于抗蝕劑應用中作為光酸生成劑的磺酸衍生化合物的制作方法
本發明涉及新穎光酸生成劑化合物(“PAG”)及包含此類PAG化合物的光致抗蝕劑組合物。具體說來,本發明的PAG化合物在有機溶劑中具有極佳溶解度并且在光刻工藝中展現比常規PAG化合物更高的敏感性及更佳的性能。
背景技術:
光致抗蝕劑是用于將圖像轉印至襯底的感光性膜。其形成負或正圖像。將光致抗蝕劑涂布于襯底上后,涂層經由圖案化光掩模曝露于例如紫外光等激活能量源,以在光致抗蝕劑涂層中形成潛像。光掩模具有對激活輻射不透明及透明的區域,這些區域界定打算轉印至底層襯底的圖像。
已證實化學增幅型光致抗蝕劑可用于在半導體制造中用以形成超微細圖案的工藝中達成高敏感性。通過將PAG與具有酸不穩定結構的聚合物基質摻合來制備這些光致抗蝕劑。根據此類光致抗蝕劑的反應機制,光酸生成劑在被光源照射時產生酸,并且在曝光或照射部分中的聚合物基質的主鏈或分支鏈在所謂“曝光后烘烤”(PEB)中與產生的酸反應并分解或交聯,使得聚合物的極性改變。此極性的改變引起在顯影溶液中被照射的曝光區域與未曝光區域之間的溶解度差異,藉此在襯底上形成遮罩的正或負圖像。酸擴散不僅對于增加光致抗蝕劑敏感性及通過量很重要,而且對于限制因散粒噪聲統計所致的線邊緣粗糙度也很重要。
在化學增幅型光致抗蝕劑中,成像所需的溶解度轉換化學物質不是直接由曝光產生;而是曝光產生在后續PEB步驟期間促進溶解度轉換化學反應的穩定催化物質。術語“化學增幅”源于以下事實:每個以光化學方式產生的催化劑分子均可促進許多溶解度轉換反應事件。轉換反應的表觀量子效率是催化劑生成的量子效率乘以平均催化鏈長。初始曝光劑量通過一系列后續化學反應事件而“擴增”。催化劑的催化鏈長可極長(多達數百個反應事件),由此提供明顯曝光增幅。
化學增幅的有利之處在于,其可大大提高抗蝕劑敏感性,但其并非沒有潛在缺點。舉例來說,由于催化劑分子圍繞數百個反應位點移動,故對曝露于成像輻射的區域無必要的限制。抗蝕劑敏感性與成像保真性之間存在潛在權衡。舉例來說,增幅型光致抗蝕劑經由光掩模曝光,在曝光區域中產生酸催化劑。通過提高PEB中的晶片溫度以允許化學反應發生,由此將第一步中產生的潛在性酸圖像轉化成可溶及不可溶區域的圖像。一些酸遷移出初始曝光區域引起“臨界尺寸偏誤”問題。烘烤后,用溶劑使圖像顯影。顯影特征寬度可由于酸自曝光區域擴散至未曝光區域中而大于標稱遮罩尺寸。對于大部分增幅型抗蝕劑的歷史,此權衡極少被關注,因為相對于印刷的特征大小,催化劑擴散距離并不重要,但由于特征大小已減小,擴散距離保持大致相同且催化劑擴散已顯現為重要問題。
為產生改變聚合物溶解度的足夠酸,需要一定曝光時間。對于已知PAG分子,如N-羥基萘二甲酰亞胺三氟甲磺酸酯(“NIT”),此曝光時間相當長(歸因于其在365nm或更長波長下的低吸收)。然而,提高此類PAG的濃度將不會產生較快曝光時間,因為PAG的溶解度為限制因素。另一可能性是增加敏化劑,所述敏化劑吸收光并將能量轉移至PAG,接著其將釋放酸。然而,此類敏化劑必須以相當高濃度使用以便能夠將能量轉移至緊密鄰近的PAG。在此類高濃度下,敏化劑常具有過高的吸收且對顯影后抗蝕劑輪廓的形狀具有負面影響。
因此,在此項技術中需要展現較佳溶解度的PAG,這意味著將較多活性分子賦予至調配物中,其中相較于自現有技術已知的光致抗蝕劑組合物,包含這些化合物的光致抗蝕劑組合物對電磁輻射,尤其對波長為200nm至500nm的電磁輻射具有高敏感性,同時允許產生具有較高分辨率的圖案化結構。
技術實現要素:
本發明通過提供由式(I)或式(II)表示的磺酸衍生化合物來滿足此需求:
其中X為氧(O)或硫(S)原子;
R1、R2、R3、R4及R5各自為氫(H)原子;
R0選自由以下組成的群組:
具有1至3的碳數的脂肪族基團,其中一或多個氫原子可經鹵素原子取代;
具有2至18的碳數的脂肪族基團,其包含至少一個選自由-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb各自獨立地為具有1至10的碳數的脂肪族基團,其可相同或不同并且可連接形成脂環基團,并且其中脂肪族基團任選包含至少一個鹵素原子;及
由式(A)表示的基團:
-R11-Ar(A),
其中
R11為單鍵或具有1至20的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;并且
Ar為芳基或雜芳基,其中一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代;
由式(B)表示的基團:
其中R21及R22各自獨立地為具有1至5的碳數的脂肪族基團;
Y21為氧(O)原子;
R23為具有1至10的碳數的脂肪族基團;并且
R24為具有1至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;
由式(C)表示的基團:
其中R31為具有2至18的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及-O-C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;
Y31為氧(O)原子;
R32為具有1至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;并且
R6選自由以下組成的群組:
具有1至18的碳數的脂肪族基團,其可經一或多個鹵素原子取代;
具有3至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,其中脂肪族基團任選包含至少一個鹵素原子;
具有4至18的碳數的芳基或雜芳基,其中一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代;及
具有4至18的碳數的芳烷基或雜芳烷基,其中芳基或雜芳基中的一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代。
本發明還提供抗蝕劑組合物,其包含成像有效量的一或多種本發明的PAG及樹脂。
本發明還提供用于形成本發明的光致抗蝕劑的凸紋圖像的方法,包括用于形成具有小于四分之一微米尺寸或更小尺寸(例如小于0.2微米或小于0.1微米尺寸)的高分辨率圖案化光致抗蝕劑圖像(例如具有基本上垂直的側壁的圖案化管)的方法。
本發明進一步提供包含襯底的制品,所述襯底例如上面已涂布本發明的光致抗蝕劑及凸紋圖像的微電子晶片或平板顯示器襯底。本發明的其它方面揭示于下文。
附圖說明
當結合附圖閱讀時,自以下實施方式更好地理解本發明。應強調的是,根據慣例,圖式的多種特征不按比例繪制。相反,為清楚起見,可任意擴大或減小多種特征的尺寸。圖式中包括以下各圖:
圖1為展示本發明的某些PAG化合物的UV吸光度與某些現有技術PAG化合物的UV吸光度的比較的圖;
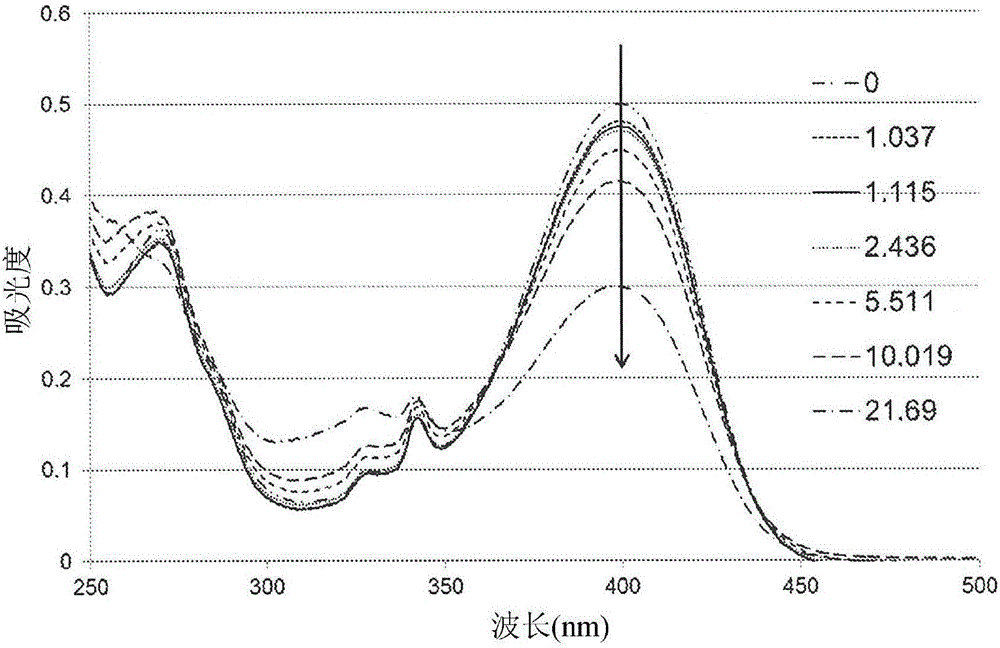
圖2為展示在增加照射的曝露劑量后S-3(本發明化合物之一)的UV-Vis光譜變化的圖,其表明在能量的曝露劑量增加的情況下光反應的進展(各光譜的能量曝露劑量(mJ/cm2)依序展示于圖例中);
圖3展示吸光度隨能量曝露劑量而變化的自然對數圖,由此得到S-3(本發明的PAG化合物)的光反應常數;及
圖4為具有多種線與間隙(L/S)大小(5、6、7、8、9及10μm)的圖案化結構的SEM顯微照片,其中采用S-3(本發明的化合物)作為PAG化合物。
具體實施方式
定義
除非另外陳述,否則以下在本申請案(包括說明書及權利要求書)中所用的術語具有下文給定的定義。應注意,除非上下文另外明確指定,否則如本說明書及隨附權利要求書中所用,單數形式“一(a/an)”及“所述(the)”包括復數個指示物。
所有數值名稱,例如重量、pH、溫度、時間、濃度及分子量,包括范圍在內,都是近似值,具有10%的變化。應理解,盡管未必總是明確陳述,但所有數值名稱的前均有術語“約”。也應理解,盡管未必總是明確陳述,但本文所述的試劑僅為例示性的且其等效物在此項技術中是已知的。
參考本發明,除非具體定義,否則本文的描述中所用的技術及科學術語將具有本領域普通技術人員通常所理解的含義。因此,以下術語意欲具有以下含義。
