聚酰胺粒料、聚酰胺粒料的制造方法、以及聚酰胺成型體的制造方法與流程
本發明涉及將二胺和二羧酸縮聚而得到的聚酰胺粒料,其制造方法,進而涉及使用該粒料的聚酰胺成型體的制造方法。
背景技術:
由包含間苯二甲胺的二胺與二羧酸的縮聚反應而得到的含間苯二甲基的聚酰胺的機械強度優異,進而相對于氧氣、二氧化碳、臭氣以及氣味等氣態物質的透過性低,因此在包裝材料、成型材料、單絲等廣泛的用途中使用。其中,由間苯二甲胺和己二酸而得到的聚酰胺(以下,稱為尼龍MXD6)為高強度、高彈性模量,阻氣性也良好,因此特別廣泛地被使用。
對于含間苯二甲基的聚酰胺,考慮處理性的難易度等,以粒料的形態而廣泛流通。對于含間苯二甲基的聚酰胺的粒料化例如可知將熔融縮聚而成的含間苯二甲基的聚酰胺抽出為線料狀并進行水冷,接著利用造粒機進行切斷。對于如此得到的聚酰胺粒料,抽出時被較快地冷卻,通常成為非晶狀態。對于聚酰胺粒料,直接進行熔融混煉而成型加工為各種制品,或者進一步需要高分子量化的情況下,有時在保持粒料的狀態下固相聚合(例如,參照專利文獻1)。固相聚合而成的聚酰胺粒料通常成為結晶狀態。
對于聚酰胺粒料,從使成型品的外觀、穩定性良好的觀點出發,謀求降低其色相。以往,為了降低色相,例如,已知將作為穩定劑的磷酸、亞磷酸、次磷酸、或它們的鹽等的含磷原子化合物在熔融縮聚時添加到反應體系(例如,參照專利文獻2)。
此外,所粒料化的樹脂材料通過擠出成型、注射成型等加工為各種成型體。各種樹脂材料的粒料被用擠出成型機、注射成型機進行成型加工時,通常,向在內部具備螺桿的料筒中,根據需要與其它的樹脂、添加劑一同投入,并且在進行增塑化、混煉之后,被擠出或注射而成型為各種成型體。作為料筒使用在內部具有2根螺桿的雙螺桿型的料筒,但由于結構簡單而廣泛使用在料筒內具有1根螺桿的單螺桿型料筒。
現有技術文獻
專利文獻
專利文獻1:日本特開2007-31475號公報
專利文獻2:日本特開2007-321035號公報
技術實現要素:
發明要解決的問題
然而,如專利文獻2那樣,即便為了降低色相而添加添加劑,有時也不能充分防止反應時的著色。此外,過度添加添加劑時,有時產生縮孔增加等其它的問題,因此謀求用除添加劑以外的方法降低聚酰胺粒料的色相。
進而,含間苯二甲基的聚酰胺的熔點比較高,并且加熱到一定溫度以上時,具有急劇軟化的性質。含間苯二甲基的聚酰胺由于那樣的性質而在成型加工時需要延長預熱時間,例如期望用單螺桿擠出成型機在比較高的壓縮比下進行混煉。然而,利用單螺桿在高壓縮比下急劇地壓縮、混煉時,有時粒料彼此緊密附著、或軟化了的粒料纏繞在螺桿壓縮區域,從而引起擠出不良,存在加工穩定性差的問題。
本發明是鑒于以上的問題而成的,其目的在于,例如,降低通過固相聚合而得到的聚酰胺粒料的色相,由此,使由該聚酰胺粒料得到的成型品的色相良好。
進而,目的還在于提供:利用具備單螺桿的料筒等混煉裝置混煉含間苯二甲基的聚酰胺時,可以以高加工穩定性進行成型加工的聚酰胺粒料。
用于解決問題的方案
