末端炔烴C?H鍵的堿催化的甲硅烷基化的制作方法
本申請要求于2014年9月2日提交的第62/044,754序列號、于2015年4月13日提交的第62/146,541序列號和于2015年6月9日提交的第62/172,969序列號的美國專利申請的優先權的權益,出于所有目的,這些專利申請的內容通過引用并入本文。
政府權利
本發明使用根據由美國國家科學基金會授予的撥款號CHE1212767的政府支持而完成。政府具有本發明中的某些權利。
技術領域
本發明涉及用于使用堿金屬氫氧化物、醇鹽或氫化物催化劑和有機硅烷試劑使炔烴基質——即含有末端炔烴C(sp)-H鍵的炔烴基質甲硅烷基化的方法。
背景
使有機部分甲硅烷基化的能力已在近年來吸引了極大關注,這歸因于甲硅烷基化材料本身、或作為用于例如在農用化學、藥學和電子材料應用中所使用的其他重要材料的中間體的效用。
在過去幾十年間,已經分配了相當大的努力來開發強大的催化劑結構以完成各種C-H官能化反應,徹底改變化學合成的邏輯并因此簡化合成化學。實現這種具有挑戰性的轉化可能常常需要使用化學計量的添加劑、苛刻的反應條件、絡合物配體,并且最值得注意的是貴金屬預催化劑。值得注意的是,使用貴金屬催化劑用于這些轉化的需求仍然是根本的和長期的限制。
用于合成乙炔基硅烷類的策略已經利用了強堿或已經依賴于化學計量的或催化性過渡金屬物質諸如Pt、Zn、Au和Ir,通常在高溫下使用多種預活化的有機硅偶聯配偶體。已經研究了廉價且可商購的含氫硅烷類(hydrosilanes),但是,此特定的硅源已經帶來新的挑戰:必需的原位Si-H鍵活化需要外源的堿、犧牲性氫受體(sacrificial hydrogen acceptor)或氧化劑,以及升高的溫度(即80℃-120℃)。另外,不希望的炔烴的氫化硅烷化可能是競爭性的,使催化劑和反應設計更加復雜。這些因素已經引起了對范圍和實際應用的重要限制。例如,值得注意的是,在以上提及的報告中不存在在藥學和天然產物應用中重要的基質種類,例如脂族胺類和含氮雜環類。盡管末端乙炔類具有固有酸性,開發用于交叉脫氫的(cross-dehydrogenative)C(sp)-Si鍵形成的溫和且通用的化學計量或催化方法仍然是本領域的長期挑戰。
本發明利用本文中引用的發現來避免與之前已知的方法相關聯的問題中的至少一些。
概述
本文公開了一種溫和、高效并且通常直接的C(sp)-H鍵甲硅烷基化。催化的交叉脫氫法避免了之前策略的限制并且在C-H甲硅烷基化反應中之前前所未有的以數克級別和高收率以及優良的化學選擇性成功地偶聯了炔烴和含氫硅烷。顯著地,催化劑可以為KOH和NaOH。
多個實施方案包括方法,每個方法包括以下或主要由以下組成:使包含末端炔基C-H鍵的至少一種有機基質與至少一種有機硅烷和堿金屬氫氧化物、醇鹽或氫化物(優選氫氧化物)的混合物在足以形成甲硅烷基化的末端炔基部分的條件下接觸。這樣的方法在基本上不存在過渡金屬化合物或其他電磁或熱的引發或增長(electromagnetic or thermal initiation or propagation)時可操作。
在一些實施方案中,有機硅烷包括式(I)、式(II)或式(III)的有機硅烷:
(R)4-mSi(H)m (I)
(R)3-m(H)mSi-(CH2)q-Or-Si(R)3-p(H)p (II)
R-[-SiH(R)-O-]n-R (III)
其中R被靈活地定義,m和p獨立地為1、2或3;q為0、1、2、3、4、5或6;r為0或1;并且n為10至100。例如,在一些實施方案中,有機硅烷獨立地為(R)3SiH、(R)2SiH2或(R)SiH3。在式(II)的一些實施方案中,q為0。在式(II)的一些實施方案中,r為0。在一些實施方案中,R獨立地為烷氧基、烷基、烯基、芳基、芳氧基、雜芳基、芳烷基或雜芳烷基。
在一些實施方案中,堿金屬氫氧化物是氫氧化鈉(NaOH)或氫氧化鉀(KOH)。在一些實施方案中,堿金屬醇鹽是醇鈉(例如NaOMe、NaOEt、NaO-t-Bu)或醇鉀(例如KOMe、KOEt、KO-t-Bu、KO-t-戊基)。在一些實施方案中,堿金屬氫化物是氫化鈉(NaH)或氫化鉀(KH)。
有機基質通常被定義為具有至少一個末端炔基C-H鍵、具有下式:
R1-C≡C-H,
其中R1包括H、任選地被取代的烷基、任選地被取代的烯基、任選地被取代的炔基、任選地被取代的芳基、任選地被取代的雜烷基、任選地被取代的雜芳基、任選地被取代的芳烷基、任選地被取代的雜芳烷基或任選地被取代的金屬茂。方法用以此方式定義的廣泛的基質是可操作的。具有兩個或更多個末端炔烴C-H鍵的化合物通常在不連續的、依序的反應中也提供甲硅烷基化的產物。基質包括單獨的化合物、寡聚物和聚合物。
在使用具有兩個或三個Si-H鍵的硅烷(例如R2SiH2或(R)SiH3)的某些實施方案中,使包含末端炔基C-H鍵的第二或第三有機基質與甲硅烷基化的末端炔基部分同時或依序地接觸,可以形成二炔基硅烷產物或三炔基硅烷產物。
一旦形成,甲硅烷基化的末端炔基部分可以經受多種已知的化學反應,并且當與本文描述的本發明的方法結合時,使用這些已知方法的方法被認為是在本發明的范圍內。例如,當與本文描述的本發明的甲硅烷基化方法結合時,以下隨后的反應中的至少一個是在范圍內的:(a)使甲硅烷基化的末端炔基部分與另一種不飽和部分在[2+2]或[4+2]環加成反應中反應以形成芳族、雜芳族、環烯基或雜環烯基部分;(b)使甲硅烷基化的末端炔基部分與第二不飽和的有機部分在交叉復分解反應(cross-metathesis reaction)中反應以形成二烯烴或聚烯烴產物;(c)使甲硅烷基化的末端炔基部分聚合;(d)使甲硅烷基化的末端炔基部分與有機疊氮化物在[3+2]疊氮化物-炔烴環加成反應中反應(通常被稱為“點擊化學”,包括1,3-偶極環加成);(e)使甲硅烷基化的末端炔烴部分氫化;(f)去除最初添加至末端炔基C-H鍵的甲硅烷基,使得甲硅烷基化充當對于其他轉化的保護基團;(g)使甲硅烷基化的末端炔基部分與芳族鹵化物在足以形成炔基-芳烴鍵的條件下反應;和(h)使甲硅烷基化的末端炔基部分與N-鹵代琥珀酰亞胺在陽離子型金催化劑的存在下反應以產生末端炔基鹵化物。
另外,在其中至少一種有機硅烷包含任選地被取代的C1-12烯基或雜烯基,使得該甲硅烷基化的末端炔基部分包含硅鍵合的任選地被取代的C1-12烯基或雜烯基的情況中,某些實施方案包括這些方法,所述方法還包括使該甲硅烷基化的末端炔基部分與醇和催化劑在產生甲硅烷基化的末端炔基部分的分子內烯丙基化的條件下反應。
另外,在其中至少一種有機硅烷包含2-吡啶基(如本文中由(Me)2(吡啶基)SiH或(i-Pr)2(吡啶基)SiH實例)的情況中,所述方法還包括使甲硅烷基化的末端炔基部分與銅碳鎂化催化劑(copper carbomagnesation catalyst)和任選地被取代的芳基鎂絡合物或任選地被取代的雜芳基鎂絡合物在足以使甲硅烷基化的末端炔基部分碳鎂化的條件下反應。仍另外的實施方案提供了使碳鎂化的甲硅烷基化的末端炔基部分與任選地被取代的芳基碘或任選地被取代的雜芳基碘在鈀催化劑的存在下反應以形成三取代的甲硅烷基化的烯烴。并且,仍另外的實施方案包括以下的實施方案:在所述實施方案中,三取代的甲硅烷基化的烯烴與BCl3和頻哪醇在足以使該化合物硼脫甲硅烷基化(borodesilylate)的條件下反應,并且然后任選地使該硼脫甲硅烷基化的化合物與第二任選地被取代的芳基碘或任選地被取代的雜芳基碘在適于使所得的C-B鍵與所述第二任選地被取代的芳基碘或任選地被取代的雜芳基碘交叉偶聯的條件下反應。
仍另外的實施方案包括用于使包含末端炔基C-H鍵的有機基質甲硅烷基化的那些體系,所述體系包括以下的混合物或主要由以下的混合物組成:(a)至少一種有機硅烷和(b)堿金屬氫氧化物、醇鹽或氫化物,以及(c)至少一種基質。以至少足以適應本文描述的方法的術語描述該體系。在一些實施方案中,該體系還包括存在來源于基質和所述至少一種有機硅烷之間的反應的甲硅烷基化的末端炔烴。
盡管主要關于用于對這些轉化起作用的方法和體系描述實施方案,應當理解,來源于這些方法和體系的任何化合物——其否則的話不能通過其他可行的手段獲得——被認為是在本發明的范圍內。
附圖簡述
當結合附圖閱讀時,進一步地理解了本申請。出于闡明主題的目的,在附圖中示出了主題的示例性實施方案;然而,本文公開的主題不限于所公開的具體的方法、裝置和體系。此外,附圖不一定按比例繪制。在附圖中:
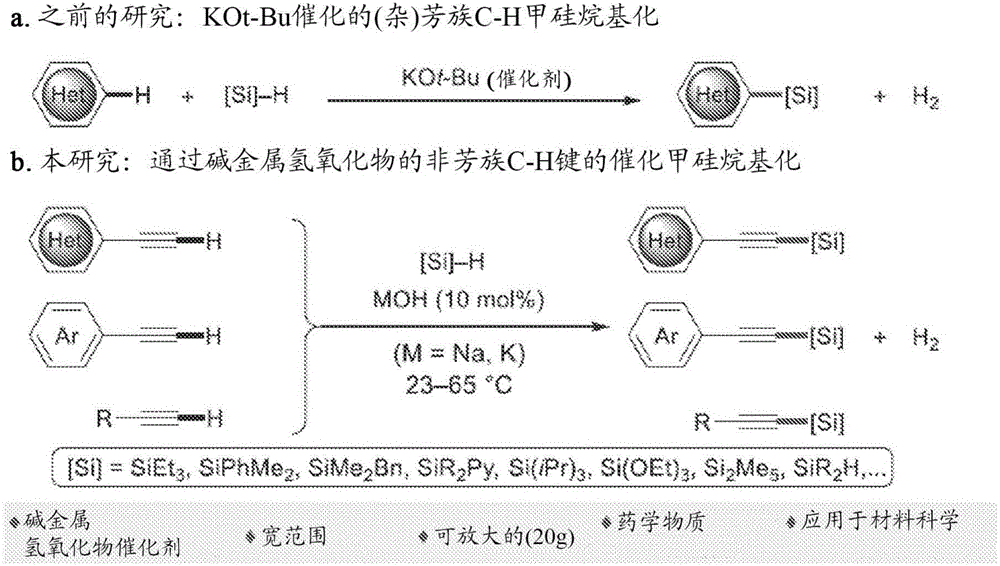
圖1圖示了目前可由地球上豐富的金屬鹽獲得的催化的C-H甲硅烷基化的范圍。a.近期公開的具有高區域控制(regiocontrol)的、KOt-Bu催化的、含有N、O和S的芳香雜環的C-H甲硅烷基化。b.堿金屬氫氧化物催化非芳族的C(sp)-H鍵的交叉脫氫甲硅烷基化。
圖2圖示了優化反應條件的子集和關于含氫硅烷的效用的范圍。對于條目1-6、8和10,通過使用十三烷作為內標的GC-FID分析確定收率。對于條目7、9和11-20,收率是分析純的分離的物質。在指定的溫度和時間下,在0.5mmol的規模用0.5mL溶劑進行反應。DME=1,2-二甲氧基乙烷;DABCO=二氮雜二環[2.2.2]辛烷;(iPr)2PySi–H=2-二異丙基甲硅烷基吡啶;Me2PySi–H=2-二甲基甲硅烷基吡啶。
圖3圖示了炔烴基質的范圍。使炔丙醇(3x)進行O-Si和C-Si二者脫氫偶聯以得到雙甲硅烷基化產物4x;相比之下,對于N-甲基炔丙胺(3w),沒有觀察到N-Si鍵形成,其得到單甲硅烷基化的4w。粗體的C-H鍵代表可以由之前開發的C-H官能化方法——包括KOt-Bu催化的甲硅烷基化——占用的位點,潛在地導致產物混合物。在規定的溫度下,用0.5mL溶劑在0.5mmol規模進行反應。收率是分析純的分離的物質。由NMR和GC確定選擇性。DME=1,2-二甲氧基乙烷。
圖4A-4E圖示了本發明的方法的合成應用中的一些。圖4A提供了用于使無偏性的(unbiased)乙炔基硅烷組成部分2a的幾克級別的合成(multigram synthesis)的方案。圖4B示出了對稱的末端二炔在烷基和芳基系列二者中的每步的反應性可以通過簡單地改變反應條件被選擇性地單官能化或雙官能化。圖4C示出了,二氫硅烷可以用NaOH作為催化劑經歷雙C(sp)-H甲硅烷基化,以產生二乙炔基硅烷。這些產物可以被容易地加工成多取代的硅雜環戊二烯(silole)。應當理解,這個和本文提供的任何具體實例應當被認為是本發明的更廣泛的實施方案的范例(例如在此情況中,形成硅雜環戊二烯和聚硅雜環戊二烯(polysilole)的范例)。圖4D示出了2-甲硅烷基吡啶定向基團可以以良好收率被安裝到簡單炔烴上。這些片段可以被進展成密集取代的烯烴。圖4E示出了藥學物質巴吉林(pargyline)和美雌醇(mestranol)的后期衍生、產生含硅治療劑(sila-therapeutic)的實例。在后者的情況下,發生O-Si和C-Si二者的脫氫偶聯。除非另有說明,用0.5mL溶劑以0.5mmol規模并且在規定的溫度下進行反應。收率是分析純的分離的物質。[Si]=PhMe2Si;DME=1,2-二甲氧基乙烷。
圖5直接比較了在另外的相同條件下KOH和NaOH催化劑的反應性,揭示了對取決于Na+或K+是否存在的反應結果的意料之外的影響。R=PhMe2Si(10a);R=H(10b)。反應條件與在圖3和圖4中為每個特定的含氫硅烷和炔烴給出的相當。除非另有說明,用0.5mL溶劑以0.5mmol規模并且在規定的溫度下進行反應。收率是分析純的分離的物質。DME=1,2-二甲氧基乙烷。
圖6直接比較了在另外的相同條件下NaOH/KOH和KO-tert-BuOH催化劑的反應性,揭示了在某些條件下使用氫氧化物對使用醇鹽的意料之外的益處。
說明性實施方案的詳述
本發明建立于一系列反應上,這些反應中的每一個都依賴于有機硅烷和強堿——包括堿金屬氫氧化物、醇鹽和氫化物(優選氫化物)——的簡單混合物,它們一起形成能夠使液相中的末端炔烴基團甲硅烷基化的原位體系(活性物質的結構和性質仍是未知的),而不存在過渡金屬催化劑、UV輻射或放電(包括等離子體放電)。這些反應與在開發用于制備對農用化學、電子學、精細化學和藥學應用重要的產品的實用方法中的重要進展有關。重要的是,該反應受到極大的關注,因為其僅產生了環境友好的硅酸鹽和二氫化物(dihydrogen)作為副產物并且可以避免正如為了這個目的對于文獻中提出的幾乎所有的其他方法將觀察到的有毒金屬廢物流。由這些體系所表現出的顯著容易性為這些領域中的化學工作者的工具箱中提供有用的工具。當與其他后續反應結合時,此效用可以被充分利用(leverage)。
本發明可以通過參照結合其全部形成本公開內容的一部分的附圖和實施例而進行的以下描述而更容易地理解。應理解,本發明不限于本文中描述的或示出的具體的產物、方法、條件或參數,并且本文中使用的術語僅用實例的方式用于描述具體實施方案的目的且不意圖是任何要求保護的發明的限制。相似地,除非另有具體說明,否則關于用于改進的作用或原因的可能的機制或模式的任何描述意味著僅是說明性的,并且本文的發明將不被用于改進的作用或原因的任何此類建議的機制或模式的正確或不正確約束。貫穿本文,意識到,該描述指的是組合物以及制造和使用所述組合物的方法。換言之,如果本公開描述或要求保護與組合物或者制造或使用組合物的方法相關聯的特征或實施方案,那么應意識到,這種描述或權利要求意圖使這些特征或實施方案延伸至這些上下文中的每個中的實施方案(即組合物、制造方法和使用方法)。
本發明包括與用于使末端炔烴甲硅烷基化的化學體系和方法相關的實施方案。具體的實施方案提供方法,每個方法包括使包含末端炔基C-H鍵的至少一種有機基質與至少一種有機硅烷和堿金屬氫氧化物、醇鹽和氫化物(優選氫氧化物)的混合物在足以形成甲硅烷基化的末端炔基部分的條件下接觸。反應在完全不存在(或基本上完全不存在)過渡金屬化合物時良好地進行。同樣地,這些方法在不存在或基本上完全不存在對于引發或增長所需的其他電磁或熱觸發劑時也是可操作的。即,這些實施方案不需要UV輻射或放電或等離子體放電條件來進行。
如本文使用的描述體系和方法的術語“有機硅烷”或“含氫硅烷”可以可互換地使用并且指的是具有至少一個硅-氫(Si-H)鍵和一個含碳部分的化合物或試劑。有機硅烷還可以含有硅-碳鍵、硅-氧鍵(即包括術語“有機硅烷”)、硅-氮鍵或其組合,并且可以是單體的或被包含在低聚物框架或聚合物框架內,包括被鏈接至異質支撐結構或均質支撐結構。在某些實施方案中,這些有機硅烷可以包括至少一種式(I)、式(II)或式(III)的化合物:
(R)4-mSi(H)m (I)
(R)3-m(H)mSi-(CH2)q-Or-Si(R)3-p(H)p (II)
R-[-SiH(R)-O-]n-R (III)
其中:m和p獨立地為1、2或3;q為0、1、2、3、4、5或6;r為0或1;n為10至100;并且每個R獨立地為鹵素(例如F、Br、Cl、I)(條件是至少一個R為包含碳)、任選地被取代的C1-12烷基或雜烷基、任選地被取代的C1-12烯基或雜烯基、任選地被取代的C1-12炔基或雜炔基、任選地被取代的C5-20芳基或C3-20雜芳基、任選地被取代的C6-30烷芳基或雜烷芳基、任選地被取代的C5-30芳烷基或雜芳烷基、任選地被取代的-O-C1-12烷基或雜烷基、任選地被取代的-O-C5-20芳基或-O-C3-20雜芳基、任選地被取代的-O-C5-30烷芳基或雜烷芳基、或任選地被取代的-O-C5-30芳烷基或雜芳烷基,并且如果被取代,取代基可以為膦酸根(phosphonato)、磷酰基(phosphoryl)、氧膦基(phosphanyl)、膦基(phosphino)、磺酸根(sulfonato)、C1-C20烷基硫烷基(alkylsulfanyl)、C5-C20芳基硫烷基、C1-C20烷基磺酰基、C5-C20芳基磺酰基、C1-C20烷基亞磺酰基、C5-C20芳基亞磺酰基、磺酰氨基、氨基、酰氨基、亞氨基、硝基、亞硝基、羥基、C1-C20烷氧基、C5-C20芳氧基、C2-C20烷氧基羰基、C5-C20芳氧基羰基、羧基、羧酸根(carboxylato)、巰基、甲酰基、C1-C20硫酯、氰基、氰酸根(cyanato)、硫代氰酸根(thiocyanato)、異氰酸酯(isocyanate)、硫代異氰酸酯(thioisocyanate)、氨基甲酰基、環氧基、苯乙烯基、甲硅烷基、甲硅氧基、硅烷基、硅氧烷氮烷基(siloxazanyl)、硼酸根(boronato)、硼基或鹵素,或含有金屬的基團或含有準金屬的基團,其中準金屬是Sn或Ge,其中取代基可以任選地向包括氧化鋁、二氧化硅或碳的不溶性或微溶性支持介質提供鏈接部(tether)。示例性的、非限制性的有機硅烷可以獨立地包括(R)3SiH或(R)2SiH2或(R)SiH3。在式(II)的一些實施方案中,q為0。在式(II)的一些實施方案中,r為0。在一些實施方案中,R獨立地為烷氧基、烷基、烯基、芳基、芳氧基、雜芳基、芳烷基或雜芳烷基。在某些實施方案中,(R)3SiH包括使用烷基、芳基、雜芳基、烷氧基或混合的烷基-芳基硅烷或烷基-雜芳基硅烷,例如EtMe2SiH、(n-Bu)3SiH、Et2SiH2、PhMe2SiH、BnMe2SiH、(EtO)3SiH、(i-Pr)3SiH、Me2(吡啶基)SiH或(i-Pr)2(吡啶基)SiH、或Me3Si-SiMe2H。涉及R2(吡啶基)SiH硅烷的實施方案(即涉及它們的方法和體系)是特別獨特的,因為發明人不知道這些已經通過任何類型的催化體系被并入。聚合物材料諸如聚甲基氫硅氧烷(PMHS)也有效。
使用具有一般結構(R)2SiH2和(R)SiH3的有機硅烷也很好地起作用,并且提供用于偶聯或橋連反應的機會,如本文描述的。在單個基質的存在下,雙-炔基硅烷已經以良好收率被分離(參見,例如實施例3.4;圖4C)。可能的是,在足夠溫和的條件下,可以獲得相應的單炔基硅烷,但有待觀察。有趣的是,R2SiH2(和(R)SiH3)有機硅烷還可以與不同的基質反應以獲得對稱的和不對稱的雙-或三-炔基硅烷(再次,參見,例如實施例3.4)。注意,實施例3.4描述了等摩爾量的兩種不同基質的反應,產生主要是交叉偶聯的(76%)產物混合物。相對于可能已經從兩種基質的純統計學組合所預期的,此交叉偶聯產物的大量富集的原因是未知的,但暗示了本發明的方法學可能提供了用于優先形成這樣的二炔基或三炔基交叉偶聯的硅烷產物的有用的工具。
另外,在R2SiH2或式(II)的硅烷的存在下使用乙炔或聚炔烴基質可能對于制備聚合的或環狀的乙炔基硅烷是有用的,所述聚合的或環狀的乙炔基硅烷的非限制性實例包括結構單元:
這些結構中的一些已經由Gleiter,R.和D.B.Werz,Chem.Rev.2010,110,4447-4488描述,其出于所有目的通過引用以其整體并入本文。
之前,發明人中的一些報道了使用醇鉀催化劑和氫氧化鉀催化劑來實現芳族基質和雜芳族基質的甲硅烷基化,并且注意到基于鉀的堿在此方面的獨特能力。在此,發明人確認,盡管關于一些基質可以使用鉀(和鈉)的醇鹽(并且在一些情況下KH),反應的范圍相對地受限制(參見,例如圖6)。在使末端炔烴C-H基團甲硅烷基化時,使用堿金屬氫氧化物,特別是氫氧化鈉(NaOH)和氫氧化鉀(KOH),提供了使更廣泛的和更多種的基質甲硅烷基化的可能性。有趣的是,并且由于未完全了解的原因,至少在本文描述的溫和的反應條件下(參見,例如圖5),某些有機硅烷-基質組合用KOH或NaOH實際操作獲得良好收率(例如EtMe2SiH、PhMe2SiH、(n-Bu)3SiH、Et2SiH2、(i-Pr)2(吡啶基)SiH或Me2(吡啶基)SiH),而其他的似乎對使用NaOH更好地響應(PhMe2SiH、BnMe2SiH、(EtO)3SiH、Me2(吡啶基)SiH或Me3Si-SiMe2H),并且另一些對使用KOH((i-Pr)3SiH或(i-Pr)2(吡啶基)SiH)更好地響應。該陽離子效應(NaOH對KOH)似乎還依賴于基質的性質(比較Me2PhSiH和圖5中的多種基質的反應性)。此獨特且以前未被認識到的陽離子效應的機械學的原因(mechanistic reason)尚未被了解。考慮到它們在其他芳基和雜芳基體系中的不易使用性(unworkability),NaOH和醇鈉(表1)影響這些甲硅烷基化的能力特別有趣。注意,提到獨立使用氫氧化鈉、醇鈉、氫氧化鉀和醇鉀時,盡管優選,但不排除在與彼此的任意組合中使用這些物質,并且這些混合物被認為是另外的實施方案。