如本文所用,術語“部分”是指分子的特定片段或官能團。化學部分為通常公認的插入或附加至分子的化學實體。
如本文所用,術語“脂肪族基團”涵蓋術語烷基、烯基、炔基,其各自如下闡述任選經取代。
如本文所用,“烷基”是指含有1-20(例如2-18、3-18、1-8、1-6、1-4或1-3)個碳原子的飽和脂肪族烴基。烷基可為直鏈、分支鏈、環狀或其任何組合。烷基的實例包括(但不限于)甲基、乙基、丙基、異丙基、丁基、異丁基、仲丁基、叔丁基、正戊基、正庚基或2-乙基己基。烷基可如下闡述經一或多個取代基取代(即,任選經取代)或可為多環的。
除非另外具體限制,否則如本文所用,術語“烷基”以及衍生術語,例如“烷氧基”及“硫烷基”在其范圍內包括直鏈、分支鏈及環狀部分。
如本文所用,“烯基”是指含有2-20(例如2-18、2-8、2-6或2-4)個碳原子及至少一個雙鍵的脂肪族碳基。與烷基相同,烯基可為直鏈、分支鏈或環狀,或其任何組合。烯基的實例包括(但不限于)烯丙基、異戊二烯基、2-丁烯基及2-己烯基。烯基可如下闡述任選經一或多個取代基取代。
如本文所用,“炔基”是指含有2-20(例如2-8、2-6或2-4)個碳原子且具有至少一個三鍵的脂肪族碳基。炔基可為直鏈、分支鏈、環狀或其任何組合。炔基的實例包括(但不限于)炔丙基及丁炔基。炔基可如下闡述任選經一或多個取代基取代。
“鹵素”為周期表第17族的原子,其包括氟、氯、溴及碘。
如本文所用,單獨或作為如“芳烷基”、“芳烷氧基”或“芳氧基烷基”的較大部分的一部分使用的“芳基”是指單環系統(例如苯基);雙環系統(例如茚基、萘基、四氫萘基、四氫茚基);及三環系統(例如芴基、四氫芴基或四氫蒽基、蒽基),其中單環系統為芳香族環,或雙環或三環系統中的至少一個環為芳香族環。雙環及三環基團包括苯并稠合2-3元碳環。舉例來說,苯并稠合基團包括與兩個或兩個以上C4-8碳環部分稠合的苯基。芳基如下闡述任選經一或多個取代基取代。
如本文所用,“芳脂肪族”基團,例如“芳烷基”是指經芳基取代的脂肪族基團(例如C1-4烷基)。“脂肪族基團”、“烷基”及“芳基”于本文中定義。芳脂肪族基團,例如芳烷基的實例為苯甲基。
如本文所用,“芳烷基”或“芳基烷基”是指經芳基取代的烷基(例如C1-4烷基)。上文已定義“烷基”及“芳基”兩者。芳烷基的實例為苯甲基。芳烷基如下闡述任選經一或多個取代基取代。
如本文所用,“環脂肪族”基團涵蓋“環烷基”及“環烯基”,其各自如下闡述任選經取代。
如本文所用,“環烷基”是指具有3-10(例如5-10)個碳原子的飽和碳環單或雙環(稠合或橋接)環。環烷基的實例包括環丙基、環丁基、環戊基、環己基、環庚基、金剛烷基、降冰片基、立方烷基(cubyl)、八氫茚基、十氫萘基、雙環[3.2.1]辛基、雙環[2.2.2]辛基、雙環[3.3.1]壬基、雙環[3.3.2.]癸基、雙環[2.2.2]辛基、金剛烷基、氮雜環烷基或((氨基羰基)環烷基)環烷基。
如本文所用,術語“雜芳基”是指具有4至18個環原子的單環、雙環或三環系統,其中一或多個環原子為雜原子(例如N、O、S或其組合)且其中單環系統為芳香族環,或雙環或三環系統中的至少一個環為芳香族環。雜芳基包括具有2至3個環的苯并稠合環系統。舉例來說,苯并稠合基團包括與一或兩個4至8元雜環脂肪族部分稠合的苯基(例如吲哚嗪基、吲哚基、異吲哚基、3H-吲哚基、吲哚啉基、苯并[b]呋喃基、苯并[b]噻吩基、喹啉基或異喹啉基)。雜芳基的一些實例為氮雜環丁烷基、吡啶基、1H-吲唑基、呋喃基、吡咯基、噻吩基、噻唑基、噁唑基、咪唑基、四唑基、苯并呋喃基、異喹啉基、苯并噻唑基、二苯并吡喃、噻噸(thioxanthene)、吩噻嗪、二氫吲哚、苯并[1,3]二氧雜環戊烯、苯并[b]呋喃基、苯并[b]噻吩基、吲唑基、苯并咪唑基、苯并噻唑基、嘌呤基、噌啉基、喹啉基、喹唑啉基、噌啉基、酞嗪基、喹唑啉基、喹喏啉基、異喹啉基、4H-喹嗪基、苯并-1,2,5-噻二唑基或1,8-萘啶基。
非限制性地,單環雜芳基包括呋喃基、噻吩基、2H-吡咯基、吡咯基、噁唑基、噻唑基、咪唑基、吡唑基、異噁唑基、異噻唑基、1,3,4-噻二唑基、2H-吡喃基、4-H-吡喃基、吡啶基、噠嗪基、嘧啶基、吡唑基、吡嗪基或1,3,5-三嗪基。單環雜芳基根據標準化學命名法編號。
非限制性地,雙環雜芳基包括吲哚嗪基、吲哚基、異吲哚基、3H-吲哚基、吲哚啉基、苯并[b]呋喃基、苯并[b]噻吩基、喹啉基、異喹啉基、吲哚嗪基、異吲哚基、吲哚基、苯并[b]呋喃基、苯并[b]噻吩基、吲唑基、苯并咪唑基、苯并噻唑基、嘌呤基、4H-喹嗪基、喹啉基、異喹啉基、噌啉基、酞嗪基、喹唑啉基、喹喏啉基、1,8-萘啶基或蝶啶基。雙環雜芳基根據標準化學命名法編號。
雜芳基如下闡述任選經一或多個取代基取代。
如本文所用,“雜芳基脂肪族基團”(例如雜芳烷基)是指經雜芳基取代的脂肪族基團(例如C1-4烷基)。上文已定義“脂肪族”、“烷基”及“雜芳基”。
如本文所用,“雜芳烷基”是指經雜芳基取代的烷基(例如C1-4烷基)。上文已定義“烷基”及“雜芳基”。雜芳烷基如下闡述任選經一或多個取代基取代。
如本文所用,“酰基”是指甲酰基或RX--C(O)--(例如-烷基-C(O)--,又稱為“烷基羰基”),其中先前已定義“烷基”。
如本文所用,術語“酰氧基”包括直鏈酰氧基、分支鏈酰氧基、環酰氧基、環狀酰氧基、未經雜原子取代的酰氧基、經雜原子取代的酰氧基、未經雜原子取代的Cn-酰氧基、經雜原子取代的Cn-酰氧基、烷基羰氧基、芳基羰氧基、烷氧基羰氧基、芳氧基羰氧基及羧酸基團。
如本文所用,“烷氧基”是指烷基-O--基團,其中先前已定義“烷基”。
如本文所用,當用作末端基團時,“羧基”是指--COOH、--COORX、--OC(O)H、--OC(O)RX;或當用作內部基團時,其是指--OC(O)-或--C(O)O--。
如本文所用,“烷氧基羰基”意謂R為如上文所定義的烷基的--COOR,例如甲氧羰基、乙氧羰基及其類似基團。
如本文所用,“磺酰基”在末端使用時是指--S(O)2--Rx及在內部使用時是指--S(O)2--。
術語“烷硫基”包括直鏈烷硫基、分支鏈烷硫基、環烷基硫基、環狀烷硫基、未經雜原子取代的烷硫基、經雜原子取代的烷硫基、未經雜原子取代的Cn-烷硫基及經雜原子取代的Cn-烷硫基。在某些實施例中,涵蓋低碳數烷硫基。
如本文所用,術語“胺”或“氨基”包括氮原子共價結合到至少一個碳或雜原子的化合物。術語“胺”或“氨基”也包括--NH2并且還包括經取代的部分。所述術語包括“烷基氨基”,其包含氮結合到至少一個另外的烷基的基團及化合物。所述術語包括“二烷基氨基”,其中氮原子結合到至少兩個另外的獨立選擇的烷基。所述術語包括“芳基氨基”及“二芳基氨基”,其中氮分別結合到至少一個或兩個獨立選擇的芳基。
術語“鹵烷基”是指經一個至最大可能數目的鹵素原子取代的烷基。術語“鹵烷氧基”及“鹵硫烷基”是指經一個至五個鹵素原子取代的烷氧基及硫烷基。
詞組“任選經取代”可與詞組“經取代或未經取代”互換使用。如本文所述,本發明的化合物可任選經一或多個取代基取代,例如上文大體上說明或如由本發明的特定類別、亞類及種類所例示。如本文所述,以上部分中的任一者或下文引入的那些部分可任選經一或多個本文所述的取代基取代。特定基團的每一取代基進一步任選經鹵基、氰基、側氧基烷氧基、羥基、氨基、硝基、芳基、鹵烷基及烷基中的一個至三個取代。舉例來說,烷基可經烷硫基取代且烷硫基可任選經鹵基、氰基、側氧基烷氧基、羥基、氨基、硝基、芳基、鹵烷基及烷基中的一個至三個取代。
一般而言,術語“經取代”無論之前是否有術語“任選地”,均是指既定結構中的氫基經指定取代基的基團置換。在上文的定義及下文有關化合物與其實例的描述中描述特定取代基。除非另外指示,否則任選經取代的基團可在基團的每一可取代位置處具有取代基,且當任何既定結構中的一個以上位置可經一個以上選自指定群組的取代基取代時,取代基在每個位置處可相同或不同。環取代基,例如雜環烷基可結合至另一環,例如環烷基,以形成螺-雙環系統,例如兩個環共有一個共用原子。本領域普通技術人員將認識到,本發明設想的取代基組合是使得形成穩定或化學上可行的化合物的組合。
預期本說明書通篇揭示的化合物的改性或衍生物均可用于本發明的方法及組合物。通過本領域技術人員已知的任何方法可制備衍生物并且可以分析此類衍生物的特性中的所要特性。在某些方面中,“衍生”是指仍保持在化學改性之前化合物的所要作用的經化學改性的化合物。