本發明人等為了解決上述問題,首先著眼于由含間苯二甲基的聚酰胺形成的聚酰胺粒料的表層部分(表皮部分)的狀態。并且,深入研究的結果,發現在表層部分密集地存在球晶的聚酰胺粒料,其表層部分恰如保護層而起作用,在固相聚合反應等中保護聚酰胺粒料,并且,可以使聚酰胺粒料的色相良好。此外,發現將由含間苯二甲基的聚酰胺形成的聚酰胺粒料的表層部分(表皮部分)制成特定的狀態,并且將聚酰胺粒料的截面積設為一定的范圍時,加工穩定性良好,從而完成以下的本發明。本發明提供以下的[1]~[18]。
[1]一種聚酰胺粒料,其由聚酰胺形成,所述聚酰胺包含二胺單元和二羧酸單元,二胺單元的50摩爾%以上源自間苯二甲胺,并且粒料的表皮部分中的球晶密度為40000~250000個/mm2。
[2]根據上述[1]記載的聚酰胺粒料,其中,核部分的球晶密度為10000~40000個/mm2。
[3]根據上述[1]或[2]記載的聚酰胺粒料,其中,前述聚酰胺的二羧酸單元的50摩爾%以上源自碳數6~12的脂肪族二羧酸。
[4]根據上述[3]記載的聚酰胺粒料,其中,前述碳數6~12的脂肪族二羧酸為己二酸、癸二酸、或它們的混合物。
[5]根據上述[1]~[4]中任一項記載的聚酰胺粒料,其中,在磷原子濃度1~100ppm下含有含磷原子化合物。
[6]根據上述[1]~[5]中任一項記載的聚酰胺粒料,其滿足以下的式(1)的條件。
-110μeq/g≤([COOH]-[NH2])≤110μeq/g (1)
(需要說明的是,式(1)中,[COOH]表示前述聚酰胺的末端羧基濃度(μeq/g)、[NH2]表示前述聚酰胺的末端氨基濃度(μeq/g)。)
[7]根據上述[1]~[6]中任一項記載的聚酰胺粒料,其相對粘度為2.0~4.2。
[8]根據上述[1]~[7]中任一項記載的聚酰胺粒料,其中,前述表皮部分的球晶密度為80000~110000個/mm2。
[9]根據上述[1]~[8]中任一項記載的聚酰胺粒料,其是將處于非晶狀態的粒料化了的聚酰胺固相聚合而得到的。
[10]根據上述[9]記載的聚酰胺粒料,前述處于非晶狀態的粒料化了的聚酰胺是通過熔融縮聚而得到的。
[11]根據上述[1]~[10]中任一項記載的聚酰胺粒料,其截面積為5~13mm2。
[12]根據上述[11]記載的聚酰胺粒料,其為用于利用壓縮比為2.0~4.0的單螺桿擠出成型機混煉、成型加工的高壓縮螺桿成型用聚酰胺粒料。
[13]一種聚酰胺成型體的制造方法,其為將上述[11]或[12]記載的聚酰胺粒料混煉之后、進行成型加工,得到的聚酰胺成型體的聚酰胺成型體的制造方法,
在內部具有單螺桿的料筒中將前述聚酰胺粒料混煉。
[14]根據上述[13]記載的聚酰胺成型體的制造方法,其中,前述料筒中的壓縮比為2.0~4.0。
[15]根據上述[13]或[14]記載的聚酰胺成型體的制造方法,其中,前述螺桿具有供給部、與前述供給部連接的壓縮部和與前述壓縮部連接的計量部,
對于前述供給部、壓縮部、計量部的長度,將它們的總計設為1時,分別為0.40~0.55、0.10~0.30、0.10~0.40。
[16]一種聚酰胺粒料的制造方法,其具備如下工序:
