400mg利用硅膠色譜進行純化(洗脫液:氯仿/甲醇=96/4~8/2(v/v)),得到了tlm-c4-dma244mg。通過1hnmr(600mhz,cdcl3)分析得到的tlm-c4-dma,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.38-1.27ppm(m,64h,ch3-ch2-ch2-ch2-,-ch=ch-ch2-ch2-ch2-ch2-ch2-ch2-),δ1.53ppm(m,8h,-ch2-ch2-o-),δ1.77ppm(quint,4h,-ch2-ch2-ch2-n(ch3)2),δ2.05ppm(q,16h,-ch2-ch=ch-ch2-ch=ch-ch2-),δ2.21ppm(s,12h,-n-(ch3)2),δ2.28ppm(t,4h,-ch2-ch2-ch2-n(ch3)2),δ2.34ppm(m,4h,-ch2-ch2-ch2-n(ch3)2),δ2.77ppm(t,8h,-ch=ch-ch2-ch=ch-),δ3.54-3.38ppm(m,14h,ch2-ch2-o-,-ch2-ch-ch-o-co-,-ch2-ch-ch-o-co-),δ5.40-5.32ppm(m,18h,-ch=ch-ch2-ch=ch-,-ch2-ch-ch-o-co-)[實施例4](tdm-c3-dma的合成)<甲磺酰化>decenyl-ms的合成向脫水甲苯50g添加癸烯醇(aldrich公司制)10.0g(64.0mol)、三乙胺7.8g(76.8mol)并使其溶解,在氮氣體氛圍中一邊攪拌一邊冷卻至10℃。以溫度為30℃以下的方式,經30分鐘滴加了甲磺酰氯8.1g(70.4mol)。滴加結束后,通過tlc分析(展開溶劑:氯仿,磷酸硫酸銅顯色)確認了癸烯醇的點已消失。添加乙醇0.9g(19.2mol),通過濾紙濾去不溶物。利用離子交換水20g洗滌濾液,并廢棄水層。再次進行水洗之后,向得到的有機層添加無水硫酸鎂5g進行脫水處理。通過濾紙濾去不溶物,用蒸發儀蒸餾除去濾液的溶劑,得到了decenyl-ms14.5g。通過1h-nmr(600mhz,cdcl3)分析得到的decenyl-ms,確認為目的物質。δ0.89ppm(t,3h,ch3-ch2-),δ1.26-1.36ppm(m,6h,ch3-ch2-ch2-ch2-),δ1.81ppm(quint,2h,-ch2-ch2-o-),δ2.02ppm(q,2h,ch3-ch2-ch2-ch2-ch=),δ2.16ppm(q,2h,=ch-ch2-ch2-ch2-o-),δ3.00ppm(s,3h,-so2-ch3),δ4.23ppm(t,2h,-ch2-o-),δ5.32ppm,δ5.45ppm(q,2h,-ch=ch-)<醚化>tdmim的合成向mim1.8g(8.1mmol)添加甲苯36g,進一步添加氫氧化鉀3.6g(64.8mmol)、decenyl-ms11.4g(48.6mmol),在25℃攪拌5分鐘。隨后升溫至80℃,攪拌14小時。通過tlc分析(展開溶劑:氯仿,磷酸硫酸銅顯色),確認decenyl-ms的殘存量變得低于10%,結束反應。向反應溶液添加甲苯42ml和離子交換水72ml,在20℃攪拌10分鐘,之后靜置10分鐘進行分層。除去水層之后,再次進行水洗。接著添加20wt%的食鹽水72ml,攪拌10分鐘,之后靜置10分鐘進行分層,除去水層。向得到的有機層添加無水硫酸鎂3.6g,進行脫水處理。通過濾紙濾去不溶成分,用蒸發儀蒸餾除去濾液的溶劑,得到了褐色液體7.5g。將得到的褐色液體7.5g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=100/0~98.5/1.5(v/v)),得到了tdmim5.3g。