磺酸衍生的光酸生成劑化合物
如下文將更詳細地解釋,本發明的磺酸衍生化合物可用作光酸生成劑。意外地,已發現本發明的PAG化合物的特征在于極佳溶解度及對電磁輻射,特別是對波長在150nm至500nm范圍內、優選在300至450nm范圍內、更優選在350nm至440nm范圍內,更優選波長為365nm(i線)、405(h線)及436nm(g線)的電磁輻射的光反應性。
本發明的磺酸衍生化合物是由式(I)或式(II)表示的N-羥基萘二甲酰亞胺磺酸酯衍生物:
其中X為氧(O)或硫(S)原子;
R1、R2、R3、R4及R5各自為氫(H)原子;
R0選自由以下組成的群組:
具有1至3的碳數的脂肪族基團,其中一或多個氫原子可經鹵素原子取代;
具有2至18的碳數的脂肪族基團,其包含至少一個選自由-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb各自獨立地為具有1至10的碳數的脂肪族基團,其可相同或不同并且可連接形成脂環基團,且其中脂肪族基團任選包含至少一個鹵素原子;及
由式(A)表示的基團:
-R11-Ar(A),
其中R11為單鍵或具有1至20的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;并且
Ar為芳基或雜芳基,其中一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代;
由式(B)表示的基團:
其中R21及R22各自獨立地為具有1至5的碳數的脂肪族基團;
Y21為氧(O)原子;
R23為具有1至10的碳數的脂肪族基團;并且
R24為具有1至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;
由式(C)表示的基團:
其中R31為具有2至18的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及-O-C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分開;
Y31為氧(O)原子;
R32為具有1至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分開;并且
R6選自由以下組成的群組:
具有1至18的碳數的脂肪族基團,其可經一或多個鹵素原子取代;
具有3至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,其中脂肪族基團任選包含至少一個鹵素原子;
具有4至18的碳數的芳基或雜芳基,其中一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代;及
具有4至18的碳數的芳烷基或雜芳烷基,其中芳基或雜芳基中的一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代。
在一些優選實施例中,式(I)及(II)中的R0為具有1至3個碳原子的脂肪族基團。優選實例包括甲基、丙基、異丙基、烯丙基、炔丙基、環丙基、丙烯基、丙炔基及乙炔基。在其它優選實施例中,式(I)及(II)中的R0為具有1至3個碳原子的脂肪族基團并且式(I)及(II)中的R6為具有1至18且優選1至6的碳數的脂肪族基團,其可經一或多個鹵素原子取代。此類優選化合物的實例包括表1中的化合物:
表1
在其它優選實施例中,式(I)及(II)中的R0為具有2至18的碳數的脂肪族基團,其包含至少一個選自由-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb各自獨立地為具有1至10的碳數的脂肪族基團,其可相同或不同并且可連接形成脂環基團,且其中脂肪族基團任選包含至少一個鹵素原子;并且式(I)及(II)中的R6為具有1至18且優選1至6的碳數的脂肪族基團,其可經一或多個鹵素原子取代。此類優選化合物的實例包括表2中的化合物:
表2
在其它優選實施例中,式(I)及(II)中的R0為由式(C)表示的基團:
其中R31為具有2至18的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及-O-C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;Y31為氧(O)原子;R32為具有1至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;并且式(I)及(II)中的R6為具有1至18且優選1至6的碳數的脂肪族基團,其可經一或多個鹵素原子取代。此類優選化合物的實例包括表3中的化合物:
表3
在其它優選實施例中,式(I)及(II)中的R0為由式(A)表示的基團:
-R11-Ar(A),
其中R11為單鍵或具有1至20的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔;Ar為芳基或雜芳基,其中一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代;并且式(I)及(II)中的R6為具有1至18且優選1至6的碳數的脂肪族基團,其可經一或多個鹵素原子取代。此類優選化合物的實例包括表4中的化合物:
表4
在優選實施例中,R11為具有1至20的碳數的脂肪族基團,其可包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-及C(=O)-NRa-組成的群組的部分,其中Ra如上文所定義,并且其中如果至少一個部分是多于一個,則其可由脂肪族基團分隔。
在其它優選實施例中,式(I)及(II)中的R6為具有3至18的碳數的脂肪族基團,其包含至少一個選自由-O-、-S-、-C(=O)-O-、-C(=O)-S-、-O-S(=O)2-、-O-C(=O)-O-、-C(=O)-NH-、-O-C(=O)-NH-、-C(=O)-NRa-、-O-C(=O)-NRa-及-C(=O)-NRaRb組成的群組的部分,其中Ra及Rb如上文所定義,其中脂肪族基團包含雙環部分,或式(I)及(II)中的R6為具有4至18的碳數的芳烷基或雜芳烷基,其中芳基或雜芳基中的一或多個氫原子可經鹵素原子、脂肪族基團、鹵烷基、烷氧基、鹵烷氧基、烷硫基、雙烷基氨基、酰氧基、酰硫基、酰胺基、烷氧基羰基、烷基磺酰基、烷基亞磺酰基、脂環基、雜環基、芳基、烷芳基、氰基或硝基取代;且R0可為如上文所定義的任何基團。此類優選化合物的實例包括表5中的化合物:
表5
在本發明的某些優選實施例中,由式(I)或式(II)表示的磺酸衍生化合物:
其中X為氧(O)或硫(S)原子;R1、R2、R3、R4及R5各自為氫(H)原子;R0為具有1至3的碳數的脂肪族基團,其中一或多個氫原子可經鹵素原子取代;并且R6為具有1至18的碳數的脂肪族基團,其可經一或多個鹵素原子取代。
舉例來說,在這些實施例中,X為S,R0為丙基或異丙基,并且R6為-CF3或-C4F9。舉例來說,在其它實施例中,X為O,R0為丙基或異丙基,并且R6為-CF3或-C4F9。在更優選實施例中,R0為異丙基。此類優選化合物的實例包括表6中的化合物:
表6
如上文中所述,本發明PAG的多個取代基可任選經取代。經取代的部分宜在一或多個可利用的位置經例如以下取代:鹵素,例如F、Cl、Br及/或I;硝基;氰基;磺酰基;烷基,包括C1-16烷基,其中優選C1-6烷基;鹵烷基,例如氟烷基(例如三氟甲基);及全鹵烷基,例如全氟C1-4烷基;烷氧基,包括具有一或多個氧鍵的C1-16烷氧基,其中優選C1-6烷氧基;烯基,包括C2-12烯基,其中優選C2-6烯基;炔基,包括C2-12炔基,其中優選C2-6炔基;芳基,例如苯基或萘基;及經取代的芳基,例如經鹵基、烷氧基、烯基、炔基及/或烷基取代的芳基,優選具有以上關于相應基團所提及的碳原子數目。
參看圖1,本發明的PAG化合物具有在有機溶劑中的極佳溶解度及在汞燈的i線、h線及g線下的強吸收。化合物S-3及S-6在400nm下展現強吸收帶,并且化合物O-12在364nm下顯示強吸收帶。其在汞燈的i線、h線及g線下的吸光度比例如NIT大得多,NIT為現有技術關于性能的商用PAG基準。因此,相對于現有技術,本發明的化合物展現在光刻中作為PAG的較高敏感性及較佳性能。