將包含50摩爾%以上的間苯二甲胺的二胺與二羧酸縮聚而得到的、處于熔融狀態的聚酰胺以線料狀送出的工序;
將被以線料狀送出的聚酰胺水冷并切斷而粒料化,之后,對粒料化了的聚酰胺進一步水冷4秒以上的工序;和
將前述水冷后的粒料化了的聚酰胺進一步固相聚合而得到聚酰胺粒料的工序。
[17]根據上述[16]記載的聚酰胺粒料的制造方法,其中,前述水冷終止時的粒料化了的聚酰胺的溫度為65℃以下。
[18]根據上述[16]或[17]記載的聚酰胺粒料的制造方法,其中,將前述被以線料狀被送出的聚酰胺在著水后2秒以內切斷而粒料化。
進而,本發明人等為了解決上述問題,著眼于由處于非晶狀態的尼龍MXD6形成的聚酰胺粒料的表層部分(表皮部分)的狀態和粒料內部(核部分)的狀態。并且,深入研究的結果,推定:通過減小由表層部分的局部熱分析測定的指針下降溫度與核部分的指針下降溫度之差、且提高表層部分的指針下降溫度,從而減小核部分與粒料部分的熱性質之差,并且還通過表層部分恰如保護層起作用,從而抑制聚酰胺粒料吸收氧氣,由此,發現可以改進由粒料得到的成型品等的色相,完成以下的發明。即,本發明還提供以下的[19]~[29]。
[19]一種聚酰胺粒料,其由聚酰胺形成,所述聚酰胺包含二胺單元和二羧酸單元,二胺單元的50摩爾%以上源自間苯二甲胺、二羧酸單元的70摩爾%以上源自己二酸,并且
由使用熱探針的局部熱分析測定的粒料的表皮部分的指針下降溫度為78~92℃、并且比粒料的核部分的指針下降溫度高,其溫度差為0.1~2.5℃。
[20]根據上述[19]記載的聚酰胺粒料,其中,觀察由0.1mol/L碘-碘化鉀溶液染色的粒料時,染色為G≤90(sRGB值)的粒料的比例不足50%。
[21]根據上述[19]或[20]記載的聚酰胺粒料,其中,前述粒料的表皮部分的指針下降溫度為80~91℃。
[22]根據上述[19]~[21]中任一項記載的聚酰胺粒料,其中,前述表皮部分的指針下降溫度與核部分的指針下降溫度的溫度差為0.2~2℃。
[23]根據上述[19]~[22]中任一項記載的聚酰胺粒料,其中,在磷原子濃度1~100ppm下含有含磷原子化合物。
[24]根據上述[19]~[23]中任一項記載的聚酰胺粒料,其滿足以下的式(1)的條件。
-110μeq/g≤([COOH]-[NH2])≤110μeq/g (1)
(需要說明的是,式(1)中,[COOH]表示前述聚酰胺的末端羧基濃度(μeq/g)、[NH2]表示前述聚酰胺的末端氨基濃度(μeq/g)。)
[25]根據上述[19]~[24]中任一項記載的聚酰胺粒料,其相對粘度為1.8~2.4。
[26]根據上述[19]~[25]中任一項記載的聚酰胺粒料,其由通過熔融縮聚得到的聚酰胺成型而成的。
[27]一種聚酰胺粒料的制造方法,其具備:將包含50摩爾%以上間苯二甲胺的二胺與包含70摩爾%以上己二酸的二羧酸縮聚而得到的、處于熔融狀態的聚酰胺以線料狀送出的工序;和
將以線料狀送出的聚酰胺水冷并切斷而粒料化,之后,對粒料化了的聚酰胺進一步水冷4秒以上的工序。