通過1h-nmr(600mhz,cdcl3)分析得到的tdmim,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.26-1.38ppm(m,24h,ch3-ch2-ch2-ch2-),δ1.38ppm(s,6h,-o-c(ch3)2-o-),δ1.63ppm(m,8h,-ch2-ch2-o-),δ2.02ppm(q,8h,ch3-ch2-ch2-ch2-ch2-),δ2.77ppm(t,8h,-ch2-ch2-ch2-o-),δ3.54-3.41ppm(m,10h,-ch2-ch2-o-,-o-ch2-ch-ch-o-c(ch3)2-),δ3.67ppm(m,4h,-o-ch2-ch-ch-o-c(ch3)2-),δ4.06ppm(d,2h,-o-ch2-ch-ch-o-c(ch3)2-),δ5.40-5.31ppm(m,16h,-ch=ch-ch2-ch=ch-)<脫保護>tdm的合成向tdmim5.0g(6.5mmol)添加乙醇50ml、離子交換水4.6g(258.0mmol)、鹽酸(4m二噁烷溶液)16.1ml(64.5mmol),在60℃攪拌3小時。通過tlc分析(展開溶劑:氯仿/甲醇=99.5/0.5(v/v),磷酸硫酸銅顯色),確認tdmim和作為中間體的單異亞丙基體消失,結束反應。向反應液添加己烷50ml,在25℃攪拌10分鐘,之后靜置10分鐘進行分層。回收上層(己烷層),向其中添加乙腈50ml。在25℃攪拌10分鐘,之后靜置10分鐘進行分層。除去乙腈層之后,再次進行乙腈洗滌。蒸餾除去得到的己烷層的溶劑,得到了微褐色液體4.1g。將得到的微褐色液體4.0g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=98/2~95/5(v/v)),得到了tdm2.6g。通過1h-nmr(600mhz,cdcl3)分析得到的tdm,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.25-1.35ppm(m,24h,ch3-(ch2)3-),δ1.63ppm(m,8h,-ch2-ch2-o-),2.01ppm(q,8h,ch3-(ch2)3-ch2-),δ2.77ppm(t,8h,-ch2-ch2-ch2-o-),3.26-3.84ppm(m,18h,-o-ch2-ch-ch-oh,-ch2-ch2-o-),5.33-5.38ppm(m,8h,-ch=ch-)<酯化>tdm二丙烯酸酯體的合成向脫水甲苯5.0g添加tdm500mg(0.68mmol)、三乙胺275mg(2.72mmol)并攪拌。向其中滴加了溶解于脫水甲苯1.0g的丙烯酰氯246mg(2.72mmol)。在25℃攪拌1h,之后過濾析出物質,得到了tdm二丙烯酸酯體的甲苯溶液。<氨基化>向tdm二丙烯酸酯體的甲苯溶液添加2.0m二甲基胺/四氫呋喃溶液1.7ml(二甲基胺3.40mmol),在70℃攪拌1h。反應溶液冷卻至25℃,添加10wt%食鹽水5.0g,攪拌10分鐘,之后靜置10分鐘進行分層。除去下層(水層),向上層(甲苯層)添加25wt%食鹽水5.0g,攪拌10分鐘,之后靜置10分鐘進行分層。除去下層(水層),利用無水硫酸鎂500mg使上層(甲苯層)脫水,過濾之后,濃縮濾液,得到了微黃色液體406mg。將得到的微黃色液體中300mg利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=99/1~95/5(v/v)),得到了241mg的tdm-c3-dma。通過1hnmr(600mhz,cdcl3)分析得到的tdm-c3-dma,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.27-1.38ppm(m,24h,ch3-(ch2)3-),δ1.63ppm(m,8h,-ch2-ch2-o-),δ2.01ppm(q,8h,ch3-(ch2)3-ch2-),δ2.23ppm(s,12h,-n-(ch3)2),δ2.49ppm(t,4h,-o-co-ch2-ch2-),δ2.60ppm(m,4h,-o-co-ch2-ch2-),δ2.77ppm(t,8h,-ch2-ch2-ch2-o-),δ3.43(m,8h,-ch2-ch2-o-),δ3.53ppm(m,6h,-o-ch2-ch-ch-o-co-),δ5.39-5.30ppm(m,10h,-ch=ch-,-o-ch2-ch-ch-o-co-)[實施例5](tlmes-c3-dma的合成)<酯化>dtbdps-tlmes的合成向氯仿45ml加入dtbdps-m4.0g(6.1mmol)、亞油酸(日油公司制,純度≥99%)9.4g(33.4mmol)、0.7g的dmap(6.1mmol),使其溶解。向其中添加7.6g的edc(39.5mmol),在30℃攪拌5小時。