表7提供包括NIT在內的不同PAG的溶解度及相對光反應性的比較。PAG的感光性完全取決于其光反應性及其產生的酸的強度。當不同PAG產生相同酸時,其相對光反應性直接反映其相對感光性。據報告,具有碳數小于4的烷氧基或烷硫基的化合物的溶解度較差(美國專利第8,680,268號)。然而,本發明人已發現碳數為3的化合物S-3展現比碳數為4的比較化合物A高很多的溶解度。盡管烷氧基或烷硫基中的碳數增加至超過8通常提高PAG化合物的溶解度,但這也導致分子量增加,由此增加用于抗蝕劑調配物中的所述化合物的量。此外,具有長碳鏈的化合物通常比具有短碳鏈的化合物生產成本高。值得注意的是,具有苯基乙炔基的比較化合物B(WO 2014/073409 A1)展現比本發明的化合物(S-3及S-11)低很多的敏感性及溶解度。具有相對光反應性及在丙二醇單甲醚乙酸酯中的溶解度的產物將兩種特性均考慮在內,允許在PAG的溶解度及反應性方面比較PAG。就此而言,本發明的化合物展現優于現有技術化合物的顯著進展。此數據有力地表明,相較于現有技術PAG化合物,本發明的化合物在光刻中作為PAG展現較高敏感性及較佳性能。
表7:
*=丙二醇單甲醚乙酸酯
**=N-羥基萘二甲酰亞胺三氟甲磺酸酯(NIT)為現用PAG商用基準
因此,本發明的PAG可對光刻工藝賦予高效率水平并且使抗蝕劑組合物的曝光區域與未曝光區域之間的對比度及分辨率提高。
本發明的PAG宜用于正性作用或負性作用化學增幅型光致抗蝕劑,即,負性作用抗蝕劑組合物,其經歷光酸促進的交聯反應以使得抗蝕劑涂層的曝光區域相較于未曝光區域不太易溶于顯影劑;及正性作用抗蝕劑組合物,其一或多種組合物組分的酸不穩定基團經歷光酸促進的脫保護反應以使得抗蝕劑涂層的曝光區域相較于未曝光區域易溶于水性顯影劑。
本發明光致抗蝕劑的優選成像波長包括小于300nm的波長,例如248nm;及小于200nm的波長,例如193nm;及EUV,更優選在200nm至500nm范圍內,優選在300nm至450nm范圍內,甚至更優選在350nm至440nm范圍內,最優選為365nm(i線)、405nm(h線)及436nm(g線)波長。
式(I)及(II)化合物的制備(其它細節見實例)
對用于產生本發明的N-羥基萘二甲酰亞胺磺酸酯衍生化合物的方法不存在特定限制,并且任何已知合成均可用于制造式(I)及(II)的化合物。用于合成4-取代的化合物的典型途徑的一些實例分別展示于流程1及2中。在3位處具有取代基的化合物可通過以3-取代的酸酐為起始物質以類似方式合成。起始酸酐(4-溴-1,8-萘二甲酸酐、3-溴-1,8-萘二甲酸酐、4-羥基-1,8-萘二甲酸酐及3-羥基-1,8-萘二甲酸酐)為市售的。
如流程1中所示,具有烷硫基取代基的化合物可容易地自4-溴-1,8-萘二甲酸酐合成。可以類似方式合成具有芳硫基或芳氧基取代基的化合物。除DBU以外的其它有機堿(例如Et3N及DABCO)對于條件(a)也很好地起作用。NaOH可代替條件(c)中的KOH使用。可根據流程2合成具有烷氧基取代基的化合物。
流程1.
條件:(a)RSH、DBU、DMF;(b)RSH、K2CO3、DMF;(c)HO-NH2·HCl、KOH、DMF;(d)HO-NH2·HCl、吡啶;(e)Tf2O、吡啶、ACN。
流程2.
條件:(f)RBr、Cs2CO3、DMF。
光致抗蝕劑組合物
本發明的光致抗蝕劑組合物包含(i)至少一種選自式(I)及(II)的光酸生成劑;(ii)至少一種可溶于堿或不可溶于堿的光致抗蝕劑聚合物或共聚物;(iii)有機溶劑;及任選使用的(iv)添加劑。
包含式(I)及(II)的光酸生成劑的本發明的光致抗蝕劑組合物適用作多種應用中的光致抗蝕劑,特別是用于制造電子裝置,包括平板顯示器(在此情況下光致抗蝕劑可為涂布的玻璃襯底或氧化銦錫層)及半導體裝置(在此情況下光致抗蝕劑可涂布至硅晶片襯底上)。可使用多種曝露輻射,包括用波長為200nm至500nm、優選在300nm至450nm范圍內、更優選在350nm至440nm范圍內、甚至更優選為365nm(i線)、436nm(g線)或405nm(h線)的電磁輻射曝露,其中特別優選波長為365nm的電磁輻射。
本發明的光致抗蝕劑組合物包含一或多種光致抗蝕劑聚合物或共聚物作為組分(ii),其可溶于或不可溶于顯影劑溶液中。本發明的光致抗蝕劑組合物可用于正型或負型組合物。在正型組合物的情況下,組分(ii)的溶解度在其與自本發明化合物釋放的酸反應后增加。在此情況下,使用具有酸不穩定基團的光致抗蝕劑聚合物或共聚物作為組分(ii),其不可溶于堿水溶液,但其在酸存在下以催化方式去保護以使得其變得可溶于溶液。在負型組合物的情況下,組分(ii)的溶解度在其與自本發明化合物釋放的酸反應后降低。在此情況下,使用光致抗蝕劑聚合物或共聚物作為組分(ii),其可溶于顯影劑溶液,但在酸存在下交聯以使得其變得不可溶于堿水溶液。因此,光致抗蝕劑聚合物或共聚物能夠在酸存在下被賦予改變的在顯影劑溶液中的溶解度。顯影劑溶液優選為水溶液,更優選為堿水溶液。
可用作正型組合物中的組分(ii)的光致抗蝕劑聚合物的實例包括(但不限于)芳香族聚合物,例如用酸不穩定基團保護的羥基苯乙烯的均聚物或共聚物;丙烯酸酯,例如聚(甲基)丙烯酸酯,其具有至少一個含有側接脂環基的單元,且具有酸不穩定基團側接聚合物主鏈及/或脂環基、環烯烴聚合物、環烯烴順丁烯二酸酐共聚物、環烯烴乙烯醚共聚物、硅氧烷;倍半硅氧烷、碳硅烷;及寡聚物,包括多面體寡聚倍半硅氧烷、碳水化合物及其它籠型化合物。視需要,前述聚合物或寡聚物宜用可溶于堿水溶液的基團、酸不穩定基團、極性官能團及含硅基團官能化。
可用作本發明的正型組合物中的組分(ii)的共聚物的實例包括(但不限于)聚(對羥基苯乙烯)-甲基金剛烷基甲基丙烯酸酯(PHS-MAdMA)、聚(對羥基苯乙烯)-2-乙基-2-金剛烷基甲基丙烯酸酯(PHS-EAdMA)、聚(對羥基苯乙烯)-2-乙基-2-環戊基甲基丙烯酸酯(PHS-ECpMA)、聚(對羥基-苯乙烯)-2-甲基-2-環戊基甲基丙烯酸酯(PHS-MCpMA)或PHS-EVE。
正型組合物中的所述至少一種組分(ii)優選為聚(羥基苯乙烯)-樹脂,其中至少一部分羥基經保護基取代。優選保護基選自由叔丁氧基羰氧基、叔丁氧基、叔戊氧基羰氧基及縮醛基組成的群組。此外,在EP 1 586 570 A1的段落[0068]至[0114]中描述為“含酸可分離基團的樹脂”的所有聚合物及共聚物適合作為組分ii)。EP 1 586 570 A1中關于這些樹脂的揭示內容以引用的方式并入本文中,并且形成本發明揭示內容的一部分。
優選負型組合物包含在暴露于酸后會固化、交聯或硬化的材料的混合物。優選負性作用組合物包含聚合物粘合劑(例如酚系或非芳香族聚合物)作為組分(ii)、交聯劑組分作為添加劑(iv)及本發明的光酸生成劑組分作為組分(i)。用于此類負型光致抗蝕劑組合物的適合聚合物粘合劑和交聯劑及其用途已揭示于EP-A-0 164 248及US 5,128,232中。用作組分(ii)的優選酚系聚合物包括酚醛清漆及聚(乙烯基苯酚)。酚醛清漆樹脂為酚與醛的熱塑性縮合產物。與醛(尤其甲醛)縮合形成酚醛清漆樹脂的適合酚的實例包括苯酚、間甲酚、鄰甲酚、對甲酚、2,4-二甲苯酚、2,5-二甲苯酚、3,4-二甲苯酚、3,5-二甲苯酚及瑞香草酚。酸催化的縮合反應導致形成適合酚醛清漆樹脂,其分子量可在約500道爾頓至100,000道爾頓間變化。聚乙烯苯酚樹脂為可在陽離子催化劑存在下通過相應單體的嵌段聚合、乳液聚合或溶液聚合形成的熱塑性聚合物。可用于制造聚乙烯苯酚樹脂的乙烯基苯酚可例如通過水解市售的香豆素或經取代香豆素,隨后使所得羥基肉桂酸脫羧基來制備。有用乙烯苯酚也可通過相應羥基烷基苯酚脫水或通過由經取代或未經取代的羥基苯甲醛與丙二酸反應產生的羥基肉桂酸脫羧基來制備。由此類乙烯苯酚制備的聚乙烯苯酚樹脂的分子量范圍優選為約2,000道爾頓至約60,000道爾頓。用作組分(iv)的優選交聯劑包括基于胺的材料,包括基于三聚氰胺、甘脲、苯并胍胺的材料,及基于脲的材料。三聚氰胺-甲醛聚合物通常特別適合。此類交聯劑為市售的,例如三聚氰胺聚合物、甘脲聚合物、基于脲的聚合物及苯并胍胺聚合物,例如氰特公司(Cytec)以商品名CymelTM301、303、1170、1171、1172、1123及1125以及BeetleTM 60、65及80出售的那些。