[28]根據上述[27]記載的聚酰胺粒料的制造方法,其中,水冷終止后的聚酰胺粒料的溫度為65℃以下。
[29]根據上述[27]或[28]記載的聚酰胺粒料的制造方法,其中,將前述以線料狀被送出的聚酰胺在著水后2秒以內切斷而粒料化。
發明的效果
本發明中,可以使聚酰胺粒料的色相良好,由聚酰胺粒料成型而成的各種成型體的色相也可以變得良好。
進而,通過將聚酰胺粒料的截面積設為規定的范圍,利用具有單螺桿的料筒等混煉裝置對粒料進行混煉時,不易產生加工不良,可以使加工穩定性優異。
附圖說明
圖1為示出本發明的一實施方式中所使用的單螺桿擠出成型機的示意性截面圖。
圖2為示出由聚酰胺粒料采取用于指針下降溫度測定的試樣的方法的示意圖。
圖3為示出指針下降溫度的測定方法的示意圖。
圖4示出對實施例1中的聚酰胺粒料的表皮部分進行離子銑削之后的擴大照片。
圖5示出對實施例1中的聚酰胺粒料的核部分進行離子銑削之后的擴大照片。
圖6示出對比較例1中的聚酰胺粒料的表皮部分進行離子銑削之后的擴大照片。
具體實施方式
以下,用實施方式對本發明進行說明。
<第1聚酰胺粒料>
本發明的第1聚酰胺粒料由聚酰胺形成,所述聚酰胺包含二胺單元和二羧酸單元,二胺單元的50摩爾%以上源自間苯二甲胺。
[二胺單元]
在第1聚酰胺粒料中,聚酰胺中的二胺單元包含50摩爾%以上的源自間苯二甲胺的構成單元、優選含有60~100摩爾%、更優選含有70~100摩爾%、進一步優選含有80~100摩爾%。
本發明中,源自間苯二甲胺的構成單元不足50摩爾%時,難以提高由第1聚酰胺粒料得到的成型品的阻隔性能,此外,難以得到本發明的聚酰胺中所謀求的各種物性。
在聚酰胺中,作為除間苯二甲胺以外的二胺,可以例示出四亞甲基二胺、五亞甲基二胺、2-甲基-1,5-戊二胺、六亞甲基二胺、七亞甲基二胺、八亞甲基二胺、九亞甲基二胺、十亞甲基二胺、十二亞甲基二胺、2,2,4-三甲基六亞甲基二胺或2,4,4-三甲基六亞甲基二胺等脂肪族二胺;1,3-雙(氨基甲基)環己烷或1,4-雙(氨基甲基)環己烷、1,3-二氨基環己烷或1,4-二氨基環己烷、雙(4-氨基環己基)甲烷、2,2-雙(4-氨基環己基)丙烷、雙(氨基甲基)萘烷、雙(氨基甲基)三環癸烷等脂環族二胺;對苯二甲胺、雙(4-氨基苯基)醚、對苯二胺、雙(氨基甲基)萘等具有芳香環的二胺類等,但不限于它們。
作為除間苯二甲胺以外的二胺,它們之中,優選使用對苯二甲胺。使用對苯二甲胺時,聚酰胺中的二胺單元包含50摩爾%以下源自對苯二甲胺的構成單元,優選含有40摩爾%以下、進一步優選含有30摩爾%以下。
[二羧酸單元]
在第1聚酰胺粒料中,聚酰胺中的二羧酸單元從結晶性的觀點出發,優選包含50摩爾%以上的脂肪族二羧酸單元,更優選包含70摩爾%以上,進一步優選包含90摩爾%以上。作為脂肪族二羧酸,具體而言,可列舉出琥珀酸、戊二酸、己二酸、庚二酸、辛二酸、壬二酸、癸二酸、1,9-十一烷二酸、1,10-癸烷二羧酸等碳數4~20的α,ω-直鏈脂肪族二羧酸。