實施例提供了對于實現期望的轉化有用的示例性的反應條件。在其他實施方案中,基質、堿金屬氫氧化物、堿金屬醇鹽和堿金屬氫化物(優選堿金屬氫氧化物)和有機硅烷可以被加熱至范圍在0℃至150℃或更高的溫度,持續范圍從24小時至數天的時間,盡管實際上,當在范圍在環境室溫(例如25℃)至約85℃的溫度下進行時,反應進行至良好的收率和選擇性。有趣的是,在此應用內示出了,通過使反應溫度分階段進行(例如,從平穩的45℃至65℃),可能的是,選擇并提供在具有2個外觀上等同的(apparently equivalent)末端炔基C-H鍵的基質上是單甲硅烷基化或二甲硅烷基化的產物(參見,例如實施例3.3,圖4B)。
這些方法通常使用烴或基于醚的溶劑,或可以不使用溶劑來操作。醚溶劑諸如四氫呋喃類(包括2-甲基四氫呋喃)、二乙醚和二甲醚、1,2-二甲氧基乙烷、二氧六環和以烷基封端的二醇類,已經示出很好地起作用。
對于基質而言,方法是相當靈活的,特別是當考慮NaOH、KOH或其混合物時。本發明的方法提供了以出色的效率使寬范圍的、具有一個、兩個、或更多個末端炔基C-H鍵的基質的甲硅烷基化。在一些實施方案中,包含末端炔基C-H鍵的有機基質根據下式描述:
R1-C≡C-H,
其中R1包括H、任選地被取代的烷基、任選地被取代的烯基、任選地被取代的炔基、任選地被取代的芳基、任選地被取代的雜烷基、任選地被取代的雜芳基、任選地被取代的芳烷基、任選地被取代的雜芳烷基或任選地被取代的金屬茂。R1可以包括單獨的分子部分或可以為寡聚的或聚合的。
獨立的實施方案包括其中R1為以下或包括以下的那些實施方案:
(a)任選地被取代的直鏈烷基、任選地被取代的支鏈烷基或任選地被取代的環烷基;
(b)任選地被取代的直鏈烯基、任選地被取代的支鏈烯基或任選地被取代的環烯基;
(c)任選地被取代的直鏈雜烷基、任選地被取代的支鏈雜烷基或任選地被取代的雜環烷基;
(d)任選地被取代的芳基、任選地被取代的芳烷基、任選地被取代的雜芳基或任選地被取代的雜芳烷基;或
(e)在(a)至(d)中列出的任意兩個或更多個類型的取代基的組合。
在更多具體的實施方案中,R1為或包括:
(a)任選地被取代的苯、聯苯、萘或蒽環結構;或
(b)任選地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、三唑、異噁唑、噁唑、噻唑、異噻唑、噁二唑、吡啶、噠嗪、嘧啶、吡嗪、三嗪酮、苯并呋喃、苯并吡咯、苯并噻吩、異苯并呋喃、異苯并吡咯、異苯并噻吩、吲哚、異吲哚、吲嗪、吲唑、氮雜吲哚、苯并異噁唑、苯并噁唑、喹啉、異喹啉、噌啉、喹唑啉、萘啶、2,3-二氫苯并呋喃、2,3-二氫苯并吡咯、2,3-二氫苯并噻吩、二苯并呋喃、呫噸(xanthene)、二苯并吡咯、或二苯并噻吩部分;或
(c)包含末端炔基C-H鍵的有機基質是聚合的。
基質和有機硅烷中的每一種代表本發明的范圍內的物質的具體實例和實施方案。
一旦形成,甲硅烷基化的末端炔基部分可以經受多種已知的化學反應,并且本發明預期,當與本文描述的本發明的方法結合時,使用這些已知方法的方法在本發明的范圍內。為了清楚的目的,此處引入術語“最初的甲硅烷基化產物”來代表本發明的方法的甲硅烷基產物,此最初的甲硅烷基化產物含有之前描述的甲硅烷基化的末端炔基部分。除了另有說明的,以下反應代表其中在終產物中并入了甲硅烷基的優異的方式。
例如,炔烴在使用Diels-Alder型[2+2]或[4+2]環加成反應形成甲硅烷基化的芳族、雜芳族、環烯基或雜環烯基部分中是有用的合成子。附接至本發明的產物的炔烴基團的甲硅烷基的預先并入提供了在此類芳族、雜芳族、環烯基或雜環烯基產物上并入甲硅烷基的有趣的可選擇的手段。因此,在本發明的一些實施方案中,由本發明的方法形成的甲硅烷基化的末端炔基部分還可以與另一種不飽和部分(例如任選地被取代的烯烴、炔烴、疊氮化物、腈、異氰酸酯、異硫氰酸酯、羰基、酰胺、脲等)反應以形成甲硅烷基化的芳族、雜芳族、環烯基或雜環烯基結構。此“另一種不飽和部分”可以作為單獨的分子實體被引入(即分子間反應),或者可以存在于最初的甲硅烷基化產物中(即分子內反應)——對于此的唯一實例,參見例如圖4C。在任一種情況下,在實現環化之前,最初的甲硅烷基化產物可以或可以不需要被分離,并且,如果是后者,反應可以在單鍋法合成中進行。
在其他實施方案中,最初的甲硅烷基化產物還可以在交叉復分解反應中與第二不飽和的有機部分(環狀的或無環的,包括任選地被取代的烯烴、炔烴、疊氮化物、腈、異氰酸酯、異硫氰酸酯、羰基、酰胺、脲等)反應以形成甲硅烷基化的二烯烴或聚烯烴產物。這樣的交叉復分解反應是公知的,并且普通技術人員將知曉如何實現這些轉化。例如,使用Grubbs型釕卡賓復分解催化劑可以被用于此目的,盡管預期的轉化不限于這些類型的催化劑。這些可以示意性地表示為:
再次,反應可以是分子內或分子間的、單鍋法或多鍋法合成,并且提供了用于在容易獲得且溫和的條件下并入甲硅烷基的另一種方法。這樣的下游轉化在例如Kim等人,J.Amer.Chem.Soc.,126(33),2004,10242-10243中描述,其出于其至少在此方面的教導通過引用并入本文。然后,這些產物可以經受以上描述的Diels-Alder型[2+2]或[4+2]環加成反應。
可選擇地或另外,使用任何合適的催化劑,最初的甲硅烷基化產物可以與任選地被取代的烯炔、二烯烴、二炔烴或環烯烴共聚合以形成甲硅烷基化聚合物。在某些實施方案中,這些反應可以包括易位聚合(metathesis polymerization),例如ROMP。這樣的易位聚合反應是公知的,并且普通技術人員將知曉如何實現它們,例如再次使用Grubbs型釕卡賓復分解催化劑,盡管預期的轉化不限于這些類型的催化劑。再次,反應可以是分子內或分子間的、單鍋法或多鍋法合成,并且提供了用于在容易獲得且溫和的條件下將甲硅烷基并入聚合物的另一種方法。例如,這樣的方法可以提供將在電子應用中有用的甲硅烷基化的導電聚乙炔聚合物。
在其他實施方案中,最初的甲硅烷基化產物可以被進一步反應以使甲硅烷基化的末端炔基部分氫化。此甲硅烷基化的末端炔基部分還可以與水、醇、氰化氫、氯化氫、鹵素單質(dihalogen)或羧酸在已知得到相應的乙烯基化合物或羰基型化合物的條件下反應。再次,技術人員將能夠實現這些轉化而不需要過多的努力。
在其他實施方案中,最初的甲硅烷基化產物可以與有機疊氮化物在[3+2]疊氮化物-炔烴環加成反應中反應,例如形成甲硅烷基化的三唑。這樣的反應也是公知的,被稱為所謂的點擊化學,其包括1,3-偶極環加成。這樣的反應可以是分子內或分子間反應,并且通常由銅、銅-鐵或含釕催化劑催化。再次,不需要過多的負擔而實現這樣的轉化是在普通技術人員的技能范圍內的。
其他實施方案提供了,炔基芳基硅烷與例如三氟甲磺酸反應,以形成硅醇。參見,例如Franz,A.K.等人,J.Med.Chem.,2013,56,388-405,其出于所有目的通過引用以其整體并入本文。因此,本發明的某些實施方案提供了最初的甲硅烷基化產物與三氟甲磺酸的進一步反應以形成相應的硅醇。因為本發明還允許并入烷氧基硅烷(例如(RO)3SiH),可以簡單地通過使最初的甲硅烷基化產物水解來制備相似的硅醇產物,其中最初的甲硅烷基化產物含有烷氧基甲硅烷基。
在本發明的這些方法中的溫和的條件以及不需要任何過渡金屬催化劑,使這些方法特別適用于藥學和醫學應用,其中甲硅烷基衍生物已經被證明特別重要。除了使用本發明的甲硅烷基衍生物用于作用方式、增加組織滲透的機械學測定、改變氫鍵作用和缺乏已知的毒性作用,有機硅化合物還被用作生物顯像劑。可以預期,本發明的甲硅烷基化的末端炔烴以其本身對于這些目的是有效的,因為方法允許在藥物合成中的幾乎任何階段并入寬范圍的甲硅烷基部分。另外,硅氧基化的末端烯烴(例如,其中甲硅烷基化的末端炔基部分包含-Si(OR)3基團)可以進一步與例如KHF2反應,以形成四氟硅酸鹽基團——即,作為R1-C≡C-SiF4-化合物(或其放射性標記的18F型式)。這樣的轉化和優勢在例如Franz,A.K.等人,J.Med.Chem.,2013,56,388-405中描述。
本發明的末端炔烴硅烷還可以被氟化物或醇鹽活化,并且相關地去除甲硅烷基以形成炔基親核試劑,所述炔基親核試劑可以然后與親電試劑反應。因此,其他實施方案還提供了甲硅烷基化的末端炔基部分在存在或不存在比HF更強的酸時與含氟的鹽(例如堿金屬鹽或四芳基銨鹽、四烷基銨鹽或混合的烷基/芳基銨鹽)反應,以產生活化的、脫甲硅烷基的炔基親核試劑,以被添加至其他基質,例如烷基鹵化物(或具有合適的離去基團的任何烷基,所述離去基團包括但不限于對甲苯磺酸酯、三氟甲磺酸酯或任何醇、胺、或羧酸保護基)、酰基鹵化物(包括酰氯)、被取代的烯烴(例如Michael加成反應受體)、烯酮(或一般地,α,β-不飽和羰基化合物)、環氧化物、酯、α-酮-酯(包括三氟丙酮酸酯或其他被取代的丙酮酸酯)等。在手性鈀催化劑——諸如(但不限于)(S)-BINAP-Pd2+催化劑——的存在下,可以完成反應以產生具有光學活性的加成產物。這樣的化學在例如Aikawa,K.等人,Org.Lett.,12,5716-5719(2010)中描述,其出于所有目的通過引用并入。
在其他實施方案中,最初的甲硅烷基化產物可以被進一步反應僅為了去除最初添加至末端炔基C-H鍵的甲硅烷基并且用氫、氘、或氚原子代替它。這可以簡單地通過例如用(同位素的)質子源使脫甲硅烷基的炔基親核試劑猝滅獲得。如果甲硅烷基被最初添加以保護否則是酸性的末端炔基C-H基團免于在基質上進行的反應而遠離該C-H基團,或充當定向基團(例如,參見以下描述的碳鎂化反應),這樣的策略將是有用的。在仍其他實施方案中,甲硅烷基化的末端炔基部分可以與N-鹵代琥珀酰亞胺在陽離子型金催化劑的存在下反應,以產生末端炔基鹵化物,其中鹵素優選為溴或碘。再次,這樣的反應是已知的并且在例如Starkov等人,Adv.Synth.Catal.,2012,354,第3217-3224頁中描述,其出于所有目的通過引用以其整體并入本文。
在其他實施方案中,最初的甲硅烷基化產物還可以與芳族鹵化物在足以形成炔基-芳烴鍵的條件下反應,例如使用Pd/Cu催化劑,諸如Pd(PPh3)2Cl2、CuI催化劑和任選地被取代的碘芳族化合物或溴芳族化合物。例如,
其中R和R1如以上所述,并且[Het]Ar-X指任選地被取代的芳基或雜芳基溴或碘。
在其他具體實施方案中,其中至少一種有機硅烷包含任選地被取代的C1-12烯基或雜烯基,使得甲硅烷基化的末端炔基部分包含硅鍵合的任選地被取代的烯基或雜烯基,甲硅烷基化的末端炔基部分還可以與醇和催化劑在產生甲硅烷基化的末端炔基部分的分子內烯丙基化的條件下反應:
其中R和R1如以上所述。這樣的反應已經在例如Park和Lee,J.Amer.Chem.Soc.2006,128,10664-10665中描述,其出于所有目的通過引用以其整體并入本文。
在其中至少一種有機硅烷包含2-吡啶基(其中(Me)2(吡啶基)SiH或(i-Pr)2(吡啶基)SiH僅是兩個非限制性實例)的具體情況下,另外的實施方案提供這樣的方法,其中該甲硅烷基化的末端炔基部分與銅碳鎂化催化劑和任選地被取代的芳基鎂絡合物或任選地被取代的雜芳基鎂絡合物在適合于且足以使甲硅烷基化的末端炔基部分碳鎂化的條件下反應。這樣的反應被詳細地記錄,例如在Itami等人,Synlett 2006,第2期,157-180中,其出于所有目的通過引用以其整體并入本文。在這樣的情況下,例如,如下示出的最初的甲硅烷基化產物與任選地被取代的芳族鎂絡合物(諸如關于[Het]Ar1–MgI所描述的)的反應,產生相應的碳鎂化產物:
其中R和R1如上所述,并且[Het]Ar1是任選地芳基或雜芳基部分,再次如上所述。對于進行這個和以下的轉化有用的條件在以上引用的Itami參考文獻中可獲得。組合的反應是形成立體專一性產物的強大的方式。
然后,碳鎂化的甲硅烷基化的末端炔基部分可以與任選地被取代的芳基碘或任選地被取代的雜芳基碘(此處被表示為[Het]Ar2)在鈀催化劑的存在下反應以形成三取代的甲硅烷基化烯烴。例如,
其中[Het]Ar2也是任選地被取代的芳基或雜芳基部分,再次如上所述,其可以與[Het]Ar1相同或不同。
然后,三取代的甲硅烷基化的烯烴可以與BCl3和頻哪醇在足以使該化合物硼脫甲硅烷基化的條件下反應。在單獨的步驟中,硼脫甲硅烷基化的化合物可以與第二任選地被取代的芳基碘或任選地被取代的雜芳基碘在適于使所得的C-B鍵與該第二任選地被取代的芳基碘或任選地被取代的雜芳基碘交叉偶聯的條件下反應。這些反應被示意性地表示為:
其中[Het]Ar3也是任選地被取代的芳基或雜芳基部分,再次如上所述,其可以與[Het]Ar1或[Het]Ar2相同或不同。硼脫甲硅烷基化反應之前已經例如在Babudri等人,Tetrahedron 1998,54,1085)中被描述為具有Suzuki-Miyaura型基于硼的交叉偶聯反應(例如,參見(a)Miyaura,N.;Suzuki,A.Chem.Rev.1995,95,2457;和(b)Miyaura,N.Top.Curr.Chem.2002,219,11)。出于全部目的,這些參考文獻中的每一個通過引用以其整體并入本文。
應當理解,使含有2-吡啶基甲硅烷基的最初的甲硅烷基化產物氫化可以產生相應的2-吡啶基甲硅烷基烯烴,并且參考Itami(即Itami等人,Synlett 2006,第2期,157-180)描述了這樣的化合物的豐富化學。至當地法律允許的程度,源自本公開內容中描述的本發明的方法的產物的這種轉化也被認為在本發明的范圍內。
至此,已經關于催化甲硅烷基化的末端炔基C(sp)-H鍵的方法描述了本發明的概念。應當理解,從這樣的方法獲得的產品(至它們通過本申請時已知的其他方式實際上不可用的程度)以及這些方法中使用的體系都被認為在本公開內容的范圍內。
再次,本發明包括用于實現本文所述的任何方法所必需的任何體系的實施方案。例如,某些實施方案提供了用于使包含末端炔基C-H鍵的有機基質甲硅烷基化的體系,每個體系包括以下的混合物或主要由以下的混合物組成:(a)至少一種有機硅烷和(b)堿金屬氫氧化物(并且在一些情況下,醇鈉或醇鉀,或氫化鈉或氫化鉀,或其混合物)以及(c)至少一種基質。這樣的體系通常包括體系在其上是可操作的基質,該基質包含至少一個末端炔基C(sp)-H部分。通常,體系大體上無過渡金屬化合物,或當存在時,該過渡金屬可以被認為是反應的旁觀者(spectator)。在一些實施方案中,該體系還包括存在來源于基質和至少一種有機硅烷之間的反應的甲硅烷基化的末端炔烴。
在這樣的體系中,該至少一種有機硅烷包括式(I)、式(II)或式(III)的有機硅烷:
(R)4-mSi(H)m (I)
(R)3-m(H)mSi-(CH2)q-Or-Si(R)3-p(H)p (II)
R-[-SiH(R)-O-]n-R (III)
其中:m、n、p、q、r和R在其他地方描述。類似地,在體系的多個獨立地實施方案中:
(a)有機硅烷是(R)3SiH、(R)2SiH2、或(R)SiH3;
(b)R獨立地包含烷氧基、烷基、烯基、芳基、芳氧基、雜芳基、芳烷基或雜芳烷基;
(c)堿金屬氫氧化物是氫氧化鈉(NaOH)、氫氧化鉀(KOH)或其組合;
(d)堿金屬醇鹽是氫氧化鈉、醇鉀(KOH)或其組合;
(e)包含末端炔基C-H鍵的有機基質具有下式;
R1-C≡C-H,
其中R1根據以上描述的任一個方法實施方案定義。
術語
在本公開內容中,單數形式“一(a)”、“一(an)”和“該(the)”包括復數指代物,并且對具體數值的指代包括至少該具體值,除非上下文另有清楚地指示。因此,例如,對“一種材料”的指代是對本領域技術人員已知的此類材料及其等同物中的至少一種的指代,等等。
當值通過使用描述符“約”被表示為近似值時,應理解,該具體值形成另一個實施方案。一般而言,術語“約”的使用指示近似值,該近似值可以取決于將通過公開的主題獲得的所尋求的期望性能而變化并且將在其中使用該主題的具體上下文中基于該主題的功能來理解。本領域技術人員將能夠使該術語解釋為常規內容。在某些情況下,用于具體值的有效數字的數可以是確定詞語“約”的程度的一個非限制性方法。在其他情況下,在一系列值中使用的等級可以用于確定預期范圍,該預期范圍對于每個值的術語“約”是可用的。如果存在,那么所有的范圍是包括端點的并且是可組合的。換言之,對范圍中陳述的值的指代包括在該范圍內的每個值。
應意識到,為了清楚起見,在本文中的單獨的實施方式的上下文中描述的本發明的某些特征也可以在單一的實施方案中被組合地提供。換言之,除非明顯地不可相容或具體地排除的,否則每個單個的實施方案被視為是與任何其他實施方案可組合的并且這種組合被認為是另一個實施方案。相反地,為了簡潔起見,單一的實施方案的上下文中描述的本發明的各種特征也可以被單獨提供或以任何子組合來提供。最后,雖然實施方案可以被描述為一系列步驟的一部分或更多通用結構的一部分,但是每個所述步驟也可以被認為是與其他實施方案可組合的自身獨立的實施方案。
過渡術語“包含”、“基本上由......組成”和“由......組成”意圖包含它們的在專利行話中的被普遍接受的意思;即,(i)與“包括”、“含有”或“以......為特征”同義的“包含”是包括端點的或開放式的并且不排除另外的、未引用的要素或方法步驟;(ii)“由......組成”排除未在權利要求中指定的要素、步驟或成分;并且(iii)“基本上由......組成”將權利要求的范圍限制于指定的材料或步驟以及不實質上影響要求保護的發明的基礎特征和新穎特征的材料或步驟。用短語“包含”(或其等同物)描述的實施方案也提供在用“由......組成”和“基本上由......組成”獨立地描述的那些作為實施方式。對于關于“主要由……組成”提供的那些實施方案,基本的和新穎的特征是所述方法僅使用列出的那些成分以有意義的收率提供甲硅烷基化的產物(或在此類方法中使用的體系以有意義的收率提供產物組合物或由其衍生的組合物的能力)以使末端炔基C(sp)-H部分甲硅烷基化的容易操作性。在提供了包括使用主要由基質、有機硅烷(可選擇地被稱為含氫硅烷)和強堿(鈉或鉀的氫氧化物、醇鹽、或氫化物)組成的混合物的體系或方法的實施方案中,它指的是這樣的事實:該體系運行以使基質在對應于本文中描述的速率下、在與本文中描述的相當的條件下,在不具有另外的(例如過渡金屬)催化劑或等離子體或UV輻射源的情況下甲硅烷基化。盡管可能存在一定水平的過渡金屬(例如,作為基質),它們對于方法的可操作性不是必需的,并且可以被認為是此反應的目的的旁觀者。實際上,進行的廣泛的實驗和分析排除了由偶然的(adventitious)過渡金屬殘余物的催化(參見實施例2.1.2,表2)。類似地,盡管其他之前的甲硅烷基化反應已經使用等離子體或UV輻射來操作,但本發明不需要這些能量源。另外存在的這些能量源不應被視為替代引起本發明的方法的可操作性的基礎。術語“有意義的產物收率”意圖反映大于50%的產物收率,但當指明時,該術語還可以指相對于最初的基質的量的10%、20%、30%、40%、50%、60%、70%、80%或90%或更高的收率。
當呈現出列表時,除非另有指示,否則應理解,該列表的每個單個的要素以及該列表的每個組合是單獨的實施方案。例如,呈現為“A、B或C”的實施方案的列表應當被解釋為包括以下實施方案:“A”、“B”、“C”、“A或B”、“A或C”、“B或C”或“A、B或C”。相似地,諸如C1-3的名稱包括C1、C2、C3、C1-2、C2-3、C1,3作為單獨的實施方案,以及C1-3。
貫穿本說明書,詞語應當被給予它們的正常意思,如相關領域技術人員將理解的。然而,為了避免誤解,某些術語的意思將被特別地定義或闡明。
如本文使用的術語“烷基”指的是直鏈的、支鏈的或環狀的飽和烴基團,通常但不一定含有1個至約24個碳原子,優選地含有1個至約12個碳原子,例如甲基、乙基、正丙基、異丙基、正丁基、異丁基、叔丁基、辛基、癸基及類似基團,以及環烷基例如環戊基、環己基及類似基團。通常,雖然再次不一定,但是本文的烷基含有1至約12個碳原子。術語“低級烷基”意指1個至6個碳原子的烷基,并且特定術語“環烷基”意指通常具有4個至8個、優選地5個至7個碳原子的環狀的烷基。術語“取代的烷基”指的是被一個或更多個取代基基團取代的烷基,并且術語“含雜原子的烷基”和“雜烷基”指的是其中至少一個碳原子被雜原子代替的烷基。如果沒有另外指明,那么術語“烷基”和“低級烷基”分別包括直鏈的、支鏈的、環狀的、未取代的、取代的和/或含雜原子的烷基和低級烷基。
如本文使用的術語“亞烷基”指的是雙官能的直鏈的、支鏈的或環狀的烷基,其中“烷基”如上文所定義。
如本文使用的術語“烯基”指的是含有至少一個雙鍵的2個至約24個碳原子的直鏈的、支鏈的或環狀的烴基,例如乙烯基、正丙烯基、異丙烯基、正丁烯基、異丁烯基、辛烯基、癸烯基、十四烯基、十六烯基、二十烯基、二十四烯基及類似基團。本文中優選的烯基含有2個至約12個碳原子。術語“低級烯基”意指2個至6個碳原子的烯基,并且特定術語“環烯基”意指優選地具有5個至8個碳原子的環狀的烯基。術語“被取代的烯基”指的是被一個或更多個取代基基團取代的烯基,并且術語“含雜原子的烯基”和“雜烯基”指的是其中至少一個碳原子被雜原子代替的烯基。如果沒有另外指明,那么術語“烯基”和“低級烯基”分別包括直鏈的、支鏈的、環狀的、未取代的、取代的和/或含雜原子的烯基和低級烯基。