作為組分(iii),本發明的組合物包含至少一種有機溶劑。有機溶劑可為能夠溶解組分(ii)及組分(i)以產生均勻溶液的任何溶劑,并且可使用選自已知用作常規化學增幅型抗蝕劑的溶劑的材料的一或多種溶劑。有機溶劑的具體實例包括酮類,例如丙酮、甲基乙基酮、環己酮、甲基異戊基酮及2-庚酮;多元醇及其衍生物,例如乙二醇、乙二醇單乙酸酯、二甘醇、二甘醇單乙酸酯、丙二醇、丙二醇單乙酸酯、二丙二醇,或二丙二醇單乙酸酯的單甲醚、單乙醚、單丙醚、單丁醚或單苯基醚;環狀醚,例如二噁烷;及酯,例如乳酸甲酯、乳酸乙酯(EL)、乙酸甲酯、乙酸乙酯、乙酸丁酯、丙酮酸甲酯、丙酮酸乙酯、甲氧基丙酸甲酯及乙氧基丙酸乙酯。這些有機溶劑可單獨使用,或作為含有兩種或兩種以上不同溶劑的混合溶劑使用。特別優選有機溶劑(iii)選自由酮、醚及酯組成的群組。
此外,本發明的組合物還可任選包含至少一種不同于組分(i)、(ii)及(iii)的添加劑。舉例來說,其它任選使用的添加劑包括光化及對比染料、抗條紋劑、塑化劑、增速劑、敏化劑等。除填充劑及染料可呈相對較大濃度,例如其量占抗蝕劑干燥組分總重量的5重量%至30重量%以外,此類任選使用的添加劑通常將在光致抗蝕劑組合物中占極小濃度。
本發明的光致抗蝕劑組合物中通常采用的一種添加劑為堿性抑止劑。堿性抑止劑的目的是中和在底層光致抗蝕劑層的表面區域中由意欲到達光致抗蝕劑層未曝光(深色)區域的雜散光產生的酸。此允許通過控制未曝光區域中不想要的脫保護反應來改良散焦區域中的聚焦深度及曝光寬容度。因此,可使所形成的抗蝕劑圖案中的輪廓不規則性(例如縮頸及T型頂)最少或得以避免。
為允許堿性抑止劑與底層光致抗蝕劑層深色區域中產生的酸之間的有效相互作用,堿性抑止劑應為非表面活性劑類型。也就是說,堿性抑止劑不應是由于例如表面自由能低于外涂層組合物其它組分而遷移至外涂層頂表面的類型。在此情況下,堿性抑止劑不會在光致抗蝕劑層界面明顯與產生的酸相互作用以防止酸脫保護。因此,堿性抑止劑應為存在于外涂層/光致抗蝕劑層界面處的類型,無論其是在外涂層上均勻分散還是在界面形成分級或分離層。此類分離層可通過選擇相對于其它外涂層組合物組分具有較高表面自由能的堿性抑止劑來獲得。
適合堿性抑止劑包括例如:直鏈及環狀酰胺及其衍生物,例如N,N-雙(2-羥基乙基)特戊酰胺、N,N-二乙基乙酰胺、N1,N1,N3,N3-四丁基丙二酰胺、1-甲基氮雜環庚-2-酮、1-烯丙基氮雜環庚-2-酮及1,3-二羥基-2-(羥基甲基)丙-2-基氨基甲酸叔丁酯;芳胺,例如吡啶和二叔丁基吡啶;脂肪族胺,例如三異丙醇胺、正叔丁基二乙醇胺、三(2-乙酰氧基-乙基)胺、2,2',2”,2”'-(乙烷-1,2-二基雙(氮三基))四乙醇及2-(二丁基氨基)乙醇、2,2',2”-氮基三乙醇;環狀脂肪族胺,例如1-(叔丁氧基羰基)-4-羥基哌啶、1-吡咯烷甲酸叔丁酯、2-乙基-1H-咪唑-1-甲酸叔丁酯、哌嗪-1,4-二甲酸二叔丁酯及N(2-乙酰氧基-乙基)嗎啉。在這些堿性抑止劑中,優選1-(叔丁氧基羰基)-4-羥基哌啶及三異丙醇胺。盡管堿性抑止劑的含量將取決于例如底層光致抗蝕劑層中光酸生成劑的含量,但以外涂層組合物的總固體計,其通常以0.1重量%至5重量%、優選0.5重量%至3重量%、更優選1重量%至3重量%的量存在。
另一觀念是將堿性部分附接至PAG分子。在此情況下,抑止劑為PAG的一部分并且緊密鄰近照射后形成的酸。這些化合物對電磁輻射,具體說來,對波長在200nm至500nm范圍內的電磁輻射,更具體說來,對波長為365nm(i線)的電磁輻射具有高敏感性,且同時允許產生相較于自含有抑止劑作為添加劑的現有技術已知的光致抗蝕劑組合物具有較高分辨率的圖案化結構。遵循此觀念的化合物為例如S-17、S-18、S-23及S-24。
本發明抗蝕劑的樹脂粘合劑組分通常以足以使抗蝕劑的曝光涂層可用例如堿性水溶液顯影的量使用。更具體說來,樹脂粘合劑宜占抗蝕劑總固體的50重量%至約90重量%。光敏性組分應以足以在抗蝕劑涂層中產生潛像的量存在。更具體說來,光敏性組分宜以占抗蝕劑總固體約1重量%至40重量%的量存在。通常,較小量的光敏性組分應適用于化學增幅型抗蝕劑。
根據一個優選實施例,本發明的組合物包含:
(i)0.05重量%至15重量%、優選0.1重量%至12.5重量%且最優選1重量%至10重量%的至少一種式(I)或(II)的光酸生成劑化合物;
(ii)5重量%至50重量%、優選7.5重量%至45重量%且最優選10重量%至40重量%的至少一種光致抗蝕劑聚合物或共聚物,其可為可溶于堿或不可溶于堿的;及
(iv)0重量%至10重量%、優選0.01重量%至7.5重量%且最優選0.1重量%至5重量%的其它添加劑,其中組合物中的剩余部分為有機溶劑(iii)。
如在本發明的化合物中,充當曝露于電磁輻射后釋放的酸基的抑止劑的官能性堿性基團為光酸生成劑化合物的一部分,不需要添加單獨堿性組分作為抑止劑(如在自現有技術已知的光致抗蝕劑組合物中其為需要的)。根據本發明組合物的一個優選實施例,此組合物優選包含小于5重量%、更優選小于1重量%、甚至更優選小于0.1重量%且最優選0重量%的不同于組分(i)至(iv)的堿性化合物,例如氫氧化物、羧酸鹽、胺、亞胺及酰胺。
本發明的光致抗蝕劑一般遵循已知程序制備,不過用本發明的PAG取代用于此類光致抗蝕劑調配物中的先前光敏性化合物。舉例來說,可通過將光致抗蝕劑組分溶解于適合溶劑中來制備呈涂布組合物形式的本發明的抗蝕劑,所述溶劑為例如二醇醚,例如2-甲氧基乙基醚(二乙二醇二甲醚)、乙二醇單甲醚、丙二醇單甲醚;乳酸酯,例如乳酸乙酯或乳酸甲酯,其中優選乳酸乙酯;丙酸酯,尤其丙酸甲酯及丙酸乙酯;賽珞蘇酯(Cellosolve ester),例如賽珞蘇乙酸甲酯;芳香烴,例如甲苯或二甲苯;或酮,例如甲基乙基酮、環己酮及2-庚酮。光致抗蝕劑的固體含量通常在光致抗蝕劑組合物總重量的5重量%與35重量%之間變化。
本發明的光致抗蝕劑可根據已知程序使用。盡管本發明的光致抗蝕劑可涂覆為干膜形式,但其優選以液體涂布組合物形式涂覆于襯底上,通過加熱干燥以移除溶劑,優選直至涂層無粘性,經由光掩模曝露于激活輻射,任選進行曝露后烘烤以產生或提高抗蝕劑涂層的曝露區域與非曝露區域之間的溶解度差異,并且接著優選用堿性顯影劑水溶液顯影以形成凸紋圖像。經本發明抗蝕劑涂覆和處理的襯底可適宜地為用于涉及光致抗蝕劑的工藝中的任何襯底,例如微電子晶片。舉例來說,襯底可為硅、二氧化硅或鋁-氧化鋁微電子晶片。也可采用砷化鎵、陶瓷、石英或銅襯底。還宜采用用于液晶顯示器及其它平板顯示器應用的襯底,例如玻璃襯底、氧化銦錫涂布的襯底及其類似襯底。液體涂布抗蝕劑組合物可通過任何標準方式涂覆,例如旋涂、浸漬或滾涂。曝露能量應足以有效激活輻射敏感性系統中的光敏性組分以在抗蝕劑涂層中產生圖案化圖像。適合曝露能量通常在約1mJ/cm2至300mJ/cm2范圍內。如上所述,優選曝露波長包括小于200nm,例如193nm。適合曝露后烘烤溫度為約50℃或更高,更具體說來為約50℃至140℃。對于酸硬化的負性作用抗蝕劑,視需要可在約100℃至150℃的溫度下采用顯影后烘烤,持續數分鐘或更長時間以使顯影時形成的凸紋圖像進一步固化。在顯影及任何顯影后固化之后,通過顯影而露出的襯底表面接著可經選擇性加工,例如根據此項技術中已知的程序化學蝕刻或鍍敷光致抗蝕劑露出的襯底區域。適合蝕刻劑包括氫氟酸蝕刻溶液及等離子氣體蝕刻,例如氧等離子蝕刻。
復合物
本發明提供用于制造復合物的方法,所述復合物包含襯底及以圖案化結構涂覆至所述襯底上的涂層,所述方法包含以下步驟:
(a)將一層本發明組合物涂覆至襯底表面上并且至少部分地移除有機溶劑(iii);
(b)將所述層的選定區域曝露于電磁輻射,藉此將酸自化合物(i)釋放于曝露于電磁輻射的區域中;
(c)任選加熱所述層以在釋放酸的區域中賦予化合物(ii)改變的在水溶液中的溶解度;及
(d)至少部分地移除所述層。
在方法步驟(a)中,將一層本發明組合物涂布至襯底表面上,隨后至少部分地移除有機溶劑(iii)。