通過tlc分析(展開溶劑:氯仿,過磷酸硫酸銅顯色),確認dtbdps-m及作為中間體的單~三酯體消失,結束反應。從反應溶液蒸餾除去溶劑之后,溶解于己烷60ml中。向己烷溶液添加乙腈30ml,在25℃攪拌10分鐘,之后靜置10分鐘,進行分層。回收己烷層,通過蒸餾除去溶劑,得到微黃色液體11.1g。將得到的微黃色液體10.0g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=99.5/0.5~99/1(v/v)),得到了6.8g的dtbdps-tlmes。通過1h-nmr(600mhz,cdcl3)分析得到的dtbdps-tlmes,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.03ppm(s,18h,(ch3-)3c-),δ1.25-1.37ppm(m,64h,ch3-(ch2)3-,=ch-(ch2)4-ch2-ch2-),δ1.47-1.54ppm(m,8h,-ch2-ch2-co-o-),δ2.04ppm(q,16h,-ch2-ch2-ch=ch-),δ2.11-2.32(m,8h,ch2-co-o-),δ2.77ppm(t,8h,=ch-ch2-ch=),δ3.62-3.74ppm(m,4h,-o-ch2-ch-),δ4.99ppm(m,2h,-o-ch2-ch-ch-),δ5.32-5.39ppm(m,16h,-ch=ch-ch2-ch=ch-),δ5.57ppm(m,2h,-o-ch2-ch-ch-),δ7.33-7.63ppm(m,20h,tbu-si(-ph)2-)<脫保護>tlmes的合成將3.00g的dtbdps-tlmes(1.8mmol)溶解于四氫呋喃中,一邊攪拌一邊冷卻至5℃。通過以不超過10℃的方式進行滴加,依次添加了乙酸(關東化學株式會社制)0.7g(12.3mmol)、及tbaf(1m四氫呋喃溶液)(東京化成工業株式會社制)10.5ml(10.5mmol)。滴加后,在25℃攪拌7小時。通過tlc分析(展開溶劑:氯仿,磷酸硫酸銅顯色),確認dtbdps-tlmes及作為中間體的單甲硅烷基體消失,結束反應。用氯仿30ml稀釋反應溶液之后,添加5wt%碳酸氫鈉水溶液30ml,在25℃攪拌10分鐘。攪拌后靜置10分鐘進行分層,回收有機層。得到的有機層進一步利用離子交換水30ml進行水洗,之后蒸餾除去溶劑,得到微黃色液體3.0g。將得到的微黃色液體2.8g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=97/3~80/20(v/v)),得到了1.9g的tlmes。通過1h-nmr(600mhz,cdcl3)分析得到的tlmes,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.25-1.37ppm(m,64h,ch3-(ch2)3-,=ch-(ch2)4-ch2-ch2-),δ1.61ppm(m,8h,-ch2-ch2-co-o-),δ2.04ppm(q,16h,-ch2-ch2-ch=ch-),δ2.26-2.37(m,8h,-ch2-co-o-),δ2.77ppm(t,8h,=ch-ch2-ch=),δ2.90-5.23ppm(m,8h,-o-ch2-ch-ch-),δ5.32-5.39ppm(m,16h,-ch=ch-ch2-ch=ch-)<酯化>tlmes二丙烯酸酯體的合成向脫水甲苯17g添加1.7g的tlmes(1.4mmol)、三乙胺0.6g(5.5mmol),在25℃攪拌。向其中滴加了溶解于脫水甲苯3.4g的丙烯酰氯0.5g(5.5mmol)。在25℃攪拌1h之后,過濾析出物質,得到了tlmes二丙烯酸酯體的甲苯溶液。<氨基化>tlmes-c3-dma的合成向tlmes二丙烯酸酯體的甲苯溶液添加2.0m二甲基胺/四氫呋喃溶液6.9ml(13.8mmol),在70℃攪拌1h。將反應溶液冷卻至25℃,添加10wt%食鹽水17g攪拌10分鐘,之后靜置10分鐘進行分層。除去下層(水層),向上層(甲苯層)添加25wt%食鹽水17g,攪拌10分鐘,之后靜置10分鐘進行分層。除去下層(水層),利用無水硫酸鎂1.0g使上層(甲苯層)脫水,過濾之后,濃縮濾液,得到了微黃色液體1.4g。