如本文使用的術語“亞烯基”指的是雙官能的直鏈的、支鏈的或環狀的烯基,其中“烯基”如上文所定義。
如本文使用的術語“炔基”指的是含有至少一個三鍵的2個至約24個碳原子的直鏈的或支鏈的烴基,例如乙炔基、正丙炔基及類似基團。本文中優選的炔基含有2個至約12個碳原子。術語“低級炔基”意指2個至6個碳原子的炔基。術語“被取代的炔基”指的是被一個或更多個取代基基團取代的炔基,并且術語“含雜原子的炔基”和“雜炔基”指的是其中至少一個碳原子被雜原子代替的炔基。如果沒有另外指明,那么術語“炔基”和“低級炔基”分別包括直鏈的、支鏈的、未取代的、取代的和/或含雜原子的炔基和低級炔基。
如本文使用的術語“烷氧基”意指通過單獨的、末端醚鍵結合的烷基;即,“烷氧基”基團可以表示為-O-烷基,其中烷基是如上文定義的。“低級烷氧基”指含有1個至6個碳原子的烷氧基。類似地,“烯氧基”和“低級烯氧基”分別指的是通過單獨的、末端醚鍵結合的烯基和低級烯基,并且“炔氧基”和“低級炔氧基”分別是指通過單獨的、末端醚鍵結合的炔基和低級炔基。
術語“芳族的”指的是滿足芳香性的Hückel 4n+2規則的環部分,并且包括芳基(即,碳環的)結構和雜芳基(也被稱為雜芳族的)結構二者,這些結構包括芳基、芳烷基、烷芳基、雜芳基、雜芳烷基、或烷雜芳基的部分、或預聚合物(例如,單體、二聚體)、或其低聚物類似物或聚合物類似物。
如本文使用的術語“芳基”,并且除非另有指定,否則指的是含有單個芳環或稠合在一起、直接連接或間接連接(使得不同的芳環被結合到共用基團例如亞甲基部分或亞乙基部分)的多個芳環的芳族取代基或結構。除非另有修飾,否則術語“芳基”指的是碳環結構。優選的芳基含有5個至24個碳原子,并且特別優選的芳基含有5個至14個碳原子。示例性芳基含有一個芳環或兩個稠合或連接的芳環,例如苯基、萘基、聯苯基、二苯醚、二苯胺、二苯甲酮等。“被取代的芳基”是指被一個或更多個取代基取代的芳基部分,并且術語“含雜原子的芳基”和“雜芳基”是指其中至少一個碳原子被雜原子代替的芳基取代基,如下文將進一步詳細描述。
如本文使用的術語“芳氧基”指的是通過單獨的、末端醚鍵結合的芳基,其中“芳基”是如上文定義的。“芳氧基”基團可以表示為-O-芳基,其中芳基是如上文定義的。優選的芳氧基含有5個至24個碳原子,并且特別優選的芳氧基含有5個至14個碳原子。芳氧基的實例包括但不限于苯氧基、鄰-鹵代-苯氧基、間-鹵代-苯氧基、對-鹵代-苯氧基、鄰-甲氧基-苯氧基、間-甲氧基-苯氧基、對-甲氧基-苯氧基、2,4-二甲氧基-苯氧基、3,4,5-三甲氧基-苯氧基及類似基團。
術語“烷芳基”指的是具有烷基取代基的芳基,并且術語“芳烷基”指的是具有芳基取代基的烷基,其中“芳基”和“烷基”是如上文定義的。優選的烷芳基和芳烷基含有6個至24個碳原子,并且特別優選的烷芳基和芳烷基含有6個至16個碳原子。烷芳基包括,例如,對-甲基苯基、2,4-二甲基苯基、對-環己基苯基、2,7-二甲基萘基、7-環辛基萘基、3-乙基-環戊-1,4-二烯及類似基團。芳烷基的實例包括但不限于芐基、2-苯基-乙基、3-苯基-丙基、4-苯基-丁基、5-苯基-戊基、4-苯基環己基、4-芐基環己基、4-苯基環己基甲基、4-芐基環己基甲基及類似基團。術語“烷芳氧基”和“芳烷氧基”指的是式-OR的取代基,其中R分別是如剛剛定義的烷芳基或芳烷基。
術語“酰基”指的是具有式-(CO)-烷基、-(CO)-芳基、或-(CO)-芳烷基的取代基,并且術語“酰氧基”指的是具有式-O(CO)-烷基、-O(CO)-芳基或-O(CO)-芳烷基的取代基,其中“烷基”、“芳基”和“芳烷基”如上文所定義。
術語“環狀的”和“環”指的是脂環族基團或芳族基團,這些基團可以或不可以被取代和/或是含雜原子的,并且可以是單環的、雙環的或多環的。術語“脂環族的”在常規的意義上用于指代脂肪族的環狀部分,如與芳族的環狀部分相反的,并且可以是單環的、雙環的或多環的。術語“無環的”指的是其中雙鍵不被包含在環結構內的結構。
術語“鹵代”、“鹵化物”和“鹵素”在常規的意義上用于指代氯、溴、氟或碘的取代基。
“烴基”指的是含有1個至約30個碳原子、優選地1個至約24個碳原子、最優選地1個至約12個碳原子的一價烴基基團,包括直鏈的、支鏈的、環狀的、飽和的和不飽和的物質,例如烷基、烯基、芳基及類似基團。術語“低級烴基”意指1個至6個碳原子、優選地1個至4個碳原子的烴基,并且術語“亞烴基”意指含有1個至約30個碳原子、優選地1個至約24個碳原子、最優選地1個至約12個碳原子的二價烴基部分,包括直鏈的、支鏈的、環狀的、飽和的和不飽和的物質。術語“低級亞烴基”意指1個至6個碳原子的亞烴基。“被取代的烴基”指的是被一個或更多個取代基基團取代的烴基,并且術語“含雜原子的烴基”和“雜烴基”指的是其中至少一個碳原子被雜原子代替的烴基。相似地,“被取代的亞烴基”指的是被一個或更多個取代基基團取代的亞烴基,并且術語“含雜原子的亞烴基”和“雜亞烴基”指的是其中至少一個碳原子被雜原子代替的亞烴基。除非另外指明,否則術語“烴基”和“亞烴基”應分別被解釋為包括取代的和/或含雜原子的烴基部分和亞烴基部分。
如“含雜原子的烴基”中的術語“含雜原子的”指的是其中一個或更多個碳原子被除碳之外的原子例如氮、氧、硫、磷或硅、通常是氮、氧或硫代替的烴分子或烴基分子碎片。相似地,術語“雜烷基”指的是含雜原子的烷基取代基,術語“雜環的”指的是含雜原子的環狀取代基,術語“雜芳基”和“雜芳族的”分別指的是含雜原子的“芳基”和“芳族的”取代基及類似基團。應注意,“雜環的”基團或化合物可以是或可以不是芳族的,并且此外,“雜環”可以是如上文關于術語“芳基”描述的單環、雙環或多環。雜烷基的實例包括烷氧基芳基、烷基硫烷基取代的烷基、N-烷基化的氨基烷基及類似基團。雜芳基取代基的非限制性實例包括吡咯基、吡咯烷基、吡啶基、喹啉基、吲哚基、嘧啶基、咪唑基、1,2,4-三唑基、四唑基,等等,并且含雜原子的脂環族基團的實例是吡咯烷基、嗎啉基、哌嗪基、哌啶基,等等。
如本文使用的,術語“基質”或“有機基質”意圖表示離散的小分子(有時描述為“有機化合物”)和含有此類“芳族部分”的低聚物和聚合物二者。術語“芳族部分”意圖指的是具有所指示的芳族結構中的至少一種的化合物、預聚物(即能夠聚合的單體化合物)、低聚物或聚合物的那些部分。當作為結構示出時,該部分至少含有被示出的部分,以及含有另外的官能團、取代基或二者,包括但不限于本文中描述為“Fn”的官能團。
如在上文提到的定義中的某些中提到的,如“取代的烴基”、“取代的烷基”、“取代的芳基”及類似物中的“取代的”意指在烴基、烷基、芳基雜芳基或其他部分中結合至碳(或其他)原子的至少一個氫原子被一個或更多個非氫取代基代替。這樣的取代基的實例包括但不限于:在本文中被稱為“Fn”的官能團,諸如鹵素(例如F、Cl、Br、I)、羥基、氫硫基、C1-C24烷氧基、C2-C24烯氧基、C2-C24炔氧基、C5-C24芳氧基、C6-C24芳烷氧基、C6-C24烷芳氧基、酰基(包括C1-C24烷基羰基(-CO-烷基)和C6-C24芳基羰基(-CO-芳基))、酰氧基(-O-酰基,包括C2-C24烷基羰氧基(-O-CO-烷基)和C6-C24芳基羰氧基(-O-CO-芳基))、C2-C24烷氧基羰基((CO)-O-烷基)、C6-C24芳氧基羰基(-(CO)-O-芳基)、鹵代羰基((-CO)-X其中X為鹵素)、C2-C24烷基碳酸根(-O-(CO)-O-烷基)、C6-C24芳基碳酸根(-O-(CO)-O-芳基)、羧基(-COOH)、羧酸根(-COO-)、氨基甲酰基(-(CO)-NH2)、單-(C1-C24烷基)-取代的氨基甲酰基(-(CO)NH(C1-C24烷基))、二-(C1-C24烷基)-取代的氨基甲酰基(-(CO)-N(C1-C24烷基)2)、單-(C1-C24鹵代烷基)-取代的氨基甲酰基(-(CO)-NH(C1-C24烷基))、二-(C1-C24鹵代烷基)-取代的氨基甲酰基(-(CO)-N(C1-C24烷基)2)、單-(C5-C24芳基)-取代的氨基甲酰基(-(CO)-NH-芳基)、二-(C5-C24芳基)被取代的氨基甲酰基(-(CO)-N(C5-C24芳基)2)、二-N-(C1-C24烷基)、N-(C5-C24芳基)-取代的氨基甲酰基、硫代氨基甲酰基(-(CS)-NH2)、單-(C1-C24烷基)-取代的硫代氨基甲酰基(-(CO)-NH(C1-C24烷基))、二-(C1-C24烷基)-取代的硫代氨基甲酰基(-(CO)-N(C1-C24烷基)2)、單-(C5-C24芳基)取代的硫代氨基甲酰基(-(CO)-NH-芳基)、二-(C5-C24芳基)-取代的硫代氨基甲酰基(-(CO)-N(C5-C24芳基)2)、二-N-(C1-C24烷基)、N-(C5-C24芳基)-取代的硫代氨基甲酰基、脲基(-NH-(CO)-NH2)、氰基(-C≡N)、氰酸根(-O-C=N)、硫代氰酸根(-S-C=N)、甲酰基(-(CO)-H)、硫代甲酰基(-(CS)-H)、氨基(-NH2)、單-(C1-C24烷基)-取代的氨基、二-(C1-C24烷基)-取代的氨基、單-(C5-C24芳基)取代的氨基、二-(C5-C24芳基)-取代的氨基、C1-C24烷基酰氨基(-NH-(CO)-烷基)、C6-C24芳基酰氨基(-NH-(CO)-芳基)、亞氨基(-CR=NH其中R=氫、C1-C24烷基、C5-C24芳基、C6-C24烷芳基、C6-C24芳烷基等)、C2-C20烷基亞氨基(-CR=N(烷基),其中R=氫、C1-C24烷基、C5-C24芳基、C6-C24烷芳基、C6-C24芳烷基等)、芳基亞氨基(-CR=N(芳基),其中R=氫、C1-C20烷基、C5-C24芳基、C6-C24烷芳基、C6-C24芳烷基等)、硝基(-NO2)、亞硝基(-NO)、磺基(-SO2OH)、磺酸酯(SO2O-)、C1-C24烷基硫烷基(-S-烷基;也被稱為“烷基硫代”)、C5-C24芳基硫烷基(-S-芳基;也被稱為“芳基硫代”)、C1-C24烷基亞磺酰基(-(SO)-烷基)、C5-C24芳基亞磺酰基(-(SO)-芳基)、C1-C24烷基磺酰基(-SO2-烷基)、C1-C24單烷基氨基磺酰基(-SO2-N(H)烷基)、C1-C24二烷基氨基磺酰基-SO2-N(烷基)2、C5-C24芳基磺酰基(-SO2-芳基)、硼基(-BH2)、二羥硼基(-B(OH)2)、硼酸根(-B(OR)2,其中R是烷基或其他烴基)、磷酰基(-P(O)(OH)2)、膦酸根(-P(O)(O)2)、次膦酸根(P(O)(O-))、二氧膦基(-PO2)和膦基(-PH2);以及烴基部分C1-C24烷基(優選地C1-Cl2烷基,更優選地C1-C6烷基)、C2-C24烯基(優選地C2-C12烯基,更優選地C2-C6烯基)、C2-C24炔基(優選地C2-C12炔基,更優選地C2-C6炔基)、C5-C24芳基(優選地C5-C24芳基)、C6-C24烷芳基(優選地C6-C16烷芳基)和C6-C24芳烷基(優選地C6-C16芳烷基)。在這些取代基結構內,“烷基”、“亞烷基”、“烯基”、“亞烯基”、“炔基”、“亞炔基”、“烷氧基”、“芳族的”、“芳基”、“芳氧基”、“烷芳基”和“芳烷基”部分可以任選地是氟化的或全氟化的。此外,提到的醇、醛、胺、羧酸、酮或其他相似的反應性官能團還包括它們的被保護的類似物。例如,提到的羥基或醇還包括其中羥基被以下保護的那些取代基:乙酰基(Ac)、苯甲酰基(Bz)、芐基(Bn、Bnl)、β-甲氧基乙氧基甲醚(MEM)、二甲氧基三苯甲基、[雙-(4-甲氧基苯基)苯基甲基](DMT)、甲氧基甲醚(MOM)、甲氧基三苯甲基[(4-甲氧基苯基)二苯基甲基、MMT)、對-甲氧基芐基醚(PMB)、甲基硫代甲醚、新戊酰(Piv)、四氫吡喃基(THP)、四氫呋喃(THF)、三苯甲基(三苯基甲基、Tr)、硅醚(最常用的硅醚包括三甲基甲硅烷基(TMS)、叔丁基二甲基甲硅烷基(TBDMS)、三異丙基甲硅氧基甲基(TOM)、和三異丙基甲硅烷基(TIPS)醚)、乙氧基乙基醚(EE)。提到的胺還包括其中胺被以下保護的那些取代基:BOC甘氨酸、芐氧羰基(Cbz)、對-甲氧基芐基羰基(Moz或MeOZ)、叔丁基氧羰基(BOC)、9-芴基甲氧基羰基(FMOC)、乙酰基(Ac)、苯甲酰基(Bz)、芐基(Bn)、氨基甲酸酯、對-甲氧基芐基(PMB)、3,4-二甲氧基芐基(DMPM)、對-甲氧基苯基(PMP)、甲苯磺酰基(Ts)基團或磺酰胺(Nosyl和Nps)基團。提到的含有羰基的取代基還包括其中羰基被以下保護的取代基:縮醛基或縮酮基、縮羰酯基或代森基(diathane group)。提到的含有羧酸或羧酸酯基團的取代基還包括其中羧酸或羧酸酯基團被以下保護的取代基:其甲酯、芐酯、叔丁酯、2,6-二取代的苯酚(例如2,6-二甲基苯酚、2,6-二異丙基苯酚、2,6-二叔丁基苯酚)的酯、甲硅烷基酯、原酸酯或噁唑啉。優選的取代基是本文中被鑒定為不影響或很少影響甲硅烷基化化學的那些,例如,包括包含烷基的那些取代基;醇鹽、芳氧化物、芳烷基醇鹽、被保護的羰基基團類;任選地被F、Cl、-CF3取代的芳基類;環氧化物;N-烷基吖丙啶類;順式和反式烯烴類;乙炔類;吡啶類、伯胺類、仲胺類和叔胺類;膦類;和氫氧化物。
如“官能化的烴基”、“官能化的烷基”、“官能化的烯烴”、“官能化的環烯烴”及類似物中的“官能化的”意指在烴基、烷基、芳基、雜芳基、烯烴、環烯烴或其他部分中結合于碳(或其他)原子的至少一個氫原子被一個或更多個官能團例如在這里和上文描述的那些代替。術語“官能團”意指包括適合于本文描述的用途的任何官能物質。特別地,如本文使用的,官能團將必需具備與基質表面上的相應的官能團反應或鍵合的能力。
此外,如果特定的基團允許的話,那么前面提到的官能團可以被一個或更多個另外的官能團或被一個或更多個烴基部分例如上文具體地列舉的那些進一步地取代。類似地,上文提到的烴基部分可以被一個或更多個官能團或另外的烴基部分例如具體地列舉的那些進一步地取代。
“任選的”或“任選地”意指后續描述的情況可以或可以不發生,使得該描述包括其中該情況發生的例子和其中該情況不發生的例子。例如,短語“任選地被取代的”意指非氫取代基可以或可以不存在于給定的原子上,并且,因此,該描述包括其中存在非氫取代基的結構和其中不存在非氫取代基的結構。
如本文使用的,術語“甲硅烷基化”指的是通常使碳-硅鍵形成于之前被碳-氫鍵、通常是未活化的C-H鍵占據的位置中。甲硅烷基化可以被視為偶聯C-H鍵和Si-H鍵以形成C-Si鍵。在本文描述的條件下,用C-Si鍵直接地代替C-H鍵的能力被認為是無先例的。
如本文使用的,術語“大體上無過渡金屬化合物”意圖反映出,在本文所述的相對溫和的條件下該體系對于其使末端炔烴C-H鍵甲硅烷基化的預期目的是有效的,即使在不存在任何外源的(即,有意地加入的或以其他方式加入的)過渡金屬催化劑時。雖然某些實施方案提供了包括能夠催化硅烷基化反應的那些的過渡金屬可以以與此催化活性通常相關聯的水平存在于本文描述的體系或方法內(例如,在其中基質包含金屬茂的情況中),但是此準金屬(作為催化劑或旁觀化合物)的存在不是必需的并且在許多情況下不是合意的。據此,在優選的實施方案中,體系和方法是“大體上無過渡金屬化合物”。除非另有聲明,否則那么,術語“大體上無過渡金屬化合物”定義成反映出,甲硅烷基化體系內的過渡金屬的總水平獨立地或在有機基質的存在下如在下面實施例2.1.2,表2中描述的由ICP-MS測量的小于約5ppm。當這樣表述時,另外的實施方案還提供了過渡金屬的濃度小于約10wt%、5wt%、1wt%、100ppm、50ppm、30ppm、25ppm、20ppm、15ppm、10ppm、或5ppm至約1ppm或0ppm。如本文使用的,術語“過渡金屬”定義成包括d區元素,例如Ag、Au、Co、Cr、Rh、Ir、Fe、Ru、Os、Ni、Pd、Pt、Cu、Zn或其組合。在另外的具體的獨立的實施方案中,如由ICP-MS測量的Ni的濃度小于25ppm、小于10ppm、小于5ppm或小于1ppm。
雖然可以不必限制該體系向水和氧氣的暴露,但是,在某些實施方案中,化學體系和方法在大體上無水、氧氣、或水和氧氣二者的環境中進行。在其他實施方案中,存在空氣和/或水。除非另有指定,否則術語“大體上無水”指的是水的水平小于約500ppm且“大體上無氧氣”指的是氧氣的水平對應于小于1托的分壓。如果聲明的話,那么另外的獨立的實施方案可以提供:“大體上無水”指的是水的水平小于1.5%、1%、0.5%、1000ppm、500ppm、250ppm、100ppm、50ppm、10ppm、或1ppm并且“大體上無氧氣”指的是對應于小于50托、10托、5托、1托、500毫托、250毫托、100毫托、50毫托、或10毫托的分壓的氧氣水平。在本文描述的一般程序中,除非另有規定,否則做出有意的努力以排除水和氧氣二者。
以下實施方案的列表意圖補充而非代替或替換之前的描述。
實施方案1.一種方法,所述方法包括使包含末端炔基C-H鍵的至少一種有機基質與至少一種有機硅烷和堿金屬氫氧化物(或醇鹽或氫化物)的混合物在基本上不存在過渡金屬化合物下、在足以形成甲硅烷基化的末端炔基部分的條件下接觸。與此相關,此方法還包括在基本上不存在過渡金屬化合物或其他電磁或熱的引發或增長的條件下操作。
實施方案2.如實施方案1所述的方法,其中所述過渡金屬化合物以相對于總體系的重量的小于1ppm存在。
實施方案3.如實施方案1或2所述的方法,其中至少一種有機硅烷包括式(I)、式(II)或式(III)的有機硅烷:
(R)4-mSi(H)m (I)
(R)3-m(H)mSi-(CH2)q-Or-Si(R)3-p(H)p (II)
R-[-SiH(R)-O-]n-R (III)
其中:m和p獨立地為1、2或3;q為0、1、2、3、4、5或6;r為0或1;n為10至100;并且每個R獨立地為鹵素(例如F、Br、Cl、I)(條件是至少一個R為包含碳)、任選地被取代的C1-12烷基或雜烷基、任選地被取代的C1-12烯基或雜烯基、任選地被取代的C1-12炔基或雜炔基、任選地被取代的C5-20芳基或C3-20雜芳基、任選地被取代的C6-30烷芳基或雜烷芳基、任選地被取代的C5-30芳烷基或雜芳烷基、任選地被取代的-O-C1-12烷基或雜烷基、任選地被取代的-O-C5-20芳基或-O-C3-20雜芳基、任選地被取代的-O-C5-30烷芳基或雜烷芳基、或任選地被取代的-O-C5-30芳烷基或雜芳烷基,并且如果被取代,取代基可以為膦酸根、磷酰基、氧膦基、膦基、磺酸根、C1-C20烷基硫烷基、C5-C20芳基硫烷基、C1-C20烷基磺酰基、C5-C20芳基磺酰基、C1-C20烷基亞磺酰基、C5-C20芳基亞磺酰基、磺酰氨基、氨基、酰氨基、亞氨基、硝基、亞硝基、羥基、C1-C20烷氧基、C5-C20芳氧基、C2-C20烷氧基羰基、C5-C20芳氧基羰基、羧基、羧酸根、巰基、甲酰基、C1-C20硫酯、氰基、氰酸根、硫代氰酸根、異氰酸酯、硫代異氰酸酯、氨基甲酰基、環氧基、苯乙烯基、甲硅烷基、甲硅氧基、硅烷基、硅氧烷氮烷基、硼酸根、硼基或鹵素,或含有金屬的基團或含有準金屬的基團,其中準金屬是Sn或Ge,其中所述取代基可以任選地向包括氧化鋁、二氧化硅或碳的不溶性或微溶性支持介質提供鏈接部。在式(II)的一些實施方案中,q為0。在式(II)的一些實施方案中,r為0。當在這些方法或體系中使用時,在此公開內容中描述的任何硅烷還被認為是單獨的實施方案。
實施方案4.如權利要求3所述的方法,其中所述有機硅烷是(R)3SiH、(R)2SiH2或(R)SiH3。在這些實施方案中的一些中,R獨立地為烷氧基、烷基、烯基、芳基、芳氧基、雜芳基、芳烷基或雜芳烷基。
實施方案5.如實施方案1-4中任一項所述的方法,其中所述堿金屬氫氧化物是氫氧化鈉(NaOH)(或所述堿金屬醇鹽是醇鈉)。
實施方案6.如實施方案5所述的方法,其中所述有機硅烷是EtMe2SiH、PhMe2SiH、BnMe2SiH、(n-Bu)3SiH、Et2SiH2、(EtO)3SiH、Me2(吡啶基)SiH或Me3Si-SiMe2H。
實施方案7.如實施方案1-4中任一項所述的方法,其中所述堿金屬氫氧化物是氫氧化鉀(KOH)(或所述堿金屬醇鹽是醇鉀)。
實施方案8.如實施方案7所述的方法,其中所述有機硅烷是EtMe2SiH、PhMe2SiH、(n-Bu)3SiH、Et2SiH2、(i-Pr)3SiH或(i-Pr)2(吡啶基)SiH。