襯底可為任何尺寸及形狀,并且優選為可用于光刻的那些,例如硅、二氧化硅、絕緣體上硅(silicon-on-insulator,SOI)、應變硅(strained silicon)、砷化鎵;涂布的襯底,包括涂有氮化硅、氮氧化硅、氮化鈦、氮化鉭的那些;超薄柵氧化物,例如氧化鉿;金屬或經金屬涂布的襯底,包括涂有鈦、鉭、銅、鋁、鎢、其合金的那些;及其組合。優選地,本文中的襯底的表面包括待圖案化的臨界尺寸層,包括例如一或多個閘極層,或用于半導體制造的襯底上的其它臨界尺寸層。此類襯底優選可包括硅、SOI、應變硅及其它此類襯底材料,其形成為具有例如20cm、30cm或更大直徑的尺寸或可用于晶片制造生產的其它尺寸的圓形晶片。
將本發明的組合物可通過任何適合方法涂覆至襯底上,包括旋涂、噴涂、浸涂、刮涂或其類似方法。涂覆光致抗蝕劑層優選通過使用涂布軌道(coating track)旋涂光致抗蝕劑來完成,在涂布軌道中將光致抗蝕劑分配于旋轉晶片上。在旋涂工藝期間,晶片可以至多4,000rpm、優選約500rpm至3,000rpm且更優選1,000rpm至2,500rpm的速度旋轉。旋轉經涂布的晶片以移除有機溶劑(iii),并且在加熱板上烘烤以自膜移除殘留溶劑及自由體積來使其均勻地致密。
在方法步驟(b)中,將所述層的選定區域曝露于電磁輻射,藉此將酸自化合物(i)釋放至曝露于電磁輻射的區域中。如上所述,可使用多種曝露輻射,包括用波長為365nm(i線)、436nm(g線)或405nm(h線)的電磁輻射曝露,其中特別優選波長為365nm的電磁輻射。
此類逐圖案曝露可使用例如步進器等曝露工具來實施,在所述工具中經由圖案遮罩照射膜且由此進行逐圖案曝露。所述方法優選使用產生能夠具有高分辨率的波長的激活輻射,包括遠紫外光(EUV)或電子束輻射的高級曝露工具。應了解,使用激活輻射進行的曝露分解包含在光致抗蝕劑層曝露區域中的本發明組分并產生酸及分解副產物,并且酸接著實現聚合物化合物(ii)的化學變化(使酸敏感性基團去保護以產生堿溶性基團,或者催化曝露區域中的交聯反應)。此類曝露工具的分辨率可小于30nm。
在方法步驟(c)中,所述層可任選經加熱以在釋放酸的區域中賦予化合物(ii)改變的在水溶液中的溶解度。在此所謂“曝露后烘烤”中,產生或提高涂層曝露區域與未曝露區域之間的溶解度差異。曝露后烘烤條件通常包括約50℃或更高的溫度,更具體說來,在約50℃至約160℃范圍內的溫度,持續10秒至30分鐘,優選30秒至200秒。根據本發明方法的特定實施例,在方法步驟(b)之后且在(d)之前不進行熱處理。
在方法步驟(d)中,用水溶液、優選堿水溶液至少部分地移除所述層。此可通過用適合顯影劑處理經曝露的光致抗蝕劑層來完成,所述適合顯影劑能夠選擇性地移除膜的曝露部分(其中光致抗蝕劑為正型的)或移除膜的未曝露部分(其中光致抗蝕劑為負型的)。優選地,光致抗蝕劑是基于具有酸敏感性(可脫保護)基團的聚合物的正型光致抗蝕劑,并且顯影劑優選為無金屬離子的四烷基氫氧化銨溶液。
根據本發明制造的復合物的特征在于,其包含襯底及以圖案化結構涂覆于襯底表面上的涂層,其中所述涂層包含本發明的化合物。
使用式(i)及(II)的光酸生成劑化合物進行官能團的光誘導聚合、光誘導交聯、光誘導降解及光誘導轉化也在本發明的范圍內。本發明化合物特別適用于保護性涂層、智能卡、3D快速原型設計或添加劑制造、犧牲性涂層、粘著劑、抗反射涂層、全像圖、電流(galvano-)及鍍敷遮罩、離子注入遮罩、抗蝕劑、化學增幅型抗蝕劑、光感測應用、印刷電路板(PCB)圖案化、MEMS制造、平板顯示器上的TFT層圖案化、柔性顯示器上的TFT層圖案化、用于顯示器的像素圖案化、用于LCD的彩色濾光片或黑色基質,或封裝工藝中的半導體圖案化,及半導體制造保護性涂層、智能卡、3D快速原型設計或添加劑制造、犧牲性涂層、粘著劑、抗反射涂層、全像圖、電流及鍍敷遮罩、離子注入遮罩、抗蝕劑、化學增幅型抗蝕劑、光感測應用上或彩色濾光片中的TSV相關圖案化。
以下實例意欲說明以上本發明,且不應解釋為使本發明的范圍變窄。本領域技術人員應容易認識到,實例提出可實踐本發明的許多其它方式。應理解,可在保持于本發明的范圍內情況下進行許多變化及修改。
實例
溶解度
溶解度是PAG評估中的重要因素。高溶解度不僅使PAG純化容易,而且還使PAG能夠在光致抗蝕劑及不同的溶劑系統中以廣泛范圍的濃度使用。為測試PAG的溶解度,緩慢添加溶劑直至PAG完全溶解并且在澄清溶液中觀測不到渾濁。表8列出在20℃下一些代表性N-羥基萘二甲酰亞胺磺酸酯衍生物與NIT在多種有機溶劑中的溶解度(w/v%)的比較。所有本發明的化合物在多種溶劑中展現的溶解度都高于比較化合物A及B。除S-6及O-12以外,所有化合物展示比商用基準(NIT)高很多的溶解度。應注意,化合物S-11及S-43在三種測試溶劑中展現極高溶解度。這些結果指出本發明中的N-羥基萘二甲酰亞胺磺酸酯衍生物可以高濃度用于感光性組合物中。由于PAG的溶解度隨溫度變化而顯著變化,故高溶解度改善感光性組合物的溶液穩定性,由此可允許組合物用于廣泛范圍的操作溫度,而無需擔心組合物中的PAG再結晶。
表8
*丙二醇單甲醚乙酸酯
**γ-丁內酯
光反應性
光致抗蝕劑組合物通常包含PAG、聚合物、添加劑及溶劑。光致抗蝕劑組合物的性能主要取決于PAG及聚合物組分的特性。為調配高性能光致抗蝕劑組合物,通常選擇感光性較高的PAG。PAG的感光性通常與所產生酸的強度及PAG的光反應性直接相關。對于產生相同潛在酸的一系列PAG,其感光性僅與其光反應性相關。因此,評估PAG的感光性可通過研究其光反應性來達成。光反應性愈高,感光性愈高。可通過在低曝光強度(以避免不會產生所要酸的副反應)下使PAG在其稀溶液中光解來研究光反應性。照射后PAG濃度的變化可通過在最大吸收波長下測量PAG的吸光度來測定。
在室溫下,在空氣中于乙腈中實施PAG的光解。四溴酚藍的鈉鹽(TBPBNa;一種酸指示劑染料,其在618nm下具有最大吸收)購自阿爾德里奇公司(Aldrich)(指示劑級)并按原樣使用。使用科爾帕默(Cole-Parmer)UV 15W臺燈(EW-97605-50)在365nm下進行PAG溶液(3×10-5M)的照射。使用來自EIT有限公司的UV Power Puck II輻射計測量光強度。在賽默科技(Thermo Scientific)Evolution 201UV-可見光分光光度計上執行UV-Vis光譜。
在乙腈中檢測NIT、比較化合物A和B、S-3及S-11的光解。照射后S-3的UV-Vis光譜變化展示于圖2中。照射后,在400nm下的吸收帶逐漸減小,表明光反應隨能量曝露劑量的增加而發展。假定光反應是一級反應,則吸光度隨能量曝露劑量的變化的自然對數圖提供S-3的光反應常數(即,線性趨勢線的斜率)(圖3)。在相同照射條件下以類似方式測定其它化合物的光反應常數。將A、B、S-3及S-11的常數與標準化為一的NIT的常數相比較,得到相對光反應性(表7)。本發明的PAG(S-3及S-11)的光反應性是NIT的光反應性的13至14倍,并且是比較化合物B的光反應性的5至6倍。具有硫取代基的PAG(S-3及S-11)在測量值的誤差范圍內展現幾乎相同的光反應性。通過觀測酸指示劑TBPBNa在618nm下的光譜變化來確認照射后酸的形成。
抗蝕劑評估
遵循此通用程序制備本發明的六種不同光致抗蝕劑組合物:預混合50g PHS-EVE聚合物溶液(約30重量%含量的聚合物于PGMEA中;約35%的OH基團經EVE封端,Mw=32,000,Mw/Mn=1.88)與50g PGMEA。向此混合物中添加1.3mmol PAG(具體量參見表9)并將0.0263g(PAG的20mol%)三乙胺用作抑止劑。攪拌混合物直至固體完全溶解。接著將組合物儲存于黑暗中以待后續通過光刻進行圖案研究。
制備詳述于表9中的組合物用于評估。
表9:組合物匯總
圖案化結構的制備
使用組合物1-6,遵循此通用程序通過光刻制備圖案化結構。通過旋涂器(1500rpm,40秒,ACE-200型),在用HMDS預處理的裸硅晶片(4英寸直徑)上涂布組合物。在加熱板(Wise Therm HP-30D)上于120℃下軟烤涂層1.5分鐘且隨后使用Jesung JSM-4S,在具有用多個線與間隙(L/S)大小(5μm、6μm、7μm、8μm、9μm及10μm)圖案化的光掩模存在下,在40mJ/cm2的來自LED燈的i線照射下進行曝光。通過將晶片浸于2.38重量%四甲基氫氧化銨(TMAH)水溶液中1分鐘來移除曝露于輻射的區域中的涂層以產生圖案化結構。