將得到的微黃色液體1.4g利用硅膠柱色譜進行純化(洗脫液:己烷/乙酸乙酯=99/1~97/3(v/v)),得到了0.5g的tlmes-c3-dma。通過1h-nmr(600mhz,cdcl3)分析得到的tlmes-c3-dma,確認為目的物質。δ0.89ppm(t,12h,ch3-ch2-),δ1.25-1.37ppm(m,64h,ch3-(ch2)3-,=ch-(ch2)4-ch2-ch2-),δ1.61ppm(m,8h,-ch2-ch2-co-o-),δ2.06ppm(q,16h,-ch2-ch2-ch=ch-),δ2.22-2.36(m,20h,-(ch2)6-ch2-co-o-,(ch3)2-n-),δ2.47-2.64ppm(m,8h,(ch3)2-n-(ch2)2-co-o-),δ2.78ppm(t,8h,=ch-ch2-ch=),δ4.04ppm,δ4.30ppm,δ5.11ppm,δ5.47ppm(m,8h,-o-ch2-ch-ch-),δ5.32-5.39ppm(m,16h,-ch=ch-ch2-ch=ch-)[實施例6]各種mend的制備(1)由sirna和魚精蛋白構成的核酸靜電復合體的形成將sirna(hokkaidosystemscience株式會社)以2mg/ml、4mg/ml的方式溶解于ultrapurednase/rnase-freedistilledwater(invitrogen;thermofischerscientific公司),制備了sirna溶液。作為載體的核,將該sirna溶液、魚精蛋白溶液(calbiochem;merckmillipore公司)用10mmhepes緩沖液分別稀釋至0.3mg/ml、0.2mg/ml,一邊攪拌0.3mg/mlsirna250μl一邊每次少量滴加0.2mg/ml魚精蛋白250μl,制備了sirna與魚精蛋白的靜電復合體(以下,稱為“sirna復合體”)(n/p比=1.0)。作為針對factorvii(以下,稱為“fvii”)的sirna的序列,使用了akincetal.,moleculartherapy,17(5),872-879(may2009)中記載的序列(未經化學修飾)。(2)封裝sirna的mend的制備(作為陽離子脂質,單獨使用了tlm-c2-dma、tlm-c3-dma、tlm-c4-dma、tdm-c3-dma、tlmes-c3-dma、dlindap、dodap)將陽離子脂質(tlm-c2-dma(實施例1)、tlm-c3-dma(實施例2)、tlm-c4-dma(實施例3)、tdm-c3-dma(實施例4)、tlmes-c3-dma(實施例5)、dlindap(比較例1)、dodap(比較例2))的90%丁醇溶液和chol溶液,以陽離子脂質:chol=7:3的摩爾比,以總脂質為3000nmol的方式在1.7ml管中混合。進一步,將作為peg脂質的peg2000-dmg溶液以相對于總脂質的3mol%進行添加,分別以使得總量為400μl的方式添加90%丁醇,制成了脂質溶液。在另外的1.7ml管中,將sirna復合體(sirna含量:160μg)與包含130mmnacl的20mm檸檬酸緩沖液(ph4)進行混合,使得總計114μl,制備了sirna溶液。一邊通過渦旋震蕩混合器攪拌脂質溶液,一邊添加sirna溶液進行混合。用1ml進樣針(27g)取該混合溶液的總量,緩慢注入正在劇烈攪拌的檸檬酸緩沖液2ml中(5ml管)。用磷酸緩沖生理鹽水(以下,稱為“pbs”)進行稀釋,使用amiconultra-15100kdevice(merckmillipore公司),進行超濾(1000g,15分鐘,30℃)而濃縮。隨后,用pbs進行稀釋,再次進行超濾而濃縮。最后,用pbs調整使得形成為目標脂質濃度,得到了封裝sirna的mend。通過該操作制備的mend根據使用的陽離子脂質不同,以下稱為“tlm-c2-dmamend”、“tlm-c3-dmamend”、“tlm-c4-dmamend”、“tdm-c3-dmamend”、“tlmes-c3-dmamend”、“dlindapmend”、“dodapmend”。(3)封裝sirna的mend的制備(作為陽離子脂質,混合tlm-c2-dma、tlm-c3-dma、tlm-c4-dma中的2種進行使用)將tlm-c2-dma(實施例1)、tlm-c3-dma(實施例2)、tlm-c4-dma(實施例3)的以表4所述的摩爾比混合而成的脂質的90%丁醇溶液(tlm-cx-dmamix),分別以tlm-cx-dmamix:chol=7:3的摩爾比,以總脂質為3000nmol的方式在1.7ml管中混合。