此外,脂肪族二羧酸更優選碳數6~12的脂肪族二羧酸、進一步優選碳數6~10的脂肪族二羧酸,特別優選己二酸、癸二酸或它們的混合物。本發明中,通過使用己二酸,從而可以使成型體的阻氣性變得良好。此外,通過使用癸二酸,從而容易得到吸水性低、尺寸穩定性優異的成型體。
作為除脂肪族二羧酸單元以外的可以構成二羧酸單元的化合物,可以例示出1,3-環己烷二羧酸、1,4-環己烷二羧酸等脂環族二羧酸;對苯二甲酸、間苯二甲酸、鄰苯二甲酸、苯二甲基二酸、萘二羧酸等芳香族二羧酸等,但不限于它們。在它們之中,間苯二甲酸可以容易地得到阻隔性能優異的聚酰胺而不阻礙聚酰胺的制造時的縮聚反應,故優選。
在本發明中,作為聚酰胺,最優選的是全部的二胺單元由源自間苯二甲胺的構成單元組成、全部的二羧酸單元由脂肪族二羧酸單元組成的聚酰胺。
[球晶密度]
本發明的第1聚酰胺粒料為處于結晶狀態的聚酰胺粒料,在構成粒料表面的表皮部分中微細的球晶大量存在,并且該球晶密集地存在。其中,本發明中示出的球晶并不是指偏光顯微鏡等以往的觀察方法中所確認到的球晶,而是指如后所述利用離子束的照射進行離子銑削之后,觀察表皮部分所觀察到的源于球晶的紋路。并且,表皮部分的球晶密度用后述的測定方法進行測定時,為40000~250000個/mm2。如此,第1聚酰胺粒料中成為粒料的表皮部分的球晶密集地存在的結構,因此聚酰胺粒料被表皮部分保護,防止由熱歷程導致的色相惡化,可以使粒料的色相變得良好。此外,表面的結晶化度變高,即便進行增塑化,粒料彼此也不易緊密附著。因此,即便在單螺桿擠出成型機等中,以高壓縮力進行壓縮并且進行混煉,粒料彼此也不發生緊密附著之類的情況,可以提高加工穩定性。
另一方面,球晶密度低于上述下限值、或在離子銑削后不能充分觀察到源于球晶的紋路的情況下,在表皮部分中球晶未密集地存在、或者在表皮部分中球晶未明確地存在。因此,聚酰胺粒料未被表皮部分充分地保護,難以使粒料的色相變得良好。
進而,球晶密度低于上述下限值、或在離子銑削后不能充分地觀察到源于球晶的紋路的情況下,成為表面的結晶化度低的物質。因此,用單螺桿擠出成型機等進行增塑化、混煉時,容易產生粒料彼此緊密附著等不良。
此外,球晶密度大于上述上限值時,存在用后述的制造方法的制造變得困難的情況。
在第1聚酰胺粒料的表皮部分中,球晶密度優選為80000~110000個/mm2。通過將球晶密度設為這樣的范圍,從而聚酰胺粒料被表皮部分良好地保護,可以使其的色相變得良好,由第1聚酰胺粒料得到的成型體的色相也可以變得良好。進而,通過將球晶密度設為這樣的范圍,從而可以使第1聚酰胺粒料更不容易緊密附著,進一步提高加工穩定性。
第1聚酰胺粒料中,通常在構成粒料內部的核部分,球晶也大量存在,例如在離子銑削后觀察時,明確地看到多個球晶。其中,在核部分中,球晶比較稀疏地存在,通常第1聚酰胺粒料的核部分的球晶密度比表皮部分的球晶密度小。具體而言,核部分的球晶密度優選為10000~40000個/mm2、更優選為15000~40000個/mm2。核部的球晶密度為上述范圍時,與表皮部分的熱性質之差變小、容易使成型品的物性等穩定。