實施方案9.如實施方案1-8中任一項所述的方法,其中包含所述末端炔基C-H鍵的所述有機基質具有下式:
R1-C≡C-H,
其中R1包括H、任選地被取代的烷基、任選地被取代的烯基、任選地被取代的炔基、任選地被取代的芳基、任選地被取代的雜烷基、任選地被取代的雜芳基、任選地被取代的芳烷基、任選地被取代的雜芳烷基或任選地被取代的金屬茂。
實施方案10.如實施方案9所述的方法,其中R1為或包括任選地被取代的直鏈烷基、任選地被取代的支鏈烷基或任選地被取代的環烷基。
實施方案11.如實施方案9所述的方法,其中R1為或包括任選地被取代的直鏈烯基、任選地被取代的支鏈烯基或任選地被取代的環烯基。
實施方案12.如實施方案9所述的方法,其中R1為或包括任選地被取代的直鏈雜烷基、任選地被取代的支鏈雜烷基或任選地被取代的雜環烷基。
實施方案13.如實施方案9所述的方法,其中R1為或包括任選地被取代的芳基、任選地被取代的芳烷基、任選地被取代的雜芳基或任選地被取代的雜芳烷基。
實施方案14.如實施方案13所述的方法,其中R1為或包括任選地被取代的苯、聯苯、萘或蒽環結構。
實施方案15.如實施方案13所述的方法,其中R1為或包括任選地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、三唑、異噁唑、噁唑、噻唑、異噻唑、噁二唑、吡啶、噠嗪、嘧啶、吡嗪、三嗪酮、苯并呋喃、苯并吡咯、苯并噻吩、異苯并呋喃、異苯并吡咯、異苯并噻吩、吲哚、異吲哚、吲嗪、吲唑、氮雜吲哚、苯并異噁唑、苯并噁唑、喹啉、異喹啉、噌啉、喹唑啉、萘啶、2,3-二氫苯并呋喃、2,3-二氫苯并吡咯、2,3-二氫苯并噻吩、二苯并呋喃、呫噸、二苯并吡咯或二苯并噻吩部分。
實施方案16.如實施方案1-15中任一項所述的方法,其中包含所述末端炔基C-H鍵的所述有機基質是聚合的。
實施方案17.如實施方案3-16中任一項所述的方法,其中m=2或3,所述方法還包括使包含末端炔基C-H鍵的第二或第三有機基質與所述第一形成的甲硅烷基化的末端炔基部分接觸以形成二炔基或三炔基交叉偶聯的硅烷產物。此第二(或第三)有機基質可以與第一種相同或不同。
實施方案18.如實施方案1-17中任一項所述的方法,還包括使甲硅烷基化的末端炔基部分與另一種不飽和部分在[2+2]或[4+2]環加成反應中反應以形成芳族、雜芳族、環烯基或雜環烯基部分。所述不飽和部分可以包括烯烴、炔烴、疊氮化物、腈、異氰酸酯、異硫氰酸酯、羰基、酰胺、脲等,并且所述反應可以是分子內或分子間的。
實施方案19.如實施方案1-17中任一項所述的方法,所述方法還包括使所述甲硅烷基化的末端炔基部分與第二不飽和的有機部分在交叉復分解反應中反應,以形成二烯烴或聚烯烴產物。所述不飽和部分可以包括烯烴、炔烴、疊氮化物、腈、異氰酸酯、異硫氰酸酯、羰基、酰胺、脲等,并且所述反應可以是分子內或分子間的。在這些實施方案中的一些中,使用Grubbs型復分解反應催化劑完成復分解。
實施方案20.如實施方案1-17中任一項所述的方法,所述方法還包括使所述甲硅烷基化的末端炔基部分聚合。所述甲硅烷基化的末端炔基部分還可以與其他炔屬或烯屬化合物通過任何方式,包括復分解和自由基機制,共聚合。
實施方案21.如實施方案1-17中任一項所述的方法,所述方法還包括使甲硅烷基化的末端炔基部分與有機疊氮化物在[3+2]疊氮化物-炔烴環加成反應中反應。這所謂的點擊化學,包括1,3-偶極環加成,可以為分子間或分子內的反應,并且通常涉及使用銅、銅-鐵或含釕催化劑。
實施方案22.如實施方案1-17中任一項所述的方法,所述方法還包括使甲硅烷基化的末端炔基部分氫化。某些其他實施方案包括甲硅烷基化的末端炔基部分與水、醇、氰化氫、氯化氫、鹵素單質或羧酸的反應以產生相應的乙烯基化合物或羰基化合物。
實施方案23.如實施方案1-20中任一項所述的方法,所述方法還包括去除最初添加至末端炔基C-H鍵的甲硅烷基,使得所添加的甲硅烷基被用作用于在基質中的其他轉化的保護基團或定向基團。
實施方案24.如實施方案1-20中任一項所述的方法,所述方法還包括使甲硅烷基化的末端炔基部分與芳族鹵化物在足以形成炔基-芳烴鍵的條件下反應;例如使用Pd(PPh3)2Cl2/CuI催化劑與芳族溴或碘化合物。
實施方案25.如實施方案1-17中任一項所述的方法,所述方法還包括使甲硅烷基化的末端炔基部分與N-鹵代琥珀酰亞胺在陽離子型金催化劑的存在下反應,以產生末端炔基鹵化物,其中鹵素優選為溴或碘。
實施方案26.如實施方案1-20中任一項所述的方法,其中所述至少一種有機硅烷包含任選地被取代的C1-12烯基或雜烯基,使得所述甲硅烷基化的末端炔基部分包含硅鍵合的任選地被取代的C1-12烯基或雜烯基,所述方法還包括使甲硅烷基化的末端炔基部分與醇和催化劑在產生所述甲硅烷基化的末端炔基部分的分子內烯丙基化的條件下反應。
實施方案27.如實施方案1-20中任一項所述的方法,其中所述至少一種有機硅烷包含2-吡啶基(如本文中使用(Me)2(吡啶基)SiH或(i-Pr)2(吡啶基)SiH所代表的),所述方法還包括使所述甲硅烷基化的末端炔基部分與銅碳鎂化催化劑和任選地被取代的芳基鎂絡合物或任選地被取代的雜芳基鎂絡合物在足以使所述甲硅烷基化的末端炔基部分碳鎂化的條件下反應。
實施方案28.如實施方案27所述的方法,所述方法還包括使所述碳鎂化的甲硅烷基化的末端炔基部分與任選地被取代的芳基碘或任選地被取代的雜芳基碘在鈀催化劑的存在下反應以形成三取代的甲硅烷基化的烯烴。
實施方案29.如實施方案28所述的方法,所述方法還包括使所述三取代的甲硅烷基化的烯烴與BCl3和頻哪醇在足以使該化合物硼脫甲硅烷基化的條件下反應,并且任選地使所述硼脫甲硅烷基化的化合物與第二任選地被取代的芳基碘或任選地被取代的雜芳基碘在適于使所得的C-B鍵與所述第二任選地被取代的芳基碘或任選地被取代的雜芳基碘交叉偶聯的條件下反應。
實施方案30.一種用于使包含末端炔基C-H鍵的有機基質甲硅烷基化的體系,所述體系包括以下的混合物或主要由以下的混合物組成:(a)至少一種有機硅烷和(b)堿金屬氫氧化物(或在一些情況下,堿金屬醇鹽或堿金屬氫化物),以及(c)至少一種基質。這樣的體系可以大體上無過渡金屬化合物。這樣的體系還可以包含來源于所述至少一種有機硅烷和末端炔烴的甲硅烷基化的產物。
實施方案31.如實施方案30所述的體系,其中所述過渡金屬化合物以相對于總體系的重量的小于10ppm存在。
實施方案32.如實施方案30或31所述的體系,其中至少一種有機硅烷包括式(I)、式(II)或式(III)的有機硅烷:
(R)4-mSi(H)m (I)
(R)3-m(H)mSi-(CH2)q-Or-Si(R)3-p(H)p (II)
R-[-SiH(R)-O-]n-R (III)
其中:m和p獨立地為1、2或3;q為0、1、2、3、4、5或6;r為0或1;n為10至100;并且每個R獨立地為鹵素(例如F、Br、Cl、I)(條件是至少一個R為包含碳)、任選地被取代的C1-12烷基或雜烷基、任選地被取代的C1-12烯基或雜烯基、任選地被取代的C1-12炔基或雜炔基、任選地被取代的C5-20芳基或C3-20雜芳基、任選地被取代的C6-30烷芳基或雜烷芳基、任選地被取代的C5-30芳烷基或雜芳烷基、任選地被取代的-O-C1-12烷基或雜烷基、任選地被取代的-O-C5-20芳基或-O-C3-20雜芳基、任選地被取代的-O-C5-30烷芳基或雜烷芳基、或任選地被取代的-O-C5-30芳烷基或雜芳烷基,并且如果被取代,取代基可以為膦酸根、磷酰基、氧膦基、膦基、磺酸根、C1-C20烷基硫烷基、C5-C20芳基硫烷基、C1-C20烷基磺酰基、C5-C20芳基磺酰基、C1-C20烷基亞磺酰基、C5-C20芳基亞磺酰基、磺酰氨基、氨基、酰氨基、亞氨基、硝基、亞硝基、羥基、C1-C20烷氧基、C5-C20芳氧基、C2-C20烷氧基羰基、C5-C20芳氧基羰基、羧基、羧酸根、巰基、甲酰基、C1-C20硫酯、氰基、氰酸根、硫代氰酸根、異氰酸酯、硫代異氰酸酯、氨基甲酰基、環氧基、苯乙烯基、甲硅烷基、甲硅氧基、硅烷基、硅氧烷氮烷基、硼酸根、硼基或鹵素,或含有金屬的基團或含有準金屬的基團,其中準金屬是Sn或Ge,其中所述取代基可以任選地向包括氧化鋁、二氧化硅或碳的不溶性或微溶性支持介質提供鏈接部。在式(II)的一些實施方案中,q為0。在式(II)的一些實施方案中,r為0。
實施方案33.如實施方案32所述的體系,其中所述有機硅烷是(R)3SiH或(R)2SiH2,其中R獨立地為烷氧基、烷基、烯基、芳基、芳氧基、雜芳基、芳烷基或雜芳烷基。
實施方案34.如實施方案30-33中任一項所述的體系,其中所述堿金屬氫氧化物是氫氧化鈉(NaOH)(或所述堿金屬醇鹽是醇鈉)。
實施方案35.如實施方案34所述的體系,其中所述有機硅烷是EtMe2SiH、PhMe2SiH、(n-Bu)3SiH、Et2SiH2、PhMe2SiH、BnMe2SiH、(Me)2(吡啶基)SiH、(EtO)3SiH或Me3Si-SiMe2H。
實施方案36.如實施方案30-33中任一項所述的體系,其中所述堿金屬氫氧化物是氫氧化鉀(KOH)(或所述堿金屬醇鹽是醇鉀)。
實施方案37.如實施方案36所述的體系,其中所述有機硅烷是EtMe2SiH、PhMe2SiH、(n-Bu)3SiH、Et2SiH2、(i-Pr)3SiH或(i-Pr)2(吡啶基)SiH。
實施方案38.如實施方案30-37中任一項所述的體系,其中包含所述末端炔基C-H鍵的所述有機基質具有式:
R1-C≡C-H,
其中R1包括H、任選地被取代的烷基、任選地被取代的烯基、任選地被取代的炔基、任選地被取代的芳基、任選地被取代的雜烷基、任選地被取代的雜芳基、任選地被取代的芳烷基、任選地被取代的雜芳烷基或任選地被取代的金屬茂。
實施方案39.如實施方案38所述的體系,其中R1為或包括任選地被取代的直鏈烷基、任選地被取代的支鏈烷基或任選地被取代的環烷基。
實施方案40.如實施方案38所述的體系,其中R1為或包括任選地被取代的直鏈雜烷基、任選地被取代的支鏈雜烷基或任選地被取代的雜環烷基。
實施方案41.如實施方案38所述的體系,其中R1為或包括任選地被取代的芳基、任選地被取代的芳烷基、任選地被取代的雜芳基或任選地被取代的雜芳烷基。
實施方案42.如實施方案41所述的體系,其中R1為或包括任選地被取代的苯、聯苯、萘或蒽環結構。
實施方案43.如實施方案39所述的體系,其中R1為或包括任選地被取代的呋喃、吡咯、噻吩、吡唑、咪唑、三唑、異噁唑、噁唑、噻唑、異噻唑、噁二唑、吡啶、噠嗪、嘧啶、吡嗪、三嗪酮、苯并呋喃、苯并吡咯、苯并噻吩、異苯并呋喃、異苯并吡咯、異苯并噻吩、吲哚、異吲哚、吲嗪、吲唑、氮雜吲哚、苯并異噁唑、苯并噁唑、喹啉、異喹啉、噌啉、喹唑啉、萘啶、2,3-二氫苯并呋喃、2,3-二氫苯并吡咯、2,3-二氫苯并噻吩、二苯并呋喃、呫噸、二苯并吡咯或二苯并噻吩部分。
實施方案44.如實施方案30-43中任一項所述的體系,其中包含所述末端炔基C-H鍵的所述有機基質是聚合的。
實施方案45.如實施方案30-44中任一項所述的體系,所述體系包括至少兩種不同的有機硅烷。
實施方案46.如實施方案30-45中任一項所述的體系,所述體系包括至少兩種不同的有機基質,每種有機基質各包含末端炔基C-H鍵。
實施例
提供以下實施例以例證在本公開內容內描述的概念中的某些。雖然每個實施例被認為是提供組合物、制備方法和用途的具體的單獨的實施方案,但是無一實施例應當被認為限制本文描述的較一般的實施方案。
在以下實施例中,雖然已作出努力來確保關于所使用的數字(例如量、溫度等等)的準確度,但是某些實驗誤差和偏差應當被考慮在內。除非另有指示,否則溫度以攝氏度計,壓力在或接近大氣壓。
實施例1:實試數據的一般觀測
關于用Et3SiH使乙炔1(丙-2-炔基環己烷,圖2)甲硅烷基化的初始的研究在用于使用KOt-Bu催化的C(sp2)-H甲硅烷基化條件使雜芳烴甲硅烷基化的反應條件下進行。在此情況下,觀察到的乙炔基硅烷2a以良好的收率獲得,連同9%的不期望的炔遷移產物1-異構體(圖2,條目1)。NaOt-Bu(條目2)和LiOt-Bu(條目3)是較差的催化劑,并且一般的有機堿(條目4-6)也得到較差的結果。令人驚訝的是,在10mol%催化劑載量時,更溫和的KOH比KOt-Bu更好(條目7)。從Et3SiH改變到PhMe2SiH允許反應在環境溫度進行,同時仍保持高收率(條目8)。與之前報道的其中強鉀堿對反應性至關重要的雜芳烴C-H甲硅烷基化方案明顯不同,廉價且溫和的NaOH被證明是1的甲硅烷基化的理想催化劑,以93%收率提供2b(條目9)。LiOH(條目10)不催化反應(參見表1,以下)。
改變含氫硅烷配偶體的立體和電學性質(圖2)示出,可以產生多種新的乙炔基硅烷,包括在硅上具有合成上多功能的氫化物(2e和2f)、芐基二甲基-(2g)、三異丙基-(2h)、三乙氧基-(2i)和2-二烷基吡啶基-(2j和2k)取代基的那些(圖2b)。在過渡金屬催化劑或在親核試劑或酸的存在下被裂解的不穩定的Si-Si鍵也被良好地耐受,以95%收率產生2l。這似乎是在C-H甲硅烷基化領域迄今報道的單和二氫硅烷的最大的范圍。
多種炔烴被示出是反應性的,包括具有多電子和缺電子的芳基(4a-j)、雜芳基(4k-m)和烷基(4o-y)基團的那些(圖3)。敏感的官能團諸如芳基鹵化物(4b-d)、烷基氯化物(4v)和環丙烷(4r)被良好地耐受,沒有任何不期望的副反應。具有酸性官能團諸如炔丙胺(3w)和炔丙醇(3x)的基質也良好地反應,以高收率分別提供4w和雙甲硅烷基化的4x。前所未有的N-雜環體系(諸如吡啶3m和咪唑3k)的催化的交叉脫氫甲硅烷基化也成功地給出了相應的甲硅烷基化的組成部分4m和4k。含有對KOt-Bu-催化的甲硅烷基化敏感的C-H鍵的基質,或在其他C-H官能化化學下參與的那些,諸如苯甲醚3g、噻吩3y、甲苯3f、炔丙基醚3q和苯衍生物3t,在末端炔烴C-H鍵處都以優異的化學選擇性反應(>99:1),沒有任何觀察到的C(sp2)-H或C(sp3)-H甲硅烷基化。對Minisci-型自由基官能化(例如吡啶3m)和親電子取代反應(例如多電子體系3n和二茂鐵3h)的完全抑制表明了新穎的C-H官能化機制是可操作的。這些基質的干凈的反應概況(clean reaction profile)證明了與過渡金屬催化法和傳統的化學計量脫質子化策略相比,由NaOH催化的獨特的益處。
如由幾克級別的合成4s(圖4A)證明的,此堿金屬氫氧化物催化的甲硅烷基化反應良好地放大而不損失催化劑活性,并且可以被應用至有機合成、材料科學和后期藥物衍生的具有挑戰性的問題。例如,成功的實驗示出了,對稱的脂族或芳族二炔可以被雙甲硅烷基化(5b和6b)或選擇性地單官能化(5a和6a),產生合成上有價值的、正交活化的(orthogonally activated)炔烴組成部分(圖4B)。催化劑區分炔烴、導致單取代的5a的能力特別令人驚訝,并且考慮到起始物質中的相同炔烴之間缺乏電子交流(electronic communication),目前沒有理解這一能力。使用二氫硅烷允許通過雙重C(sp)-H甲硅烷基化制備對稱的二乙炔基硅烷(圖4C、7)。在3組分偶聯反應中同時反應2種不同的末端炔烴和二烷基硅烷以76%的收率產生不對稱的二乙炔基硅烷8,以及10%的7(由1的自偶聯(homocouple)形成)和<5%收率的自偶聯的環丙基乙炔產物。通過利用炔烴配偶體之間的速率差,獲得有利于交叉產物(cross product)的此非統計學產物分布(參見實施例3.4)。這些硅烷是官能化的硅雜環戊二烯、聚硅雜環戊二烯或硅雜環戊二烯共聚物的前體,并且為此目的,本發明的另外的實施方案提供了甲硅烷基化的產物的進一步反應。并且,在第一個催化安裝多功能的2-二甲基甲硅烷基吡啶定向基團中使用氫氧化物催化的甲硅烷基化方案,產生收率78%的2k,其可以被發展成高度取代的烯烴(圖4D)。再次,為此目的,本發明的另外的實施方案提供了甲硅烷基化的產物的進一步反應。
含硅藥物類似物正獲得來自醫學化學工作者的日益增長的關注,因為它們可以提供相對于全碳物質的改善的藥物動力學性質。并且,被安裝的有機硅部分可以充當用于隨后的加工的官能團柄狀物(handle)或充當容易去除的保護基。再次,為此目的,本發明的另外的實施方案提供了甲硅烷基化的產物的進一步反應。為了對于此類后期C-H官能化應用評價本方法,使藥學物質巴吉林和美雌醇經受催化甲硅烷基化條件,分別以96%和82%收率成功地提供了新穎的含硅藥物類似物9和10(圖4E)。
此時,堿金屬氫氧化物、醇鹽或氫化物催化的甲硅烷基化的潛在機械學的細節還未被完全了解。可以想到C-H脫質子化過程,但產生熱動力學(即,脫質子化的堿和C(sp)-H鍵之間的pKa差異)、催化劑周轉(catalyst turnover)的機制和反應性Si物質的性質的問題。初步的研究還表明,此機制與之前公開的包括KOt-Bu-催化的雜芳烴的C(sp2)-H甲硅烷基化的C(sp)-H甲硅烷基化反應不同。這基于來自自由基捕獲和抗衡陽離子螯合研究的結果(參見實施例2.1.3)、與之前報道相比的改善的收率和極大擴大的含氫硅烷和基質范圍和以下的事實:氫氧化鈉示出了與貴金屬物質、Lewis酸和KOt-Bu相比極大改善的活性。對于后者,在評價的大多數情況中,使用KOt-Bu作為用于C(sp)-H偶聯的催化劑的努力令人驚訝地失敗了(參見圖6)。并且,觀察到在僅在抗衡陽離子的身份(identity)中有區別的KOH和NaOH的反應性之間的顯著差異。考慮到在相同條件下研究的基質中缺乏可區分的反應性趨勢,明顯非良性的(non-innocence)陽離子不能通過溶解性、聚集狀態或堿性理由被簡單地合理說明(圖5)。這些數據表明,簡單的堿金屬陽離子——或作為添加劑或作為催化劑抗衡陽離子——在新穎的催化方法的發現和開發中起重要作用。
實施例2:一般信息
除非另有說明,否則在充滿氮氣的手套箱中,在烘箱干燥的全新的Fisherbrand閃爍管中或在氬氣下連接至Schlenk管線的火焰干燥的Schlenk燒瓶中,用干燥的脫氣的溶劑和全新的攪拌棒進行反應。通過在氬氣下經過活性氧化鋁柱來干燥溶劑。通過薄層色譜法(TLC)或GC-FID分析監測反應進程。使用E.Merck硅膠60F254預涂的玻璃板(0.25mm)進行TLC,并且通過UV熒光猝滅、磷鉬酸或KMnO4染色可視化。使用Silicycle SiliaFlash P60Academic硅膠(粒徑40nm-63nm)進行快速色譜法。1H NMR光譜在Varian Inova 500MHz光譜儀上、于CDCl3或THF-d8中記錄,并且分別相對于在δ7.26ppm或δ3.58ppm的殘余溶劑峰報告。13C NMR光譜在Varian Inova 500MHz光譜儀上(126MHz)、于CDCl3或THF-d8中記錄,并且分別相對于在δ77.16ppm或δ67.21ppm的殘余溶劑峰報告。對于1H NMR的數據如下報告:化學位移(δppm)(多重性,耦合常數(Hz),積分)。多重性如下報告:s=單峰,d=雙峰,t=三重峰,q=四重峰,p=五重峰,sept=七重峰,m=多重峰,br s=寬單峰,br d=寬雙峰,app=表觀的(apparent)。關于化學位移(δppm)報告13C NMR的數據。IR光譜在Perkin Elmer Spectrum BXII光譜儀上使用沉積在NaCl片上的薄膜獲得,并且以吸收的頻率(cm–1)報告。GC-FID分析在配備有HP-5(5%-苯基)-甲基聚硅氧烷毛細管柱的Agilent 6890N氣相色譜儀(Agilent)上獲得。GC-MS分析在配備有HP-5(5%-苯基)-甲基聚硅氧烷毛細管柱的Agilent 6850氣相色譜儀(Agilent)上獲得。高分辨率質譜(HRMS)從加州理工學院的質譜設備獲取。在加州理工學院的質譜設備上進行ICP-MS分析。
從Aldrich購買硅烷并在使用前蒸餾。從Aldrich購買KOt-Bu(升華級,99.99%痕量金屬基礎)并直接使用。從Aldrich購買KOH(半導體級,小球,99.99%痕量金屬基礎)并在使用前粉碎(研缽和研杵)并在真空下加熱(150℃)。從Aldrich購買NaOH(半導體級,小球,99.99%痕量金屬基礎)并在使用前粉碎(研缽和研杵)并在真空下加熱(150℃)。炔烴基質從Aldrich、TCI或Acros購買。
實施例2.1.反應優化、痕量金屬分析和初步機械學研究。
實施例2.1.1.反應優化。
用于反應條件優化的程序:在充滿氮氣的手套箱中,向配置有磁力攪拌棒的2打蘭(dram)閃爍管中加入催化劑和炔烴1a(0.1mmol,1當量)。然后,加入含氫硅烷和溶劑(0.1mL)。將該管密封,并以指定的溫度攪拌混合物持續指定的時間。然后從手套箱中取出該管,用二乙醚(1mL)稀釋并在減壓下濃縮。通過使用內標的粗混合物的1H NMR或GC分析確定收率。