進一步,將作為peg脂質的peg2000-dmg以相對于總脂質的3mol%的量進行添加,分別以使得總量為400μl的方式添加90%丁醇,制成了脂質溶液。在另外的1.7ml管中,將sirna復合體(sirna含量:160μg)與10mm的蘋果酸緩沖液(ph7.4)進行混合使得總計50μl,制備了sirna溶液。一邊用渦旋震蕩混合器攪拌脂質溶液,一邊添加sirna溶液進行混合。用1ml進樣針(27g)取該混合溶液的總量,緩慢注入正在劇烈攪拌的蘋果酸緩沖液2ml中(5ml管)。用pbs進行稀釋,使用amiconultra-15100kdevice(merckmillipore公司)進行超濾(1000g,10分鐘,30℃)而濃縮。隨后,用pbs進行稀釋,再次進行超濾而濃縮。最后,用pbs調整溶液使得形成為目標脂質濃度,得到了封裝sirna的mend。通過該操作制備的mend,如表4記載所述,根據使用的陽離子脂質的比率不同,以下稱為“第1mend”、“第2mend”、“第3mend”、“第4mend”。[表4]mend名稱脂質混合比第1mendtlm-c2-dma:tlm-c3-dma=0.21:0.79第2mendtlm-c3-dma:tlm-c4-dma=0.75:0.25第3mendtlm-c3-dma:tlm-c4-dma=0.50:0.50第4mendtlm-c3-dma:tlm-c4-dma=0.25:0.75(4)利用乙醇稀釋法的mrna封裝mend的制備(分別使用tlm-c3-dma、tdm-c3-dma)將陽離子脂質(tlm-c3-dma(實施例2)、tdm-c3-dma(實施例4))的99.5%乙醇溶液以陽離子脂質:dope:chol=3:3:4的比率,以總脂質量為131nmol的方式在5ml管中混合。另外,將作為peg脂質的peg2000-dmg以相對于總脂質的3mol%的量進行添加,使得總量為30ml。準備了這樣的4管。另外在另外準備的1.5ml管中,添加編碼熒光素酶的mrna(用mmessagemmachinet7ultratranscriptionkit(lifetechnologies公司)制備)3mg,這里添加包含30mmnacl的20mm蘋果酸緩沖液(ph3.0)使得總量為45ml,此溶液也與脂質溶液同樣準備了4管。將脂質溶液一邊渦旋震蕩一邊與mrna溶液混合,接著添加925ml的100mm2-嗎啉代乙磺酸(以下,稱為“mes”)緩沖液(ph5.5)進行混合,并潷析進入預先添加了2ml的mes緩沖液的vivaspinturbo15(sartorius公司制。以下,稱為“vivaspin”。)中。進一步向該5ml管添加2ml的mes緩沖液,并渦旋震蕩進行洗滌,相同地潷析進入vivaspin。該操作重復4次,最后向vivaspin直接添加2ml的mes緩沖液,在25℃,1000g離心進行超濾。進一步添加pbs充分稀釋,在相同條件下進行超濾。用pbs調整該溶液使其成為目標濃度,得到了mrna封裝mend。通過該操作制備的mend根據使用的陽離子脂質不同,以下稱為“tlm-c3-dmammend”、“tdm-c3-dmammend”。作為編碼熒光素酶的mrna的序列,使用了miuraetal.,nucleicacidsresearch,43(3),1317-1331(2015)的“supplementarydata”中記載的序列。[實施例7]各種mend的粒徑、多分散度、表面電位、sirna封裝率、sirna回收率的測定對各種mend的粒徑、多分散度以及表面電位用動態光散射法(zetasizernano;malverninstrumentsltd.)進行了測定。對sirna封裝率及sirna回收率而言,用ribogreen(invitrogen;thermofischerscientific公司)進行了測定。將實施例6(2)或(3)中制備的各種mend用10mmhepes緩沖液(ph7.4)稀釋成1000ng/ml,將其制成樣品溶液。另外,將mend的制備中使用的sirna復合體用10mmhepes緩沖液(ph7.4)階梯性稀釋成0~2000ng/ml,將其制成標準曲線溶液。