需要說明的是,第1聚酰胺粒料中,表皮部分是指與粒料的軸向垂直的截面中從粒料外周到60μm為止的部分,核部分是指距粒料中心為粒料半徑的70%以內的部分。需要說明的是,粒料直徑是指在粒料的上述截面中最長的直徑,粒料半徑是指粒料直徑的1/2的長度。
作為聚酰胺的聚合度的指標存在多個,通常使用相對粘度。對于第1聚酰胺粒料,其相對粘度優選為2.0~4.2。相對粘度為上述范圍的聚酰胺可以通過后述的制造方法而容易地制造。此外,由第1聚酰胺粒料成型而成的成型體的機械強度以及成型性良好。從這些觀點出發,相對粘度更優選2.0~3.6。
此外,第1聚酰胺粒料優選滿足以下的式(1)的條件。
-110μeq/g≤([COOH]-[NH2])≤110μeq/g(1)
(需要說明的是,式(1)中,[COOH]表示聚酰胺的末端羧基濃度(μeq/g)、[NH2]表示聚酰胺的末端氨基濃度(μeq/g)。)
如此,末端羧基濃度與末端氨基濃度之差小時,耐熱性良好、不易產生色劣化。此外,為了進一步抑制色劣化,([COOH]-[NH2])更優選為-80~80μeq/g。
第1聚酰胺粒料為僅具有1個熔點峰的物質。第1聚酰胺粒料的熔點沒有特別限定,優選為190~290℃、更優選為210~270℃。
第1聚酰胺粒料沒有特別限定,通常沿著其股線方向(軸向)的長度為1.0~5.0mm左右、優選為1.0~4.0mm左右、更優選為2.0~4.0mm、進一步優選為2.0~3.5mm。此外,第1聚酰胺粒料的粒料直徑通常為1.0~4.0mm左右、優選為2.0~3.5mm。粒料的形狀沒有特別限定,通常,如后述那樣以橫截股線的方式切斷而成的形狀,優選為圓柱狀、橢圓柱狀。
<第1聚酰胺粒料的制造方法>
接著,對本發明中的第1聚酰胺粒料的制造方法進行說明。
本發明中的第1聚酰胺粒料的制造方法若為可以得到上述的第1聚酰胺粒料的方法則沒有特別限定,優選對粒料化了的聚酰胺進一步進行固相聚合而得到。此外,粒料化了的聚酰胺優選例如為通過將二胺與二羧酸熔融縮聚而得到的粒料。在以下說明如此通過熔融縮聚以及固相聚合而制造本發明的聚酰胺粒料的方法的一個例子。
本發明的一實施方式中的第1聚酰胺粒料的制造方法為具備如下工序的方法:將二胺與二羧酸縮聚而得到的處于熔融狀態的聚酰胺以線料狀送出的工序;將被以線料狀送出的聚酰胺水冷并切斷而粒料化,之后,對粒料化了的聚酰胺進一步水冷4秒以上的工序;和將水冷后的粒料化了的聚酰胺進一步固相聚合而得到聚酰胺粒料的工序。
以往,在含間苯二甲基的聚酰胺的制造中,從熔融縮聚后的溫度并不那樣高的方面、進而裝置的制約出發,通常盡可能縮短水冷時間。另外,已知有將通過縮聚得到的聚酰胺抽出為線料狀,股線直接進行水冷的方法。
與之相對,在本制造方法中,以小塊化為粒料狀的狀態對聚酰胺進行水冷,并且,通過將該水冷時間如上所述延長至4秒以上,從而使粒料表面急劇降溫,由此,使表皮部分的形態成為特異的形態。因此,將用上述方法造粒后被水冷的粒料化了的聚酰胺通過固相聚合而結晶化時,如上所述,在表皮部分明確地形成球晶,其球晶密度也變高。