表1.直接C(sp)-H甲硅烷基化的條件優化。
通過使用內標的粗反應混合物的GC分析確定的收率。
來自表1的結果表明,在用于C(sp)-H甲硅烷基化反應的反應條件中存在高度的可調節性。THF、二氧六環和DME都被證明是合適的溶劑,產生少量的異構化的起始物質(分別為條目2、3、4)。用下至1mol%的KOt-Bu實現低載量的催化劑,而沒有顯著的收率損失(條目15-17)。證明了高溫(85℃)是對于用三乙基硅烷甲硅烷基化必需的(條目15、18、19);如在本文的硅烷篩選中觀察到的,當使用多種其它硅烷時獲得了較低的溫度。用更長的反應時間(72h)的廣泛的堿篩選(條目20-35)示出了對于C-H甲硅烷基化反應存在很多良好的催化劑。用更低的催化劑載量進行的精細的堿篩選(條目36-49)揭示了仍存在以令人驚訝的高效率完成的若干催化劑,但NaOH被證明是最方便且高性能的催化劑。在不存在催化劑或當使用LiOt-Bu、NaOAc、KOAc、DABCO、K2CO3、Cs2CO3或KF時沒有觀察到產物(分別為條目39、27、28、24、33、34、35)。
實施例2.1.2.通過ICP-MS進行痕量金屬分析。
全部反應組分的ICP-MS痕量金屬分析。為了提供對偶然的痕量金屬物質參與交叉脫氫C(sp)-H甲硅烷基化催化的進一步支持,在NaOH、KOH、3-環己基-1-丙炔起始物質、二甲氧基乙烷(DME)溶劑、PhMe2SiH和在手套箱中在最優條件下進行的標準反應混合物的樣品上進行電感耦合等離子體質譜法。來自定量分析的結果表明,大部分的金屬污染物以低于設備的最低檢測限存在(即以ppt范圍或更低)。表2中給出了金屬污染物的微克每升(ppb)量。
分析了NaOH(1000mg,99.99%Aldrich)、3-環己基-1-丙炔、PhMe2SiH、1,2-二甲氧基乙烷和標準反應混合物(0.5mmol規模混合物,遵循一般程序用61.1mg 3-環己基-1-丙炔、2mg NaOH、于0.5mL 1,2-二甲氧基乙烷(DME)中的204.4mg PhMe2SiH并在手套箱中攪拌48h來制備)中的每個樣品。
將每個樣品添加至50mL DigiTUBE消化管(digestion tube)(SCP Science),然后加入3.0mL Plasma Pure硝酸(SCP Science)并加熱至75℃持續36小時。在消化之后,使用Milli Q水將每個樣品稀釋至50mL并且經受痕量金屬分析。使用Agilent 8800由電感耦合等離子體質譜法確定痕量金屬濃度。進樣系統由微霧霧化器(micromist nebulizer)、scott型噴霧室和固定的注射器石英噴燈(injector quartz torch)組成。使用保護電極,并且在1500W下操作等離子體。在碰撞室(collision cell)中以單四級桿模式(single-quad mode)不用氣體或用氦來確定元素(動能歧視模式(kinetic energy discrimination mode))。使用范圍從1ppb至100ppb(微克/L)的外標溶液校準33種元素。關注的痕量元素的檢測限低于1ppb標準。另外,半定量地校準氦模式數據中的快速掃描數據。LOD表明,分析物濃度低于設備的最低檢測限。除非另有說明,否則值是以ppb。
表2.炔烴甲硅烷基化反應中的反應物的痕量金屬分析。
*ppm
實施例2.1.3.初步機械學實驗。進行多個實驗以獲得對反應機制的了解。作為第一次研究,進行實驗以確定甲硅烷基化反應實質上是極性反應還是自由基反應。在自由基阱(radical trap)TEMPO和galvinoxyl(加爾萬氧基)的存在下進行反應。兩種添加劑都不阻礙炔烴C-H甲硅烷基化:TEMPO在10%載量下不抑制反應,但在300%載量下降低甲硅烷基化收率;從10mol%移動至300mol%添加劑的galvinoxyl對反應條件的影響是出乎意料的并且目前并不了解(方案1)。
方案1.自由基捕獲劑對反應的影響
還研究了鉀和鈉螯合劑影響甲硅烷基化的作用以研究陽離子在催化中的重要性。當18-冠-6和15-冠-5被添加至反應,使用KOH和NaOH分別作為催化劑時,仍然在使用三乙基硅烷作為硅配偶體時觀察到定量甲硅烷基化,這表明已經發生金屬離子的無效的螯合或在此特定情況中陽離子對反應性是不必要的(方案2a)。然而,這可能是特殊情況,因為采用Et3SiH的反應使用KOH或NaOH作為催化劑同樣良好進行。
方案2.添加鉀和鈉離子螯合劑
使用不與KOH和NaOH同樣良好進行的硅配偶體還探索了鉀和鈉螯合劑影響甲硅烷基化的作用。選擇三乙氧基硅烷作為試驗硅烷,因為它僅在使用NaOH作為催化劑時顯示產物形成。在此情況中,添加鉀和鈉催化劑抑制反應性,表明鈉離子確實是用三乙氧基硅烷使炔烴甲硅烷基化所必需的(方案2b)。當添加冠醚時,僅有的產物是(EtO)4Si,這表明堿金屬陽離子從體系的多價螯合作用抑制了可能產生的C-H甲硅烷基化通路并且誘導了硅烷的歧化反應。由GC和NMR分析確定收率。
實施例2.1.4.MOH和KOt-Bu催化劑的比較。
為了比較在雜環甲硅烷基化的情況下使用的新發現的MOH(堿金屬氫氧化物)催化劑與KOt-Bu催化劑的表現,使若干乙炔基質和硅烷經受使用KOt-Bu作為催化劑的反應。結果在圖6中總結。盡管在采用環己基丙炔和三乙基硅烷的反應中,KOt-Bu成功地以中等收率(如正文中闡述的)產生了甲硅烷基化的炔烴,但在所有其他研究的情況中,KOt-Bu沒能轉化起始物質或僅產生痕量產物。似乎本文描述的炔的甲硅烷基化和之前描述的雜環的甲硅烷基化需要不同的催化劑并且可能通過不同的機制進行。
實施例3.實驗和分析。
實施例3.1.用于交叉脫氫C(sp)-H甲硅烷基化的一般程序和表征數據。
在充滿氮氣的手套箱中,向配置有磁力攪拌棒的2打蘭閃爍管中加入催化劑(0.05mmol,10mol%)和炔烴(0.5mmol,1當量),然后加入溶劑(0.5mL)和硅烷(1.5mmol,3當量)。然后將該管密封并在指定溫度下攪拌混合物持續指定時間。然后從手套箱取出該管;將反應混合物用二乙醚(2mL)稀釋、過濾通過短的硅膠的墊并在減壓下濃縮。如指示的,在高真空下加熱去除揮發物,并且如果需要,通過硅膠快速色譜法純化所得物質,得到期望的C(sp)-Si產物。
(3-環己基丙-1-炔-1-基)三乙基硅烷2a:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、Et3SiH(174mg,240μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在85℃進行反應持續48h。在高真空(45毫托,2小時)下去除溶劑后,獲得期望的產物2a的無色油(111.9mg,收率95%)。1H NMR(500MHz,CDCl3)δ2.13(d,J=6.6Hz,2H),1.84–1.76(m,2H),1.75–1.68(m,2H),1.65(dtt,J=12.9,3.4,1.5Hz,1H),1.47(dddd,J=14.8,6.8,4.7,3.4Hz,1H),1.24(tdd,J=15.9,9.4,3.4Hz,2H),1.19–1.07(m,2H),1.07–1.01(m,1H),0.98(t,J=7.9Hz,9H),0.57(q,J=7.9Hz,6H);13C NMR(126MHz,CDCl3)δ107.73,82.39,37.54,32.72,27.86,26.47,26.32,7.65,4.75。IR(純膜NaCl(Neat Film NaCl))3422,2925,2172,1645,1449,1018,802,724cm–1;對于C15H27Si[(M+H)–H2]計算的HRMS(EI+):235.1882,實測235.1881。
(3-環己基丙-1-炔-1-基)二甲基(苯基)硅烷2b:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在25℃進行反應持續48h。在45毫托下加熱至85℃持續30分鐘后,以95%純度獲得期望的產物2b(113.6mg,收率89%);隨后通過硅膠快速色譜法純化(100%己烷)獲得了分析純形式的產物2b的無色油。Rf=0.67(100%己烷);1H NMR(500MHz,CDCl3)δ7.67–7.63(m,2H),7.40–7.34(m,3H),2.19(d,J=6.6Hz,2H),1.87–1.80(m,2H),1.74(dt,J=12.8,3.3Hz,2H),1.67(dddd,J=11.3,5.2,3.3,1.6Hz,1H),1.52(ddtd,J=14.9,11.5,6.7,3.5Hz,1H),1.27(dddd,J=15.9,12.6,9.5,3.3Hz,2H),1.15(qt,J=12.7,3.3Hz,1H),1.08–0.98(m,2H),0.41(s,6H);13C NMR(126MHz,CDCl3)δ137.93,133.81,129.33,127.91,108.67,83.19,37.42,32.81,27.94,26.42,26.29,–0.38。IR(純膜NaCl)3420,2924,2852,2173,1646,1448,1427,1322,1248,1115,1071,1027,815,730cm–1;對于C17H25Si[M+H]計算的HRMS(EI+):257.1726,實測257.1720。
(3-環己基丙-1-炔-1-基)(乙基)二甲基硅烷2c:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、EtMe2SiH(132mg,198μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h。在高真空(45毫托,2小時)下去除溶劑后,獲得期望的產物2c(95.1mg,收率91%)的無色油。1H NMR(500MHz,CDCl3)δ2.12(d,J=6.6Hz,2H),1.86–1.76(m,2H),1.77–1.69(m,2H),1.66(dtd,J=12.6,3.3,1.6Hz,1H),1.53–1.40(m,1H),1.32–1.19(m,2H),1.20–1.07(m,2H),1.06–0.94(m,4H),0.57(q,J=7.9Hz,2H),0.12(s,6H);13C NMR(126MHz,CDCl3)δ107.01,84.30,37.46,32.76,27.84,26.45,26.30,8.47,7.50,–1.85。IR(純膜NaCl)3422,2922,2103,1646,1558,1260,1027,720cm–1;對于C13H23Si[(M+H)-H2]計算的HRMS(EI+):207.1569,實測207.1562。
三丁基(3-環己基丙-1-炔-1-基)硅烷2d:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、n-Bu3SiH(301mg,386μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。通過硅膠快速色譜法(100%己烷)獲得期望的產物2d(117.2mg,收率73%),收獲產物2d的無色油。Rf=0.78(100%己烷);1H NMR(500MHz,CDCl3)δ2.18(d,J=6.5Hz,2H),1.85(dddd,J=12.3,6.2,3.1,1.8Hz,2H),1.77(ddd,J=14.0,4.5,2.3Hz,2H),1.70(dddt,J=12.8,5.1,3.3,1.5Hz,1H),1.52(dddt,J=14.5,7.9,6.6,3.2Hz,1H),1.43–1.36(m,12H),1.29(qt,J=12.6,3.3Hz,2H),1.18(qt,J=12.7,3.3Hz,1H),1.11–1.02(m,2H),0.97–0.91(m,9H),0.67–0.59(m,6H);13C NMR(126MHz,CDCl3)δ107.65,83.25,37.57,32.72,27.88,26.64,26.46,26.39,26.32,13.98,13.45。IR(純膜NaCl)2955,2922,2854,2172,1449,1376,1191,1080,1029,886,758,708cm–1;對于C21H40Si[M+·]計算的HRMS(EI+):320.2899,實測320.2905。
(3-環己基丙-1-炔-1-基)二乙基硅烷2e:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、Et2SiH2(132mg,194μL,1.5mmol,3.0當量)和0.5mL四氫呋喃(THF)在25℃進行反應持續24h。在45毫托的高真空下持續30分鐘去除溶劑后,獲得90%純度的期望的產物2e(73.6mg,收率71%);隨后通過硅膠快速色譜法純化(100%己烷)獲得產物2e的無色油。Rf=0.77(100%己烷);1H NMR(500MHz,CDCl3)δ3.92(pt,J=3.2,1.2Hz,1H),2.15(dd,J=6.7,1.2Hz,2H),1.85–1.78(m,2H),1.72(ddd,J=13.9,4.5,2.2Hz,2H),1.66(dddt,J=12.7,5.1,3.3,1.5Hz,1H),1.49(ddtd,J=14.9,11.5,6.8,3.5Hz,1H),1.31–1.20(m,2H),1.15(tt,J=12.6,3.2Hz,1H),1.07–0.95(m,8H),0.70–0.64(m,4H);13C NMR(126MHz,CDCl3)δ109.00,80.24,37.39,32.76,27.91,26.41,26.28,8.09,4.23。IR(純膜NaCl)3422,2957,2174,2120,1646,1558,1457,1260,1055,804cm–1;對于C13H23Si[(M+H)-H2]計算的HRMS(EI+):207.1569,實測207.1562。
二-叔丁基(3-環己基丙-1-炔-1-基)硅烷2f:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、t-Bu2SiH2(216mg,297μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在45毫托的高真空下持續30分鐘后獲得90%純度的期望的產物2f(120.3mg,收率91%);隨后通過硅膠快速色譜法純化(100%己烷)收獲產物2f的無色油。Rf=0.88(100%己烷);1H NMR(500MHz,CDCl3)δ3.57(t,J=1.2Hz,1H),2.17(dd,J=6.5,1.2Hz,2H),1.84–1.78(m,2H),1.76–1.70(m,2H),1.66(dddt,J=12.8,5.1,3.3,1.5Hz,1H),1.50(dddt,J=14.5,7.8,6.5,3.1Hz,1H),1.26(qt,J=12.7,3.4Hz,3H),1.19–1.09(m,2H),1.06(s,18H);13C NMR(126MHz,CDCl3)δ108.94,79.54,37.51,32.75,28.28,27.88,26.44,26.29,18.63。IR(純膜NaCl)2958,2927,2855,2173,2111,1469,1449,1363,1028,1012,810,793,617cm–1;對于C17H31Si[(M+H)-H2]計算的HRMS(EI+):263.2195,實測263.2206。
芐基(3-環己基丙-1-炔-1-基)二甲基硅烷2g:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、BnMe2SiH(150mg,238μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。通過硅膠快速色譜法(100%己烷)獲得期望的產物2g的無色油(101.9mg,收率75%)。Rf=0.51(100%己烷);1H NMR(500MHz,CDCl3)δ7.25–7.21(m,2H),7.12–7.08(m,3H),2.20(s,2H),2.14(d,J=6.8Hz,2H),1.81(ddd,J=13.3,3.5,1.5Hz,2H),1.75(dt,J=12.7,3.2Hz,2H),1.69(dddd,J=11.3,5.3,3.4,1.7Hz,1H),1.49(tdt,J=11.4,6.7,3.3Hz,1H),1.28(qt,J=12.6,3.3Hz,2H),1.16(qt,J=12.7,3.3Hz,1H),1.06–0.94(m,2H),0.13(s,6H);13C NMR(126MHz,CDCl3)δ139.44,128.51,128.19,124.32,108.08,83.69,37.38,32.77,27.86,26.71,26.41,26.29,–1.69。IR(純膜NaCl)3081,3060,3024,2999,2922,2851,2664,2173,1936,1600,1493,1449,1422,1408,1368,1322,1249,1207,1155,1056,1029,947,839,761,697cm–1;對于C18H26Si[M+·]計算的HRMS(EI+):270.1804,實測270.1810。
(3-環己基丙-1-炔-1-基)三異丙基硅烷2h:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、i-Pr3SiH(238mg,307μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在85℃進行反應持續48h。通過硅膠快速色譜法(100%己烷)獲得期望的產物2h的無色油(95.6mg,收率69%)。Rf=0.79(100%己烷);1H NMR(500MHz,CDCl3)δ2.16(d,J=6.4Hz,2H),1.84–1.77(m,2H),1.73(dt,J=12.8,3.4Hz,2H),1.66(dtd,J=12.7,3.3,1.6Hz,1H),1.48(ddtd,J=14.6,11.2,6.5,3.4Hz,1H),1.25(qt,J=12.6,3.4Hz,2H),1.15(tt,J=12.6,3.3Hz,1H),1.10–0.99(m,23H);13C NMR(126MHz,CDCl3)δ108.17,80.94,37.64,32.71,27.87,26.49,26.33,18.80,11.48。IR(純膜NaCl)2924,2864,2170,2463,1449,1264,1025,995,883,743,676,633cm-1;對于C18H33Si[(M+H)-H2]計算的HRMS(EI+):277.2352,實測277.2349。
(3-環己基丙-1-炔-1-基)三乙氧基硅烷2i:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、(EtO)3SiH(246mg,277μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。通過硅膠快速色譜法(于己烷中的5%Et2O)獲得期望的產物2i的無色油(97.1mg,收率68%)。Rf=0.41(于己烷中的5%Et2O);1H NMR(500MHz,CDCl3)δ3.87(q,J=7.0Hz,6H),2.16(d,J=6.6Hz,2H),1.84–1.78(m,2H),1.72(dp,J=12.6,3.7Hz,2H),1.66(dddt,J=12.8,5.1,3.3,1.5Hz,1H),1.52(ddtd,J=14.9,11.5,6.8,3.5Hz,1H),1.26(t,J=7.0Hz,9H),1.24–1.19(m,2H),1.13(qt,J=12.7,3.3Hz,1H),1.02(qd,J=12.7,3.5Hz,2H);13C NMR(126MHz,CDCl3)δ106.50,76.85,59.02,37.10,32.74,27.55,26.33,26.20,18.18。IR(純膜NaCl)2974,2925,2852,2182,1449,1390,1168,1101,1079,1036,964,790,721cm–1;對于C15H29O3Si[M+H]計算的HRMS(EI+):285.1886,實測285.1889。