在這些溶液之外,準備了用10mmhepes緩沖液將葡聚糖硫酸、tritonx-100、ribogreen分別稀釋成0.08mg/ml、0.4%、5μl/ml而成的測定溶液。另外,也準備了用10mmhepes緩沖液替換tritonx-100而成的溶液。向96孔板添加標準曲線溶液或樣品溶液50μl,進一步分別加入含有或者不含tritonx-100的測定溶液50μl并混合,以700rpm攪拌5分鐘之后,測定了在激發波長500nm及觀測波長525nm的熒光強度。通過用含有tritonx-100的條件下測定時的sirna量除以1000ng/ml,算出了sirna回收率。再從以含有tritonx-100的條件測定時的sirna量減去以不含tritonx-100的條件測定時的sirna量,通過用含有tritonx-100的條件測定時的sirna量除以該值,算出了sirna封裝率。結果示于表5及表6。[表5][表6][實施例8]mend的pka評價準備了符合在ph3.0~10.0的范圍的各種ph的、包含終濃度150mm的nacl的20mm的檸檬酸緩沖液、磷酸鈉緩沖液及trishcl緩沖液。向這些緩沖液中添加了實施例6(2)或(3)中制備的mend使得脂質濃度為30μm,進一步以使得為6μm的方式添加了6-(對甲苯胺基)-2-萘磺酸鈉鹽,使最終容量為100μl。隨后,在37℃測定了在激發波長321nm及觀測波長447nm的熒光強度。以各mend中熒光強度的最大值為100%,最小值為0%,以百分率計,算出相對熒光強度。另外,將相對熒光強度為50%的ph作為pka。結果示于表7及表8。[表7]mend名稱mend的pkatlm-c2-dmamend4.67tlm-c3-dmamend5.89tlm-c4-dmamend6.83tdm-c3-dmamend6.14tlmes-c3-dmamend5.78dlindapmend5.34dodapmend5.44[表8]mend名稱mend的pka第1mend5.81第2mend6.28第3mend6.56第4mend6.74[實施例9]tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend的膜融合能力試驗從雄性icr小鼠采血,回收紅細胞,懸浮于生理鹽水。將包含一定量的紅細胞的生理鹽水,添加至pbs(ph7.4)或10mm磷酸-10mm蘋果酸緩沖生理鹽水(ph6.5、5.5)。接下來,添加含有實施例6(2)制備的mend的pbs溶液,使脂質終濃度為300μmol/l。另外,以添加了不含有mend的相同量的pbs的溶液作為陰性對照(nc),將以下液體作為陽性對照(pc):添加了不含有mend的相同量的pbs之后,添加tritonx-100并使得終濃度為0.02%(w/v)、使紅細胞溶解的液體。將這些在37℃溫育45分鐘,之后在4℃,400×g的條件下離心分離5分鐘,回收上清,通過測定在545nm的吸光度,定量了血紅蛋白從紅細胞的滲出量。接下來,以pc的測定值為100%,以百分率表示各樣品的測定值(溶血活性)。該百分率越高,顯示膜融合能力越高。將結果示于圖1。和dodapmend、dlindapmend比較,tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend顯示了高膜融合能力。尤其是,tlm-c3-dmamend、tlmes-c3-dmamend僅在ph5.5顯示了高膜融合能力。[實施例10]基于第1mend、第2mend、第3mend、第4mend的膜融合能力試驗從雄性icr小鼠采血,回收紅細胞,懸浮于生理鹽水。將pbs調整為ph7.4、6.5、5.5后,添加了包含一定量的紅細胞的生理鹽水。接下來,分別添加了包含實施例6(2)中制備的tlm-c3-dmamend及tlm-c4-dmamend、實施例6(3)中制備的mend的pbs3.3μl、10μl及30μl。另外,以添加了不含有mend的相同量的pbs的溶液作為陰性對照(nc),將添加了不含有mend的相同量的pbs之后添加tritonx-100至0.5%(w/v)的、使紅細胞溶解的液體作為陽性對照(pc)。將這些在37℃溫育30分鐘,之后在4℃,400×g的條件下離心分離5分鐘,回收上清,通過測定在545nm的吸光度,測定了血紅蛋白的量。接下來,以pc的測定值為100%,以百分率表示各測定值(溶血活性)。