另外,推定粒料的核部分的冷卻速度雖然稍慢于表皮部分,但核部分依然被驟冷,雖然核部分的形態與表皮部分存在少許差別,但仍成為特異的形態。因此,將由上述方法而粒料化了的聚酰胺通過固相聚合而結晶化時,在核部分明確地形成球晶,可以使該球晶密度如上所述為較高。
以下,對于本制造方法進行詳細地說明。
在本制造方法中,二胺與二羧酸的縮聚優選利用熔融縮聚法進行。
作為熔融縮聚法的適宜的例子,可以列舉出將二胺直接加入到熔融的二羧酸進行縮聚的所謂直接聚合法。更具體而言,在反應槽中邊攪拌處于熔融狀態的二羧酸邊連續地或者間斷地添加二胺,邊去除縮合水邊進行縮聚,使反應溫度上升以使溫度在添加二胺期間不低于生成的聚酰胺的熔點。此外,二胺添加終止之后,也控制溫度使其不低于生成的聚酰胺的熔點并進一步繼續反應即可。對于以上的反應,可以在常壓、加壓的任一條件下實施。之后,可以逐步減壓而成為低于常壓的壓力,進一步繼續進行一定時間的反應。需要說明的是,對于本制造方法中的反應溫度的上限值,通常控制在所得到的非晶狀態的聚酰胺的熔點+70℃左右以下,優選控制在所得到的非晶狀態的聚酰胺的熔點+20℃左右以下。
本制造方法中所使用的二胺以及二羧酸若能夠得到上述的聚酰胺即可,例如,所使用的全二胺中的各二胺(間苯二甲胺等)的每種種類的含有比例(摩爾%)與上述的聚酰胺中的二胺單元中的源自各二胺的每種種類的構成單元的比例(摩爾%)相同。對于二羧酸也同樣。
此外,熔融縮聚法并不限于直接聚合法,也可以進行在水的存在下、加壓下將由二羧酸和二胺形成的尼龍鹽加熱而進行的尼龍鹽法。
進而,縮聚反應也可以由如下方法進行:用擠出機對由二胺以及二羧酸形成的聚酰胺的低聚物進行熔融混煉并使其反應的反應擠出法。對于反應擠出法,為了使其充分地反應,優選使用適于反應擠出的螺桿,使用L/D較大的雙螺桿擠出機。
對于通過縮聚得到的處于熔融狀態的聚酰胺,例如由在反應槽的底部所具備的線料模頭抽出為線料狀。需要說明的是,抽出聚酰胺時,反應槽內部通常利用氮氣等進行加壓。其中,線料模頭的口模直徑根據所得到的粒料的粒料直徑、截面積而設定。此外,對于抽出股線時的聚酰胺的溫度,以聚酰胺保持在熔融狀態的方式為該聚酰胺的熔點以上即可,優選熔點以上且(熔點+70℃)以下的溫度,更優選為熔點以上且(熔點+20℃)以下的溫度。
抽出為線料狀的熔融狀態的聚酰胺被水冷并被粒料化。更具體而言,抽出為線料狀的聚酰胺邊被浸漬到水槽中、在水中被冷卻,邊以規定的牽引速度被牽引并且利用造粒機的刀具將股線以橫截的方式切斷。其中,造粒機的刀具牽引速度沒有特別限定,例如為100~300m/分鐘、優選為120~280m/分鐘。
抽出為線料狀的熔融狀態的聚酰胺優選在著水之后立即切斷,具體而言,優選在著水后2秒以內、更優選在著水后1秒以內切斷而粒料化。聚酰胺在著水后立即切斷而粒料化時,在小塊化的狀態下立即冷卻,從而容易驟冷。
對于上述那樣粒料化了的聚酰胺,接著送到水槽內并被水冷之后離水,從水槽取出。其中,粒料化之后至離水為止的時間(以下,稱為“粒料水冷時間”)為4秒以上、優選為5秒以上。粒料水冷時間不足4秒時,擔心聚酰胺未被充分地冷卻,不能使表皮部分的球晶密度致密。