通過高分辨率顯微鏡小心地分析所獲得的組合物1-6的圖案化結構(參見圖4)以獲得實際CD圖案尺寸(臨界尺寸或線寬),其與光掩模的10μm圖案匹配。應注意,包括三種現有技術化合物在內的全部六種組合物都展現良好圖案,表明這些類型的PAG可在i線照射下產生三氟甲磺酸并且產生的酸也與用于這些組合物中的PHS-EVE聚合物相容。如表9中所示,所有CD圖案大小(12.5μm至12.91μm)都大于光掩模的間隙大小(10μm),表明酸擴散至未曝光區域。一般而言,較大的圖案大小意謂較大量的所產生的酸擴散至未曝光區域,由此表明PAG的較高敏感性。由于所研究的全部六種PAG在其組合物中以相同摩爾濃度使用并且也產生相同三氟甲磺酸,展現較大CD圖案大小的本發明的PAG(S-5、S-3及O-41)因此具有比CD圖案大小較小的現有技術化合物(NIT、A及B)更高的敏感性。因此,本發明的化合物獨特地展現高溶解度及高敏感性兩者。
PAG化合物的制備
實例3、6、7、8、11、12、13、14、16、19、20、21及22描述合成本發明的磺酸衍生物的實例。
實例1
化合物A1的合成。
在3L燒瓶中裝入4-溴-1,8-萘二甲酸酐(300g,1.08mol)、1L DMAc及1-丙基硫醇(90.7g,1.19mol)。將DBU(181.3g,1.19mol)逐滴添加至此漿料混合物中,并將溫度保持在70℃。完成添加之后,在70℃下攪拌反應混合物過夜。將反應混合物冷卻至室溫,且接著添加1L的去離子水與MeOH的1:1混合物。過濾混合物得到黃色固體,在真空下于50℃下將其干燥過夜,得到255g酸酐A1(產率:86%)。Mp:156-7℃。應注意,A1不經進一步純化即用于后續反應。
實例2
化合物H1的合成。
向5L燒瓶裝入A1(255g,0.93mol)、2.5L DMAc及H2NOH·HCl(71.7g,1.03mol)。向漿料混合物中逐滴添加48%NaOH溶液(41.2g,1.03mol),且在添加期間將溫度保持在25℃。完成添加之后,在室溫下攪拌反應混合物過夜。接著將混合物加熱至80℃并于相同溫度保持3小時。將反應混合物冷卻至室溫,且接著添加1L的MeOH與去離子水的1:3混合物。在室溫下攪拌混合物2小時。過濾得到固體,用100mL MeOH及100mL CH2Cl2洗滌。在真空下于70℃下干燥黃色固體過夜,得到265g羥基酰亞胺H1(產率:99%)。Mp:191-3℃。應注意,H1不經進一步純化即用于后續反應。
實例3
化合物S-1的合成。
向2L燒瓶裝入H1(100g,0.35mol)、乙腈(600g)及吡啶(68.9g,0.87mol)。將混合物冷卻至4℃且接著在低于10℃下逐滴添加三氟甲磺酸酐(127.7g,0.453mol)。添加之后,將反應混合物加熱至60℃并在60℃下攪拌過夜。將反應混合物冷卻至室溫,且接著添加1L去離子水。在室溫下攪拌混合物2小時。過濾得到黃色固體。將固體溶解于1L CH2Cl2中,并使溶液通過硅膠墊。移除CH2Cl2并自500mL乙腈再結晶得到黃色固體,使其在真空下于50℃下干燥過夜,得到120g S-1(產率:82%)。Mp:163-4℃。1H NMR(300MHz,CDCl3)δ:8.60(dd,1H),8.57(dd,1H),8.43(d,1H),7.72(t,1H),7.48(d,1H),3.10(t,2H),1.80(六重峰,2H),1.08(t,3H)。
實例4
化合物A3的合成。
在3L燒瓶中裝入4-溴-1,8-萘二甲酸酐(230g,0.83mol)、1L DMAc及2-丙基硫醇(68.3g,0.897mol)。將DBU(136.7g,0.897mol)逐滴添加至此漿料混合物中,且將溫度保持在60℃。完成添加之后,在55℃下攪拌反應混合物過夜。將反應混合物冷卻至室溫,且接著添加1L的去離子水與MeOH的1:1混合物。過濾混合物以得到黃色固體,在真空下于50℃下將其干燥過夜,得到212g酸酐A3(產率:93%)。Mp:122-9℃。應注意,A3不經進一步純化即用于后續反應。
實例5
化合物H3的合成。
向5L燒瓶裝入A3(212g,0.78mol)、1L DMAc及H2NOH·HCl(57.0g,0.82mol)。向漿料混合物逐滴添加48%NaOH溶液(32.8g,0.82mol),且在添加期間將溫度保持在25℃。完成添加之后,在室溫下攪拌反應混合物過夜。接著將混合物加熱至80℃并于相同溫度保持3小時。將反應混合物冷卻至室溫,且接著添加1L的MeOH與去離子水的1:3混合物。在室溫下攪拌混合物2小時。過濾得到固體,用100mL MeOH及100mL CH2Cl2洗滌。在真空下于50℃下干燥黃色固體過夜,得到178g羥基酰亞胺H3(產率:80%)。Mp:179-182℃。應注意,H3不經進一步純化即用于后續反應。
實例6
化合物S-3的合成。
向2L燒瓶裝入H3(142g,0.495mol)、乙腈(830g)及吡啶(117.5g,1.48mol)。將混合物冷卻至0℃且接著在低于5℃下逐滴添加三氟甲磺酸酐(188.5g,0.669mol)。添加之后,使反應混合物升溫至室溫并攪拌過夜。將反應混合物加熱以使所有固體溶解,接著冷卻至室溫。接著添加1L的1.0M HCl溶液。在室溫下攪拌混合物30分鐘。過濾得到黃色固體。將固體溶解于600g CH2Cl2中,并使溶液通過硅膠墊。移除CH2Cl2并自300g的異丙醇與乙腈的1:1混合物再結晶,得到黃色固體,將其在真空下于50℃下干燥過夜,得到154.5g S-3(產率:74%)。Mp:125.5-126℃。1H NMR(300MHz,CDCl3)δ:8.62(dd,1H),8.58(dd,1H),8.44(d,1H),7.71(t,1H),7.57(d,1H),3.70(七重峰,1H),1.42(d,6H)。
實例7
化合物S-2的合成。
通過使羥基酰亞胺H1與全氟丁烷試劑反應來合成九氟丁磺酸酯S-2,產率72%。Mp:156-7℃。1H NMR(300MHz,CDCl3)δ:8.59(dd,1H),8.55(dd,1H),8.42(d,1H),7.71(t,1H),7.46(d,1H),3.10(t,2H),1.80(六重峰,2H),1.08(t,3H)。
實例8
化合物S-4的合成。
通過使羥基酰亞胺H3與全氟丁烷試劑反應來合成九氟丁磺酸酯S-4,產率73%。Mp:148.5-149℃。1H NMR(300MHz,CDCl3)δ:8.63(dd,1H),8.60(dd,1H),8.46(d,1H),7.73(t,1H),7.59(d,1H),3.71(七重峰,1H),1.42(d,6H)。
實例9
化合物A6的合成。
在500mL燒瓶中裝入4-溴-1,8-萘二甲酸酐(27.7g,100mmol)、250mL DMF、叔丁基硫酚(20g,120mmol)及K2CO3(6.9g,50mmol)。將混合物加熱至回流,保持3小時。將反應混合物冷卻至室溫,且接著添加450mL去離子水。攪拌混合物1小時。過濾并用MeOH(100mL×3)洗滌,得到黃色產物,將其自CH2Cl2與乙腈的混合物再結晶,得到32.1g酸酐A6(產率:89%)。Mp:194-5℃。
實例10
化合物H6的合成。
向500mL燒瓶裝入A6(10.0g,27.6mmol)、H2NOH·HCl(1.96g,30.4mmol)及吡啶(21.8g,276mmol)。將反應混合物加熱至回流,保持1.5小時。在旋轉蒸發器上移除吡啶。將20mL DMF及100mL去離子水添加至殘留物中。過濾得到固體,將其在真空下于50℃下干燥過夜,得到8.4g羥基酰亞胺H6(產率:81%)。Mp:225-6℃。應注意,H6不經進一步純化即用于后續反應。
實例11
化合物S-6的合成。
向250mL燒瓶裝入H6(8.2g,21.7mmol)、乙腈(50mL)及吡啶(2.57g,32.5mmol)。將混合物冷卻至4℃,且接著在半小時內逐滴添加三氟甲磺酸酐(6.74g,23.9mmol)。添加之后,在室溫下攪拌反應混合物過夜,接著將其加熱至回流,保持2小時。添加200mL去離子水。過濾得到黃色固體,將其溶解于100mL CH2Cl2中并使其通過硅膠墊。移除CH2Cl2并用100mL MeOH洗滌,得到8.4g S-6(產率:76%)。Mp:183-5℃。1H NMR(300MHz,CDCl3)δ:8.75(dd,1H),8.70(dd,1H),8.35(d,1H),7.85(t,1H),7.54(s,4H),7.14(d,1H),1.39(s,9H).13H NMR(75.5MHz,CDCl3)δ:31.2,35.0,117.7,122.0,124.3,125.2,127.1,127.5,127.7,129.0,131.6,132.3,133.2,135.1,150.1,153.9,158.8,158.9。