2-((3-環己基丙-1-炔-1-基)二異丙基甲硅烷基)吡啶2j:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、i-Pr2(Pyr)SiH(290mg,322μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。通過硅膠快速色譜法(于己烷中的10%EtOAc)獲得期望的產物2j的無色油(122.5mg,收率78%)。Rf=0.47(于己烷中的10%EtOAc);1H NMR(500MHz,THF-d8)δ8.65(ddd,J=4.8,1.7,1.1Hz,1H),7.76(dt,J=7.5,1.3Hz,1H),7.59(td,J=7.6,1.8Hz,1H),7.19(ddd,J=7.7,4.8,1.4Hz,1H),2.26(d,J=6.4Hz,2H),1.95–1.84(m,2H),1.78–1.73(m,2H),1.67(dtt,J=13.0,3.4,1.6Hz,1H),1.55(ddtd,J=14.9,11.4,6.6,3.5Hz,1H),1.37–1.26(m,4H),1.21–1.16(m,1H),1.16–1.11(m,2H),1.09(d,J=7.4Hz,6H),0.99(d,J=7.3Hz,6H);13C NMR(126MHz,THF-d8)δ164.80,150.76,134.42,132.12,123.73,110.50,80.33,38.63,33.66,28.41,27.38,27.23,18.46,18.40,12.71。IR(純膜NaCl)2924,2862,2170,1573,1462,1449,1417,1136,1081,1028,995,882,747,723cm–1;對于C20H32NSi[M+H]計算的HRMS(EI+):314.2304,實測314.2311。
2-((3-環己基丙-1-炔-1-基)二甲基甲硅烷基)吡啶2k:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、Me2(Py)SiH(206mg,225μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。通過硅膠快速色譜法(于己烷中的10%EtOAc)獲得期望的產物2k的無色油(99.9mg,收率78%)。Rf=0.42(與己烷中的10%EtOAc);1H NMR(500MHz,THF-d8)δ8.65(ddd,J=4.8,1.8,1.1Hz,1H),7.74(dt,J=7.5,1.2Hz,1H),7.59(td,J=7.6,1.8Hz,1H),7.18(ddd,J=7.7,4.8,1.4Hz,1H),2.19(d,J=6.6Hz,2H),1.88–1.81(m,2H),1.73–1.70(m,2H),1.66(dddd,J=12.7,5.1,3.2,1.5Hz,1H),1.50(dddt,J=14.7,7.9,6.7,3.2Hz,1H),1.28(tdd,J=16.0,9.4,3.4Hz,2H),1.17(qt,J=12.7,3.3Hz,1H),1.05(qd,J=12.8,3.4Hz,2H),0.36(s,6H);13C NMR(126MHz,THF-d8)δ166.55,150.96,134.69,130.13,123.84,109.23,83.58,38.47,33.68,28.42,27.34,27.22,–1.00。IR(純膜NaCl)3423,2924,2852,2175,1646,1449,1255,1044,832,797,676cm–1;對于C16H24NSi[M+H]計算的HRMS(EI+):258.1678,實測258.1672。
1-(3-環己基丙-1-炔-1-基)-1,1,2,2,2-五甲基二硅烷2l:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、Me5Si2H(246mg,277μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在25℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物2l的混濁的無色油(120.0mg,收率95%)。1H NMR(500MHz,THF-d8)δ2.11(d,J=6.5Hz,2H),1.81(dddd,J=13.1,6.1,3.1,1.9Hz,2H),1.73–1.69(m,2H),1.65(dddt,J=12.7,5.1,3.2,1.5Hz,1H),1.44(dddt,J=14.6,8.0,6.7,3.2Hz,1H),1.33–1.21(m,2H),1.15(qt,J=12.7,3.2Hz,1H),1.03(qd,J=12.8,3.5Hz,2H),0.15(s,6H),0.11(s,9H);13C NMR(126MHz,THF-d8)δ109.11,84.06,38.62,33.61,28.52,27.37,27.22,–2.25,–2.35。IR(純膜NaCl)2923,2852,2168,1449,1259,1244,1077,1027,871,833,799,765,725,691,667cm–1;對于C14H28Si2[M+·]計算的HRMS(EI+):252.1730,實測252.1737。
二甲基(苯基)(苯基乙炔基)硅烷4a:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、乙炔苯(52mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在45毫托下加熱至85℃持續30分鐘后獲得95%純度的期望的產物4a(105.7mg,收率89%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4a的無色油。Rf=0.38(100%己烷);1H NMR(500MHz,THF-d8)δ7.71–7.65(m,2H),7.49–7.44(m,2H),7.38–7.28(m,6H),0.46(s,6H)。13C NMR(126MHz,THF-d8)δ137.86,134.66,132.88,130.35,129.75,129.28,128.79,124.15,107.86,92.55,–0.50。IR(純膜NaCl)3068,3051,2959,2899,2158,1592,1488,1442,1428,1278,1250,1219,1118,1068,1026,846,807,780,731,690cm–1;對于C16H17Si[M+H]計算的HRMS(EI+):237.1100,實測237.1101。
((4-氟苯基)乙炔基)二甲基(苯基)硅烷吡啶4b:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-乙炔基-4-氟苯(60mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4b(111.9mg,收率88%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4b的無色油。Rf=0.49(100%己烷);1H NMR(500MHz,THF-d8)δ7.68–7.65(m,2H),7.53–7.48(m,2H),7.34(dd,J=4.9,1.9Hz,3H),7.08(t,J=8.8Hz,2H),0.46(s,6H);13C NMR(126MHz,THF-d8)δ163.93(d,J=248.7Hz),137.74,135.10(d,J=8.5Hz),134.65,130.39,128.81,120.43(d,J=3.5Hz),116.51(d,J=22.4Hz),106.68,92.43(d,J=1.3Hz),–0.56。IR(純膜NaCl)3420,3069,2961,2160,1653,1600,1505,1428,1251,1233,1155,1117,1092,857,835,816,781,731,698cm–1;對于C16H16FSi[M+H]計算的HRMS(EI+):255.1005,實測255.1000。
((4-溴苯基)乙炔基)二甲基(苯基)硅烷4c:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-溴-4-乙炔基苯(90mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得80%純度的期望的產物4c(81.3mg,收率52%);隨后通過硅膠快速色譜法純化(100%己烷)收獲與二苯基四甲基二硅氧烷9:1混合物中的產物4c的無色晶體。Rf=0.54(100%己烷);1H NMR(500MHz,THF-d8)δ7.69–7.63(m,2H),7.51(d,J=8.5Hz,2H),7.39(d,J=8.5Hz,2H),7.36–7.30(m,3H),0.46(s,6H);13C NMR(126MHz,THF-d8)δ137.55,134.65,134.53,132.66,130.44,128.83,123.94,123.19,106.51,94.19,–0.66。IR(純膜NaCl)3068,2958,2159,1653,1540,1484,1473,1457,1427,1249,1214,1114,1071,1010,846,830,780,730,698cm–1;對于C16H16Si18Br[M+H]計算的HRMS(EI+):317.0184,實測317.0180。
((3-氯苯基)乙炔基)二甲基(苯基)硅烷4d:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-氯-3-乙炔基苯(68mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4d(121.6mg,收率90%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4d的無色油。Rf=0.42(100%己烷);1H NMR(500MHz,CDCl3)δ7.70–7.66(m,2H),7.49(ddd,J=2.1,1.5,0.5Hz,1H),7.40(dd,J=5.0,1.9Hz,3H),7.38(dt,J=7.6,1.4Hz,1H),7.31(ddd,J=8.1,2.1,1.2Hz,1H),7.24(ddd,J=8.0,7.6,0.5Hz,1H),0.51(s,6H);13C NMR(126MHz,CDCl3)δ136.75,134.21,133.86,132.05,130.29,129.70,129.61,129.12,128.10,124.77,105.13,93.82,–0.79。IR(純膜NaCl)3420,2163,1684,1647,1559,1521,1507,1457,1249,1117,1091,884,781,681cm–1;對于C16H16ClSi[M+H]計算的HRMS(EI+):271.0710,實測271.0710。
4-((二甲基(苯基)甲硅烷基)乙炔基)-N,N-二甲基苯胺4e:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、4-乙炔基-N,N-二甲基苯胺(73mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4e的無色晶體(139.4mg,收率100%)。1H NMR(500MHz,CDCl3)δ7.73–7.68(m,2H),7.41–7.36(m,5H),6.61(d,J=8.9Hz,2H),2.98(s,6H),0.48(s,6H);13C NMR(126MHz,CDCl3)δ150.46,137.88,133.93,133.38,129.36,127.94,111.69,109.78,108.49,89.19,40.32,-0.39。IR(純膜NaCl)3067,2957,2147,1682,1607,1519,1487,1427,1360,1248,1186,1115,945,850,817,779,730,699,653cm–1;對于C18H21NSi[M+·]計算的HRMS(EI+):279.1443,實測279.1445。
二甲基(苯基)(對甲苯基乙炔基)硅烷4f:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-乙炔基-4-甲基苯(58mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4f的淡黃色油(115.5mg,收率92%)。1H NMR(500MHz,CDCl3)δ7.71(ddt,J=6.0,2.4,1.1Hz,2H),7.41(ddq,J=5.8,3.0,0.9Hz,5H),7.16–7.10(m,2H),2.37(s,3H),0.51(d,J=1.1Hz,6H);13C NMR(126MHz,CDCl3)δ139.02,137.33,133.90,132.10,129.52,129.12,128.02,120.00,107.18,91.28,21.69,–0.59。IR(純膜NaCl)3420,3068,3049,2959,2920,2156,1507,1428,1408,1249,1223,1117,1020,851,816,780,731,700,656cm–1;對于C17H19Si[M+H]計算的HRMS(EI+):251.1256,實測251.1257。
((4-甲氧基苯基)乙炔基)二甲基(苯基)硅烷4g:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-乙炔基-4-甲氧基苯(66mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4g(121.6mg,收率91%);隨后通過硅膠快速色譜法純化(100%己烷→于己烷中的5%EtOAc)收獲分析純形式的產物4g的黃色油。Rf=0.27(100%己烷);1H NMR(500MHz,CDCl3)δ7.71(dd,J=6.5,3.0Hz,2H),7.46(d,J=8.9Hz,2H),7.43–7.38(m,3H),6.84(d,J=8.9Hz,2H),3.82(s,3H),0.51(s,6H);13C NMR(126MHz,CDCl3)δ160.02,137.42,133.89,133.73,129.50,128.01,115.20,113.96,107.03,90.47,55.42,-0.56。IR(純膜NaCl)3068,2959,2154,1605,1507,1441,1293,1249,1171,1116,1032,853,832,812,779,755,731,699cm–1;對于C17H18OSi[M+·]計算的HRMS(EI+):266.1127,實測266.1135。
((3,5-二甲氧基苯基)乙炔基)二甲基(苯基)硅烷4h:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-乙炔基-3,5-二甲氧基苯(81mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4h的淡黃色油(140.6mg,收率95%)。1H NMR(500MHz,CDCl3)δ7.70(ddd,J=5.5,2.7,1.2Hz,2H),7.41(dd,J=4.6,2.1Hz,3H),6.67(d,J=2.3Hz,2H),6.47(t,J=2.3Hz,1H),3.79(s,6H),0.52(d,J=1.5Hz,6H);13C NMR(126MHz,CDCl3)δ160.56,137.05,133.90,129.61,128.05,124.29,109.87,106.78,102.53,91.75,55.57,-0.68。IR(純膜NaCl)3421,3069,3001,2959,2837,2160,1596,1456,1419,1348,1298,1250,1205,1155,1116,1064,979,964,817,753,732,681cm–1;對于C18H21O2Si[M+H]計算的HRMS(EI+):297.1311,實測297.1309。
2-乙炔基-1,3,5-三甲基苯4i:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、2-乙炔基-1,3,5-三甲基苯(57mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在25℃進行反應持續24h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4i的無色油(119.1mg,收率86%)。1H NMR(500MHz,CDCl3)δ7.73(ddt,J=4.5,3.2,0.8Hz,3H),7.40(dd,J=2.5,0.8Hz,2H),6.88–6.86(m,2H),2.42(s,6H),2.29(s,3H),0.52(t,J=0.7Hz,6H);13C NMR(126MHz,CDCl3)δ140.86,138.23,137.66,133.89,129.45,127.99,127.67,119.94,104.95,99.66,21.51,21.15,–0.34。IR(純膜NaCl)3440,3068,2959,2146,1646,1610,1474,1428,1224,1117,841,825,779,753,698cm–1;對于C19H23Si[M+H]計算的HRMS(EI+):279.1569,實測279.1561。
((6-甲氧基萘-2-基)乙炔基)二甲基(苯基)硅烷4j:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、2-乙炔基-6-甲氧基萘(91mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4j的無色油(134.8mg,收率85%)。此產物在二氧化硅上分解。1H NMR(500MHz,CDCl3)δ7.99(dd,J=1.5,0.7Hz,1H),7.78–7.72(m,2H),7.70(d,J=9.0Hz,1H),7.68(d,J=8.2Hz,1H),7.53(dd,J=8.4,1.6Hz,1H),7.46–7.40(m,3H),7.17(dd,J=8.9,2.5Hz,1H),7.11(d,J=2.6Hz,1H),3.93(s,3H),0.56(s,6H);13C NMR(126MHz,CDCl3)δ158.57,137.30,134.48,133.93,132.17,129.56,129.34,128.44,128.05,126.85,122.76,119.59,117.93,107.50,105.91,91.68,55.50,-0.57。IR(純膜NaCl)3422,2959,2152,1631,1601,1499,1481,1461,1390,1267,1232,1161,1117,1031,937,890,814,780,731,703,656cm–1;對于C21H20OSi[M+·]計算的HRMS(EI+):316.1284,實測316.1296。
5-((二甲基(苯基)甲硅烷基)乙炔基)-1-甲基-1H-咪唑4k:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、5-乙炔基-1-甲基-1H-咪唑(53mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4k(98.7mg,收率82%);隨后通過硅膠快速色譜法純化(100%EtOAc)收獲分析純形式的產物4k的無色油。Rf=0.45(100%EtOAc);1H NMR(500MHz,CDCl3)δ7.68–7.65(m,2H),7.40(m,4H),7.31(d,J=1.0Hz,1H),3.68–3.65(m,3H),0.52(s,6H);13C NMR(126MHz,CDCl3)δ138.37,136.49,135.29,133.74,129.73,128.09,116.28,100.60,94.11,32.11,-0.85。IR(純膜NaCl)3417,2960,2157,1646,1533,1489,1428,1274,1250,1227,1116,924,823,782,732,702,661cm–1;對于C14H17N2Si[M+H]計算的HRMS(EI+):241.1161,實測241.1169。