該百分率越高,顯示膜融合能力越高。將結果示于圖2。在第2mend、第3mend、第4mend中顯示了比tlm-c3-dmamend高的膜融合能力。尤其是,與第3mend、第4mend比較,第2mend僅在ph5.5顯示了高的膜融合能力。[實施例11]tlm-c2-dmamend、tlm-c3-dmamend、tlm-c4-dmamend、dlindapmend、dodapmend的體內敲低活性試驗將包含以實施例6(2)所示的方法制備的mend的溶液,以0.5mg/kg從尾靜脈給予4周齡的雄性小鼠。24小時后采血,通過將該血液樣品以1000g,10分鐘,4℃進行離心,回收上清得到了血漿。對血漿中的factorvii(fvii)的量而言,使用biophenfviichromogenicassay(sysmexbiomed公司)進行定量,將未處理組(nt)的fvii表達量作為1,以相對值(血漿中的相對fvii量)示出了mend給予組的fvii表達量。將結果示于圖3。可觀察到與dlindapmend、dodapmend比較,tlm-c3-dmamend、tlm-c4-dmamend的fvii表達量降低。尤其是,tlm-c3-dmamend顯示了最高的敲低活性。[實施例12]tlm-c3-dmamend、tdm-c3-dmamend、tlmes-c3-dmamend的體內敲低活性試驗將包含以實施例6(2)所示的方法制備的mend的溶液,以0.1mg/kg從尾靜脈給予4周齡的雄性小鼠。24小時后采血,通過將該血液樣品以1000g,10分鐘,4℃進行離心,回收上清得到了血漿。對血漿中factorvii(fvii)的量而言,使用biophenfviichromogenicassay(hyphenbiomed公司)進行定量,將未處理組(nt)的fvii表達量作為1,以相對值(血漿中的相對fvii量)示出了mend給予組的fvii表達量。將結果示于圖4。可見tdm-c3-dmamend、tlmes-c3-dmamend的fvii表達量也降低,與tlm-c3-dmamend比較,顯示出了基本相同的敲低活性。[實施例13]tlm-c3-dmamend、tlm-c4-dmamend、及第1mend、第2mend、第3mend、第4mend的體內敲低活性試驗將以實施例6(2)所示的方法制備的tlm-c3-dmamend的溶液及tlm-c4-dmamend溶液、以實施例6(3)所示的方法制備的mend溶液,以0.1mg/kg從尾靜脈給予4周齡的雄性小鼠。24小時后采血,通過將該血液樣品以1000g,10分鐘,4℃進行離心,回收上清得到了血漿。對血漿中factorvii(fvii)的量而言,使用biophenfviichromogenicassay(sysmexbiomed公司)進行定量,將未處理組(nt)的fvii表達量作為1,以相對值(血漿中的相對fvii量)示出了mend給予組的fvii表達量。將結果示于圖5。在第1mend、第2mend中顯示出了高于tlm-c3-dmamend的敲低活性,第2mend顯示了較高較大的敲低活性。[實施例14]體內的mrna表達試驗將以實施例6(4)所示的方法制備的tlm-c3-dmammend溶液、tdm-c3-dmammend溶液用pbs稀釋至使得mrna為1mg/100ml,將其在頸部進行皮下給予6周齡的雌性icr小鼠。在5.5小時后,將預先制備使得為3mg/200ml/匹的熒光素(vivoglotmluciferin,invivograde,promega公司制)的生理鹽水溶液,從腹腔內給予各小鼠,30分鐘后,利用ivistmluminaii(caliperlifesciences公司)觀察mrna給予部位的發光并進行定量。將結果示于圖6。在tlm-c3-dmammend及tdm-c3-dmammend中均確認到發光,均顯示了mrna表達活性。其中,在tdm-c3-dmammend中顯示最強的發光量,顯示mrna表達活性較高。工業實用性本發明的試劑可以高效率地將功能性核酸遞送至細胞質內,因此對核酸醫藥品開發、生化學實驗有用。本申請以在日本申請的日本特愿2014-166041(申請日:2014年8月18日)為基礎,并將其內容全部包含于本說明書。當前第1頁12
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1