實例12
化合物S-5的合成。
在如實例9、10及11中所述的程序中,自4-溴-1,8-萘二甲酸酐以類似方式合成三氟甲磺酸酯S-5,總產率69%。Mp:166-8℃。1H NMR(300MHz,CDCl3)δ:8.65(dd,1H),8.61(dd,1H),8.27(d,1H),7.76(t,1H),7.50(m,5H),7.09(d,1H)。
實例13
化合物S-7的合成。
在如實例9、10及11中所述的程序中,自4-溴-1,8-萘二甲酸酐以類似方式合成三氟甲磺酸酯S-7,總產率27%。Mp:209-211℃。1H NMR(300MHz,DMSO)δ:8.56(dd,1H),8.52(dd,1H),8.38(d,1H),8.35(d,1H),8.05(m,4H),7.65(m,3H),7.37(d,1H)。
實例14
化合物S-8的合成。
在如實例9、10及11中所述的程序中,自4-溴-1,8-萘二甲酸酐以類似方式合成三氟甲磺酸酯S-8,總產率61%。Mp:204-6℃。1H NMR(300MHz,CDCl3)δ:8.86(dd,1H),8.71(dd,1H),8.38(d,1H),7.67(t,1H),7.52(d,1H),7.30(s,4H),4.30(s,2H),1.24(s,9H)。
實例15
化合物A9的合成。
在250mL燒瓶中裝入4-溴-1,8-萘二甲酸酐(10g,36.1mmol)、20mL DMF及DABCO(4.45g,39.7mmol)。將HSCH2CH2OH(3.24g,41.5mmol)逐滴添加至此漿料混合物中,并將溫度保持在28℃。添加之后,將反應混合物在室溫下攪拌過夜。接著添加130mL去離子水。過濾混合物,得到黃色固體,將其在真空下于50℃下干燥過夜,得到8.0g酸酐(產率:80%)。
向100mL燒瓶裝入以上酸酐(8.0g,29.2mmol)、20mL THF、Et3N(3.25g,39.7mmol)、DMAP(0.178g,1.46mmol)及鄰甲苯基氯(4.96g,32.1mmol)。在室溫下攪拌反應混合物過夜且接著加熱至回流過夜。將混合物冷卻至室溫,且接著添加100mL去離子水。過濾得到固體,將其自200mL乙腈再結晶。在真空下于60℃下干燥黃色固體過夜,得到7.4g酸酐A9(產率:65%)。應注意,A9不經進一步純化即用于后續反應。
實例16
化合物S-9的合成。
向100mL燒瓶裝入A9(3.7g,9.4mmol)、H2NOH·HCl(0.67g,10.4mmol)及吡啶(7.45g,94.3mmol)。將反應混合物加熱至回流,保持1小時。接著將反應混合物冷卻至室溫,并添加5mL CH2Cl2。使用冰-鹽浴將混合物冷卻至0℃。在低于5℃下將三氟甲磺酸酐(5.85g,20.7mmol)逐滴添加至混合物中。在室溫下攪拌混合物過夜。在旋轉蒸發器上移除溶劑,并添加50mL去離子水。過濾得到固體,使用CH2Cl2/EA(10:1)使其通過硅膠墊。移除溶劑得到呈黃色固體的4.0g S-9(產率:78%)。Mp:145-7℃。1H NMR(300MHz,DMSO)δ:8.66(dd,1H),8.60(dd,1H),8.43(d,1H),8.00(d,1H),7.91(t,1H),7.62(d,1H),7.39(t,1H),7.18(m,2H),4.59(t,2H),4.59(t,2H),3.75(t,2H),2.42(s,3H)。
實例17
化合物A11的合成。
在500mL燒瓶中裝入4-溴-1,8-萘二甲酸酐(13.8g,49.5mmol)、60g DMF、2-巰基丙酸3-甲氧基丁酯(10g,52mmol)及K2CO3(3.44g,24.8mmol)。將混合物加熱至回流,保持3小時。將反應混合物冷卻至室溫,且接著添加200mL去離子水。將棕色油狀物與水層分離。分離油狀物之后,用CH2Cl2(20mL×2)萃取水層。合并有機層,并在旋轉蒸發器上移除CH2Cl2,得到棕色油狀產物,將其在真空下于50℃下干燥過夜,得到11.5g酸酐A11(產率:60%)。應注意,A11不經進一步純化即用于后續反應。
實例18
化合物H11的合成。
向500mL燒瓶裝入A9(8.0g,20.6mmol)、H2NOH·HCl(1.58g,22.7mmol)及吡啶(16.3g,206mmol)。將反應混合物加熱至回流,保持1小時。接著將反應混合物冷卻至室溫,且接著添加200mL去離子水。過濾得到固體,將其在真空下于50℃下干燥過夜,得到8.0g羥基酰亞胺H11(產率:96%)。應注意,H11不經進一步純化即用于后續反應。
實例19
化合物S-11的合成。
向100mL燒瓶裝入H11(1.0g,2.5mmol)、乙腈(10g)及吡啶(0.267g,3.4mmol)。將混合物冷卻至4℃,且接著逐滴添加三氟甲磺酸酐(0.84g,0.3mmol)。添加之后,在室溫下攪拌反應混合物2小時。添加50mL去離子水。用CH2Cl2(3×20mL)萃取混合物。合并有機層,并在旋轉蒸發器下移除溶劑。使用CH2Cl2使殘留物通過硅膠墊。移除CH2Cl2并使其自30mL異丙醇再結晶,得到1.0g呈黃色固體的S-11(產率:75%)。Mp:60-3℃。1H NMR(300MHz,CDCl3)δ:8.61(d,1H),8.55(d,1H),8.45(d,1H),7.75(t,1H),7.54(d,1H),4.18(t,2H),3.29-3.43(m,3H),3.21(s,3H),2.72(t,2H),1.71(m,2H),1.09(d,3H)。
實例20
化合物S-43的合成。
向100mL燒瓶裝入H11(5.0g,12.5mmol)、甲苯磺酰氯(2.62g,13.8mmol)、CH2Cl2(25g)及Et3N(1.39g,13.8mmol)。在室溫下攪拌混合物4小時。添加50mL去離子水,并分離有機層。用CH2Cl2(2×10mL)萃取水層。合并有機層,并在旋轉蒸發器上移除溶劑。使用CH2Cl2作為洗脫劑使殘留物通過硅膠墊。移除CH2Cl2并使其自30mL EtOAc再結晶,得到5.1g呈黃色固體的S-43(產率:73%)。1H NMR(300MHz,CDCl3)δ:8.55(d,1H),8.47(d,1H),8.35(d,1H),7.95(t,2H),7.65(t,1H),7.46(d,1H),7.32(d,1H),4.15(t,2H),3.35(m,3H),3.20(s,3H),2.71(t,2H),2.41(s,3H),1.71(m,2H),1.05(d,3H)。
實例21
化合物O-12的合成。
在如實例9、10及11中所述的程序中,自4-溴-1,8-萘二甲酸酐以類似方式合成三氟甲磺酸酯O-12,總產率77%。Mp:188-9℃。1H NMR(300MHz,CDCl3)δ:8.76(dd,1H),8.59(dd,1H),8.39(d,1H),7.75(t,1H),7.45(m,2H),7.27(m,1H),7.14(m,2H),6.83(d,1H)。
實例22
化合物O-41的合成。
在如實例9、2及11中所述的程序中,自4-溴-1,8-萘二甲酸酐以類似方式合成三氟甲磺酸酯O-41,總產率27%。Mp:204-6℃。1H NMR(300MHz,DMSO)δ:8.88(dd,1H),8.72(dd,1H),8.55(d,1H),8.03(t,1H),7.56(d,2H),7.25(d,2H),7.03(d,1H),1.34(s,9H)。
盡管根據某些特定實施例和實例說明并描述上文,但本發明并不意欲限于所展示的細節。相反地,可在權利要求書的等效物的范疇與范圍內并且在不背離本發明精神的情況下對細節作出多種修改。舉例來說,明確預期在本文件中廣泛列舉的所有范圍在其范圍內包括在較寬范圍內的所有較窄范圍。此外,一個實施例的特征可并入另一實施例中。
- 還沒有人留言評論。精彩留言會獲得點贊!