二甲基(苯基)(噻吩-3-基乙炔基)硅烷4l:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、3-乙炔基噻吩(54mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續60h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4l(113.2mg,收率93%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4l的無色油。Rf=0.39(100%己烷);1H NMR(500MHz,CDCl3)δ7.72–7.68(m,2H),7.53(dd,J=3.0,1.2Hz,1H),7.43–7.39(m,3H),7.27–7.24(m,1H),7.17(dd,J=5.0,1.2Hz,1H),0.51(s,6H);13C NMR(126MHz,CDCl3)δ137.08,133.88,130.26,130.11,129.59,128.04,125.36,122.32,101.67,91.93,-0.68。IR(純膜NaCl)3107,3068,2959,2152,1427,1356,1249,1163,1116,944,870,781,753,698cm–1;對于C14H14SSi[M+·]計算的HRMS(EI+):242.0586,實測242.0576。
3-((二甲基(苯基)甲硅烷基)乙炔基)吡啶4m:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、3-乙炔基吡啶(52mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4m(91.8mg,收率77%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4m的無色油。Rf=0.31(100%己烷);1H NMR(500MHz,CDCl3)δ8.74(dd,J=2.1,0.9Hz,1H),8.54(dd,J=4.9,1.7Hz,1H),7.77(ddd,J=7.9,2.1,1.7Hz,1H),7.71–7.67(m,2H),7.42(dd,J=4.9,1.9Hz,3H),7.24(ddd,J=7.9,4.9,0.9Hz,1H),0.54(s,6H);13C NMR(126MHz,CDCl3)δ152.82,149.02,139.01,136.49,133.81,129.74,128.11,123.00,120.21,103.14,96.34,-0.88。IR(純膜NaCl)3420,3069,3048,3025,2960,2161,1559,1474,1406,1250,1184,1119,1022,847,781,754,703,670cm–1;對于C15H16NSi[M+H]計算的HRMS(EI+):238.1052,實測238.1049。
((二甲基(苯基)甲硅烷基)乙炔基)二茂鐵4n:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、乙炔基二茂鐵(105mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4n的紅色結晶固體(170.1mg,收率99%)。1H NMR(500MHz,CDCl3)δ7.70(dd,J=6.1,3.1Hz,2H),7.43–7.37(m,3H),4.48(s,2H),4.21(m,7H),0.47(s,6H);13C NMR(126MHz,CDCl3)δ137.71,133.89,129.44,127.98,106.30,88.52,72.02,70.26,69.00,64.64,–0.40。IR(純膜NaCl)2958,2147,1428,1248,1106,1024,1001,925,819,779,753,730,699cm–1;對于C20H20FeSi[M+·]計算的HRMS(EI+):344.0684,實測344.0696。
(環己-1-烯-1-基乙炔基)二甲基(苯基)硅烷4o:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、1-乙炔基環己-1-烯(53mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續15分鐘去除溶劑后,獲得95%純度的期望的產物4o(102.7mg,收率85%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4o的無色油。Rf=0.50(100%己烷);1H NMR(500MHz,CDCl3)δ7.67–7.63(m,2H),7.39–7.36(m,3H),6.24(tt,J=3.9,1.8Hz,1H),2.17(tdd,J=6.0,2.7,1.8Hz,2H),2.11(tdd,J=6.4,4.6,2.5Hz,2H),1.68–1.55(m,4H),0.43(s,6H);13C NMR(126MHz,CDCl3)δ137.59,136.90,133.84,129.40,127.94,120.82,109.17,88.79,29.14,25.81,22.33,21.54,-0.51。IR(純膜NaCl)3422,2937,2145,1647,1428,1249,1116,863,819,779,730,698cm–1;對于C16H21Si[M+H]計算的HRMS(EI+):241.1413,實測241.1402。
(環己基乙炔基)二甲基(苯基)硅烷4p:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、乙炔基環己烷(54mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在25℃進行反應持續48h。在85℃、45毫托下持續15分鐘去除溶劑后,獲得80%純度的期望的產物4p(97.4mg,收率80%);隨后通過硅膠快速色譜法純化(100%己烷)收獲與二苯基四甲基二硅氧烷9:1混合物中的產物4p的無色油。Rf=0.53(100%己烷);1H NMR(500MHz,CDCl3)δ7.65(ddd,J=5.4,2.4,1.7Hz,2H),7.37(ddq,J=4.0,1.9,0.8Hz,3H),2.47(tt,J=9.0,3.8Hz,1H),1.89–1.79(m,2H),1.73(ddd,J=9.8,6.2,3.1Hz,2H),1.52(td,J=9.7,9.2,3.8Hz,3H),1.38–1.26(m,3H),0.40(d,J=1.0Hz,6H);13C NMR(126MHz,CDCl3)δ133.82,133.13,129.29,127.89,113.93,81.74,32.70,30.23,26.00,24.93,-0.30。IR(純膜NaCl)2931,2854,2173,1448,1427,1248,1116,1076,843,834,816,779,729,698cm–1;對于C16H21Si[(M+H)-H2]計算的HRMS(EI+):241.1413,實測241.1419。
(3-甲氧基丙-1-炔-1-基)二甲基(苯基)硅烷4q:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、3-甲氧基丙-1-炔(35mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續15分鐘去除溶劑后,獲得95%純度的期望的產物4q(61.0mg,收率60%);仔細地加熱是必要的,因為產物在這些條件下是揮發性的。隨后通過硅膠快速色譜法純化(1:1DCM:己烷)收獲分析純形式的產物4q的無色油。Rf=0.38(1:1DCM:己烷);1H NMR(500MHz,CDCl3)δ7.65–7.62(m,2H),7.41–7.36(m,3H),4.16(s,2H),3.41(s,3H),0.45(s,6H);13C NMR(126MHz,CDCl3)δ136.63,133.66,129.49,127.90,103.05,89.53,60.48,57.67,–0.97。IR(純膜NaCl)3423,2925,2173,1640,1428,1353,1250,1186,1103,1007,990,903,838,817,781,731,698cm–1;對于C12H16OSi[M+·]計算的HRMS(EI+):204.0971,實測204.0977。
(環丙基乙炔基)二甲基(苯基)硅烷4r:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、乙炔基環丙烷(33mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在25℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4r(70.1mg,收率70%);仔細地加熱是必要的,因為該產物在這些條件下是揮發性的。隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4r的無色油。Rf=0.38(100%己烷);1H NMR(500MHz,CDCl3)δ7.64–7.61(m,2H),7.39–7.36(m,3H),1.40–1.30(m,1H),0.87–0.75(m,4H),0.40(s,6H);13C NMR(126MHz,CDCl3)δ137.77,133.79,129.36,127.92,112.40,77.65,8.97,0.70,–0.45。IR(純膜NaCl)3423,3068,2960,2172,2158,1646,1428,1348,1249,1114,1028,839,779,730,659cm–1;對于C13H16Si[M+·]計算的HRMS(EI+):200.1021,實測200.1031。
二甲基(辛-1-炔-1-基)(苯基)硅烷4s:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、辛-1-炔(55mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在25℃進行反應持續48h。在85℃、45毫托下持續15分鐘去除溶劑后,獲得95%純度的期望的產物4s(101.0mg,收率83%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4s的無色油。Rf=0.53(100%己烷);1H NMR(500MHz,CDCl3)δ7.67–7.62(m,2H),7.40–7.35(m,3H),2.28(t,J=7.1Hz,2H),1.59–1.53(m,2H),1.47–1.39(m,2H),1.35–1.27(m,4H),0.91(t,J=6.9Hz,3H),0.40(s,6H);13C NMR(126MHz,CDCl3)δ137.86,133.80,129.35,127.92,109.85,82.31,31.43,28.68,28.64,22.69,20.12,14.19,–0.44。IR(純膜NaCl)3422,3069,2957,2931,2858,2174,1647,1428,1248,1115,836,815,779,729,699cm–1;對于C16H23Si[M+H]計算的HRMS(EI+):245.1726,實測245.1727。
二甲基(苯基)(4-苯基丁-1-炔-1-基)硅烷4t:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、丁-3-炔-1-基苯(65mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4t的淡黃色油(130.0mg,收率98%)。1H NMR(500MHz,CDCl3)δ7.64–7.59(m,2H),7.42–7.37(m,3H),7.31(dd,J=8.0,6.8Hz,2H),7.28–7.23(m,3H),2.90(t,J=7.5Hz,2H),2.60(t,J=7.5Hz,2H),0.42(d,J=0.6Hz,6H);13C NMR(126MHz,CDCl3)δ140.63,137.56,133.80,129.39,128.68,128.47,127.93,126.43,108.62,83.39,35.10,22.38,–0.56。IR(純膜NaCl)3423,3086,3067,3027,2959,2174,1647,1602,1495,1453,1427,1248,1114,1077,1042,869,811,779,729,696,661cm–1;對于C18H19Si[(M+H)-H2]計算的HRMS(EI+):263.1256,實測263.1258。
癸-1,5-二炔-1-基二甲基(苯基)硅烷4u:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、癸-1,5-二炔(67mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4u的無色油(131.3mg,收率98%)。1H NMR(500MHz,CDCl3)δ7.68–7.64(m,2H),7.38(dd,J=5.0,1.9Hz,3H),2.49(ddd,J=7.7,6.1,1.7Hz,2H),2.46–2.39(m,2H),2.18(tt,J=7.0,2.3Hz,2H),1.52–1.39(m,4H),0.92(t,J=7.2Hz,3H),0.42(s,6H);13C NMR(126MHz,CDCl3)δ137.56,133.81,129.40,127.92,107.79,83.33,81.59,78.33,31.20,22.05,20.79,19.16,18.54,13.77,–0.54。IR(純膜NaCl)2958,2932,2872,2177,1465,1428,1336,1249,1115,1042,870,837,816,780,754,731,700,662cm–1;對于C18H23Si[(M+H)-H2]計算的HRMS(EI+):267.1569,實測267.1565。
(5-氯戊-1-炔-1-基)二甲基(苯基)硅烷4v:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、5-氯戊-1-炔(51mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物4v(93.3mg,收率79%);仔細地加熱是必要的,因為該產物在這些條件下是揮發性的。隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物4v的無色油。Rf=0.31(100%己烷);1H NMR(500MHz,CDCl3)δ7.65–7.60(m,2H),7.38(dd,J=4.9,1.9Hz,3H),3.67(t,J=6.4Hz,2H),2.49(t,J=6.8Hz,2H),2.01(p,J=6.6Hz,2H),0.41(s,6H);13C NMR(126MHz,CDCl3)δ137.46,133.75,129.48,127.99,107.20,83.81,43.77,31.40,17.57,–0.56。IR(純膜NaCl)3420,3069,2960,2928,2174,1646,1428,1249,1114,1041,837,816,780,731,701,665cm–1;對于C13H16ClSi[(M+H)-H2]計算的HRMS(EI+):235.0710,實測235.0713。
3-(二甲基(苯基)甲硅烷基)-N-甲基丙-2-炔-1-胺4w:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、N-甲基丙-2-炔-1-胺(69mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續15分鐘去除溶劑后,獲得95%純度的期望的產物4w(81.8mg,收率80%);仔細地加熱是必要的,因為產物在這些條件下是揮發性的。隨后通過硅膠快速色譜法純化(100%EtOAc)收獲分析純形式的產物4w的無色油。Rf=0.32(100%EtOAc);1H NMR(500MHz,THF-d8)δ7.63–7.59(m,2H),7.33–7.29(m,3H),3.36(s,2H),2.39(s,3H),0.36(s,6H);13C NMR(126MHz,THF-d8)δ138.26,134.58,130.18,128.67,108.45,85.45,41.75,35.64,–0.33。IR(純膜NaCl)3416,3068,2957,2165,1725,1651,1427,1250,1116,1044,836,817,730,699cm–1;對于C12H18NSi[M+H]計算的HRMS(EI+):204.1208,實測204.1214。
(3-((二甲基(苯基)甲硅烷基)氧基)丙-1-炔-1-基)二甲基(苯基)硅烷4x:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、丙-2-炔-1-醇(28mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物4x的無色油(142.9mg,收率88%)。仔細地加熱是必要的,因為產物在這些條件下是揮發性的。1H NMR(500MHz,CDCl3)δ7.62(ddt,J=6.4,1.8,0.9Hz,4H),7.44–7.36(m,6H),4.35(s,2H),0.48(s,6H),0.43(s,6H);13C NMR(126MHz,CDCl3)δ137.08,136.80,133.82,133.73,129.93,129.57,128.01,127.98,105.77,88.23,52.27,–0.93,–1.36。IR(純膜NaCl)3069,3049,2959,2177,1428,1363,1250,1117,1085,1043,1004,817,782,731,698cm–1;對于C19H23OSi2[(M+H)–H2]計算的HRMS(EI+):323.1288,實測323.1297。
5-(丙-2-炔-1-基)-4,5,6,7-四氫噻吩并[3,2-c]吡啶3y:向四氫噻吩并[3,2-c]吡啶鹽酸鹽(1.40g,10mmol,1當量)和K2CO3(2.76g,20mmol,2當量)于DMF(30ml)的混合物中加入1-丙炔-3-溴(1.18g,10mmol,1當量),并且將混合物在室溫攪拌16h。過濾混合物并在減壓下去除溶劑以獲得棕色油。用20mL二乙醚稀釋該油,并用20mL水然后用20mL鹽水洗滌,然后經無水Na2SO4干燥。在真空中去除溶劑,并用硅膠上的柱色譜法純化殘余物(10:1己烷:Et2O),獲得產物3y的黃色液體(1.27g,收率72%)。Rf=0.35(于己烷中的10%EtOAc);1H NMR(500MHz,CDCl3)δ7.08(dt,J=5.1,0.7Hz,1H),6.73(d,J=5.1Hz,1H),3.69(t,J=1.7Hz,2H),3.53(d,J=2.4Hz,2H),2.95–2.91(m,2H),2.91–2.88(m,2H),2.29(t,J=2.4Hz,1H);13C NMR(126MHz,CDCl3)δ133.55,132.89,125.19,122.83,78.78,73.39,51.50,49.70,46.37,25.57。IR(純膜NaCl)3937,3626,3390,3289,3103,3065,2910,2816,2101,2651,1614,1565,1461,1428,1405,1328,1275,1219,1191,1166,1130,1109,1079,1051,1017,983,902,835,789,703cm–1;對于C10H12NS[M+H]計算的HRMS(EI+):178.0690,實測178.0689。
5-(3-(二甲基(苯基)甲硅烷基)丙-2-炔-1-基)-4,5,6,7-四氫噻吩并[3,2-c]吡啶4y:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、5-(丙-2-炔-1-基)-4,5,6,7-四氫噻吩并[3,2-c]吡啶(89mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續15分鐘去除溶劑后,獲得95%純度的期望的產物4y(120.4mg,收率77%);隨后通過硅膠快速色譜法純化(于己烷中的10%EtOAc)收獲分析純形式的產物4y的黃色油。Rf=0.40(于己烷中的10%EtOAc);1H NMR(500MHz,CDCl3)δ7.67–7.61(m,2H),7.43–7.34(m,3H),7.09(dd,J=5.1,0.8Hz,1H),6.75(d,J=5.1Hz,1H),3.72(t,J=1.6Hz,2H),3.61(d,J=0.7Hz,2H),2.99–2.88(m,4H),0.43(s,6H);13C NMR(126MHz,CDCl3)δ137.12,133.78,133.16,133.06,129.54,128.00,125.36,122.92,102.74,88.35,51.69,49.89,47.60,25.69,–0.60。IR(純膜NaCl)3067,2957,2906,2814,2163,1427,1327,1249,1166,1115,1034,1016,975,836,817,780,731,699cm–1;對于C18H20NSSi[(M+H)-H2]計算的HRMS(EI+):310.1086,實測310.1087。
實施例3.2.用于4s的幾克級別合成的程序:
二甲基(辛-1-炔-1-基)(苯基)硅烷4s:將配置有攪拌棒并用橡膠隔片塞住的500mL烘箱干燥的Schlenk燒瓶排空并用氬氣再次再充滿。在工作臺上稱量出NaOH(364mg,9.1mmol,10mol%)并在氬氣的強氣流下添加至燒瓶。然后,將被裝載的燒瓶排空,并用熱風槍在真空下加熱2分鐘,然后再充滿氬氣。通過注射器經該隔片添加1,2-二甲氧基乙烷(DME)(經脫氣的,90mL)、1-辛炔(13.4mL,90.7mmol,1.0當量)和PhMe2SiH(20.9mL,136.1mmol,1.5當量)。然后用設置在45℃的加熱套加熱燒瓶并攪拌60小時。從加熱移除含有所得的混濁的棕褐色溶液的燒瓶,并允許其冷卻至室溫,用無水Et2O(50mL)稀釋,并過濾通過短的二氧化硅的墊以去除固體殘余物。在真空中去除溶劑后,加入攪拌棒,并在高真空(100毫托)下攪拌透明的深琥珀色溶液數小時以去除殘留的揮發物。然后使混合物在真空下經受蒸餾:
a)加熱浴至80℃,當少量液滴濃縮至初餾物中時將真空穩定在200毫托。初餾物以無色液體落下。溫度計讀數為22℃。
b)真空保持在200毫托。當最后的殘余的硅烷煮掉時將加熱浴設置在85℃。
c)使加熱浴溫度升高至125℃。溶液開始緩慢地沸騰。溫度計讀數為60℃。真空保持在200毫托。
d)使溫度升高至130℃,真空在200毫托以蒸出期望的二甲基(辛-1-炔-1-基)(苯基)硅烷(無色油)。溫度計讀數85℃。以無色油獲得期望的產物4s(19.0g,收率86%)。
實施例3.3.單-和雙-甲硅烷基化的二炔的合成。
癸-1,9-二炔-1-基二甲基(苯基)硅烷5a:遵循一般程序。用KOH(5.6mg,0.1mmol,20mol%)、癸-1,9-二炔(67mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h。在85℃、45毫托下持續20分鐘去除溶劑后,獲得95%純度的期望的產物5a(126.2mg,收率94%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物5a的無色油。Rf=0.48(100%己烷);1H NMR(500MHz,CDCl3)δ7.66–7.61(m,2H),7.40–7.35(m,3H),2.29(t,J=7.1Hz,2H),2.20(td,J=7.1,2.6Hz,2H),1.96(t,J=2.6Hz,1H),1.57(dtd,J=9.6,7.1,4.5Hz,4H),1.47–1.42(m,4H),0.40(s,6H);13C NMR(126MHz,CDCl3)δ137.79,133.78,129.37,127.93,109.55,84.74,82.51,68.34,28.51,28.45,28.39,28.31,20.04,18.48,–0.46。IR(純膜NaCl)3420,3306,3068,2936,2859,2173,2117,1646,1457,1428,1325,1248,1114,1026,836,816,754,731,700,661cm–1;對于C18H23Si[(M+H)-H2]計算的HRMS(EI+):267.1569,實測267.1556。
1,10-雙(二甲基(苯基)甲硅烷基)癸-1,9-二炔5b:遵循一般程序。用KOH(5.6mg,0.1mmol,20mol%)、癸-1,9-二炔(67mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物5b(190.9mg,收率95%);隨后通過硅膠快速色譜法純化(100%己烷)收獲分析純形式的產物5b的無色油。Rf=0.43(100%己烷);1H NMR(500MHz,CDCl3)δ7.65(ddt,J=5.4,3.0,1.4Hz,4H),7.38(ddt,J=4.4,2.2,1.1Hz,6H),2.30(td,J=7.2,1.1Hz,4H),1.59(t,J=6.8Hz,4H),1.49–1.42(m,4H),0.43–0.40(s,12H);13C NMR(126MHz,CDCl3)δ137.79,133.78,129.37,127.93,109.58,82.49,28.53,28.38,20.04,–0.45。IR(純膜NaCl)3423,3068,2937,2858,2173,1647,1428,1248,1114,836,815,753,730,699,661cm–1;對于C26H33Si2[(M+H)-H2]計算的HRMS(EI+)。401.2121,實測401.2120。
((3-乙炔基苯基)乙炔基)二甲基(苯基)硅烷6a:遵循一般程序。用NaOH(4.0mg,0.1mmol,20mol%)、1,3-二乙炔基苯(63mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑并且隨后通過硅膠快速色譜法純化(100%己烷→于己烷中的3%EtOAc)獲得期望的產物6a的無色油(99.2mg,收率76%)。Rf=0.27(100%己烷);1H NMR(500MHz,CDCl3)δ7.70–7.68(m,2H),7.64(t,J=1.6Hz,1H),7.46(ddt,J=14.2,7.8,1.4Hz,2H),7.41(dd,J=4.9,1.9Hz,3H),7.29(dd,J=7.7,0.6Hz,1H),3.09(s,1H),0.51(d,J=0.5Hz,6H);13C NMR(126MHz,CDCl3)δ136.86,135.71,133.86,132.38,132.35,129.66,128.48,128.09,123.43,122.51,105.63,93.20,82.78,77.99,–0.75。IR(純膜NaCl)3294,2950,2152,1474,1428,1249,1118,924,838,818,781,731,698cm–1;對于C18H17Si[M+H]計算的HRMS(EI+):261.1100,實測261.1093。
1,3-雙((二甲基(苯基)甲硅烷基)乙炔基)苯6b:遵循一般程序。用NaOH(4.0mg,0.1mmol,20mol%)、1,3-二乙炔基苯(63mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在65℃進行反應持續48h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得95%純度的期望的產物6b(173.5mg,收率88%);隨后通過硅膠快速色譜法純化(100%己烷→于己烷中的3%EtOAc)收獲分析純形式的產物6b的淡黃色油。Rf=0.26(100%己烷);1H NMR(500MHz,CDCl3)δ7.73–7.70(m,4H),7.69(t,J=1.7Hz,1H),7.47(dd,J=7.8,1.7Hz,2H),7.44–7.41(m,6H),7.28(ddd,J=8.0,7.4,0.5Hz,1H),0.53(s,12H);13C NMR(126MHz,CDCl3)δ136.88,135.69,133.86,132.23,129.64,128.40,128.08,123.33,105.73,93.08,–0.74。IR(純膜NaCl)3068,2959,2153,1589,1474,1428,1405,1249,1164,1118,944,838,816,780,753,730,702,685cm–1;對于C26H27Si2[M+H]計算的HRMS(EI+):395.1651,實測395.1659。
實施例3.4.對稱和不對稱的二乙炔基硅烷的合成。
雙(3-環己基丙-1-炔-1-基)二乙基硅烷7:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、Et2SiH2(24mg,36μL,0.275mmol,0.55當量)和0.5mL四氫呋喃(THF)在45℃進行反應持續48h。在45毫托的高真空下持續30分鐘去除溶劑后,獲得90%純度的期望的產物7(125.0mg,收率76%);隨后通過硅膠快速色譜法純化(100%己烷)獲得產物7的無色油。Rf=0.51(100%己烷);1H NMR(500MHz,CDCl3)δ2.15(d,J=6.6Hz,4H),1.81(ddd,J=13.6,4.0,1.8Hz,4H),1.72(dt,J=12.7,3.2Hz,4H),1.65(dddt,J=12.7,5.1,3.3,1.5Hz,2H),1.49(dddt,J=14.6,8.0,6.7,3.2Hz,2H),1.25(qt,J=12.7,3.4Hz,4H),1.15(tt,J=12.6,3.2Hz,4H),1.05(t,J=7.8Hz,6H),1.03–0.98(m,2H),0.67(q,J=7.8Hz,4H)。13C NMR(126MHz,CDCl3)δ108.03,80.73,37.36,32.76,27.95,26.43,26.29,7.47,7.02。IR(純膜NaCl)2923,2873,2852,2175,1448,1031,725,688cm–1;對于C22H37Si[M+H]計算的HRMS(EI+):329.2665,實測329.2661。
(3-環己基丙-1-炔-1-基)(環丙基乙炔基)二乙基硅烷8:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、環己基丙炔(61mg,0.5mmol,1.0當量)、環丙基乙炔(36mg,0.55mmol,1.1當量)、Et2SiH2(49mg,71μL,0.55mmol,1.1當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h,然后在65℃反應持續48h。在45毫托的高真空下持續30分鐘去除溶劑后,獲得90%純度的期望的產物8(102.8mg,收率76%);隨后通過硅膠快速色譜法純化(100%己烷)獲得產物8的無色油。還分離了收率10%的自偶聯的3-環己基-1-丙炔產物7;通過GC-MS鑒定了<5%的自偶聯的環丙基乙炔8-SI。在2步過程中,可以以相當的收率獲得此相同產物8(106.4mg,收率78%),在該2步過程中,首先分離甲硅烷基化的環己基丙炔2e,并且然后將此預甲硅烷基化的產物與環丙基乙炔(1.1當量)和NaOH(10mol%)結合。Rf=0.34(100%己烷);1H NMR(500MHz,CDCl3)δ2.14(d,J=6.6Hz,2H),1.83–1.77(m,2H),1.71(dt,J=12.7,3.2Hz,2H),1.65(dddt,J=12.8,5.1,3.3,1.5Hz,1H),1.48(ddtd,J=15.0,11.6,6.8,3.6Hz,1H),1.33–1.28(m,1H),1.28–1.19(m,2H),1.13(qt,J=12.8,3.3Hz,1H),1.03(t,J=7.9Hz,6H),1.01–0.95(m,2H),0.81–0.73(m,4H),0.65(q,J=7.9Hz,4H);13C NMR(126MHz,CDCl3)δ111.83,108.11,80.57,75.08,37.34,32.76,27.94,26.41,26.27,8.98,7.43,6.98,0.73。IR(純膜NaCl)3422,3094,3012,2955,1923,2852,2174,2105,1641,1449,1424,1376,1348,1322,1275,1232,1130,1073,1052,1028,979,891,873,828,779,725,688,642cm–1;對于C18H29Si[M+H]計算的HRMS(EI+):273.2039,實測273.2025。
實施例3.5.藥物的后期甲硅烷基化。
N-芐基-3-(二甲基(苯基)甲硅烷基)-N-甲基丙-2-炔-1-胺9:遵循一般程序。用NaOH(2.0mg,0.05mmol,10mol%)、巴吉林(N-芐基-N-甲基丙-2-炔-1-胺)(80mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h。在85℃、45毫托下持續30分鐘去除溶劑后,獲得期望的產物9的淡黃色油(140.4mg,收率96%)。1H NMR(500MHz,CDCl3)δ7.69(dq,J=6.8,3.4,2.7Hz,2H),7.40(dt,J=4.3,2.1Hz,3H),7.35–7.31(m,4H),7.30–7.26(m,1H),3.60(d,J=3.0Hz,2H),3.38(d,J=3.1Hz,2H),2.38(d,J=3.2Hz,3H),0.47(d,J=3.4Hz,6H);13C NMR(126MHz,CDCl3)δ138.47,137.34,133.82,129.54,129.39,128.45,128.01,127.35,102.95,88.41,60.17,46.08,42.09,–0.49。IR(純膜NaCl)3067,3026,2958,2793,2162,1494,1453,1428,1366,1249,1115,1026,980,837,817,780,732,698cm–1;對于C19H24NSi[M+H]計算的HRMS(EI+):294.1678,實測294.1689。
(((8R,9S,13S,14S,17S)-17-((二甲基(苯基)甲硅烷基)乙炔基)-3-甲氧基-13-甲基-7,8,9,11,12,13,14,15,16,17-十氫-6H-環戊并[a]菲-17-基)氧基)二甲基(苯基)硅烷10a:遵循一般程序。用KOH(2.8mg,0.05mmol,10mol%)、美雌醇((8R,9S,13S,14S,17R)-17-乙炔基-3-甲氧基-13-甲基-7,8,9,11,12,13,14,15,16,17-十氫-6H-環戊并[a]菲-17-醇)(155mg,0.5mmol,1.0當量)、PhMe2SiH(204mg,230μL,1.5mmol,3.0當量)和0.5mL 1,2-二甲氧基乙烷(DME)在45℃進行反應持續24h,然后在65℃進行反應持續48h。通過硅膠快速色譜法(于己烷中的1%→5%EtOAc)獲得產物10a的無色油(185.5mg,收率64%)。Rf=0.50(于己烷中的5%EtOAc);1H NMR(500MHz,THF-d8)δ7.62–7.56(m,4H),7.30(dtq,J=9.6,5.1,2.2Hz,6H),7.16(d,J=8.6Hz,1H),6.63(dd,J=8.5,2.7Hz,1H),6.59–6.55(m,1H),3.69(d,J=1.0Hz,3H),2.88–2.75(m,2H),2.42–2.23(m,2H),2.18(qd,J=10.8,10.1,3.5Hz,1H),2.11–1.95(m,2H),1.94–1.85(m,1H),1.83–1.74(m,2H),1.54–1.38(m,4H),1.34(ddt,J=24.2,12.3,5.9Hz,1H),0.94(d,J=2.0Hz,3H),0.52–0.43(m,6H),0.38–0.32(m,6H).13C NMR(126MHz,THF-d8)δ158.87,140.78,138.42,134.65,134.38,133.09,130.31,129.98,128.73,128.45,127.12,114.47,112.88,112.37,90.44,82.68,55.34,51.46,49.86,45.16,41.66,40.95,34.17,30.86,28.64,27.69,24.01,17.10,13.81,1.44,–0.61。IR(純膜NaCl)3417,3068,3048,2946,2869,2234,2160,2081,1610,1575,1500,1465,1427,1279,1252,1136,1117,1088,1045,929,886,818,783,730,699,642cm–1;對于C37H47O2Si2[M+H]計算的HRMS(EI+):579.3115,實測579.3109。
(8R,9S,13S,14S,17S)-17-((二甲基(苯基)甲硅烷基)乙炔基)-3-甲氧基-13-甲基-7,8,9,11,12,13,14,15,16,17-十氫-6H-環戊并[a]菲-17-醇10b:通過硅膠快速色譜法(于己烷中的1%→5%EtOAc)還從此反應獲得與10a 9:1混合物中的期望的產物10b的白色固體泡沫(40.0mg,收率18%)。Rf=0.39(于己烷中的5%EtOAc);1H NMR(500MHz,THF-d8)δ7.62–7.56(m,2H),7.32–7.27(m,3H),7.05(dd,J=8.8,0.9Hz,1H),6.60(dd,J=8.5,2.8Hz,1H),6.57(d,J=2.7Hz,1H),5.68(s,1H),3.71(s,3H),2.82–2.78(m,2H),2.33–2.26(m,1H),2.24–2.16(m,3H),2.00(ddd,J=13.3,11.9,4.1Hz,1H),1.90–1.81(m,3H),1.68–1.60(m,1H),1.40–1.27(m,4H),0.91(s,3H),0.44(d,J=1.5Hz,6H);13C NMR(126MHz,THF-d8)δ158.91,140.86,138.41,134.21,132.92,130.05,128.59,127.30,114.45,112.45,91.04,82.87,55.37,49.97,48.26,45.20,40.94,40.81,37.16,34.18,30.91,28.64,27.77,24.00,14.21,1.45。IR(純膜NaCl)3421,2932,2869,1609,1500,1464,1427,1979,1253,1138,1117,1099,1035,888,829,783,742,699cm–1;對于C29H37O2Si[M+H]計算的HRMS(EI+):445.2563,實測445.2575。
如本領域技術人員將理解的,本發明的許多修改和變化形式根據這些教導是可能的,并且據此設想到了所有的此類修改和變化形式。例如,除本文描述的實施方案之外,本發明設想并且要求保護源自本文中引用的本發明的特征的組合的那些發明以及補充本發明的特征的引用的現有技術參考文獻的那些。相似地,將明白,任何所描述的材料、特征或制品都可以與任何其他的材料、特征或制品組合使用,并且此類組合被認為是在本發明的范圍內。
- 還沒有人留言評論。精彩留言會獲得點贊!