此外,粒料水冷時間的上限沒有特別限定,從效率良好地制造聚酰胺粒料的觀點出發,通常為30秒以下、優選為10秒以下。
此外,水冷終止時(即、離水時)的粒料化了的聚酰胺的溫度優選為65℃以下、更優選為60℃以下、進一步優選為50℃以下。此外,水冷終止時的聚酰胺的溫度的下限值沒有特別限定,為了使工序高效化,優選20℃以上、進一步優選30℃以上、進一步優選35℃以上。
從水槽取出的粒料化了的聚酰胺可以被自然干燥,通過利用干燥機的吹風,從而可以強制性地去除粒料表面的水。
此外,水槽的溫度例如為0~50℃、優選為10~40℃、更優選為15~30℃。
經過以上的工序而得到的粒料化了的聚酰胺(以下,為了方便也稱為“縮聚聚酰胺粒料”)通常成為非晶狀態,并且通過后述的固相聚合從而成為結晶狀態。需要說明的是,本說明書中,非晶狀態的聚酰胺是指結晶化度不足25%的聚酰胺,結晶狀態的聚酰胺是指結晶化度為25%以上的聚酰胺。結晶化度根據后述的實施例中的測定方法而測定。
需要說明的是,本制造方法中,例如,通過調整造粒機中的牽引速度、線料模頭的口模直徑、口模閥開度、或將聚酰胺從反應槽抽出時的反應槽內部的壓力的任一者以上,從而可以適宜調整粒料的截面積以及粒料的直徑。
此外,在上述縮聚反應中,二羧酸成分與二胺成分可以在含磷原子化合物存在下縮聚。如此,通過存在含磷原子化合物,從而可以使聚酰胺的聚合性良好,并且可以防止聚酰胺的著色。
作為含磷原子化合物,可以列舉出二甲基次膦酸、苯基甲基次膦酸等次膦酸化合物;次磷酸、次磷酸鈉、次磷酸鉀、次磷酸鋰、次磷酸鎂、次磷酸鈣、次磷酸乙酯等二亞磷酸化合物;膦酸、膦酸鈉、膦酸鉀、膦酸鋰、膦酸鉀、膦酸鎂、膦酸鈣、苯基膦酸、乙基膦酸、苯基膦酸鈉、苯基膦酸鉀、苯基膦酸鋰、苯基膦酸二乙酯、乙基膦酸鈉、乙基膦酸鉀等膦酸化合物;亞膦酸、亞膦酸鈉、亞膦酸鋰、亞膦酸鉀、亞膦酸鎂、亞膦酸鈣、苯基亞膦酸、苯基亞膦酸鈉、苯基亞膦酸鉀、苯基亞膦酸鋰、苯基亞膦酸乙酯等亞膦酸化合物;亞磷酸、亞磷酸氫鈉、亞磷酸鈉、亞磷酸鋰、亞磷酸鉀、亞磷酸鎂、亞磷酸鈣、亞磷酸三乙酯、亞磷酸三苯酯、焦亞磷酸等亞磷酸化合物等。
它們之中,特別是次磷酸鈉、次磷酸鈣、次磷酸鉀、次磷酸鋰等次磷酸金屬鹽的促進縮聚反應的效果高并且抗著色效果優異,因此優選使用,特別優選次磷酸鈉。需要說明的是,本發明中可以使用的含磷原子化合物并不限于這些化合物。
含磷原子化合物優選以所得到的聚酰胺粒料中所含有的磷原子濃度換算計為1~350ppm的方式配合,更優選以成為1~200ppm的方式配合、進一步優選為1~100ppm、更進一步優選為1~80ppm、最優選為2~80ppm。若為1ppm以上,則以適合的速度推進縮聚反應并且在縮聚反應中不易產生著色。若為350ppm以下,則所得到的聚酰胺難以凝膠化,此外,也可以降低認為由含磷原子化合物引起的縮孔向成型品中的混入,成型品的外觀良好。進而,本發明中,所得到的聚酰胺粒料不易著色,即便為100ppm或80ppm以下程度的少量的配合量,聚酰胺粒料的色相也不易惡化。
縮聚反應在含磷原子化合物、以及堿金屬化合物存在下進行即可。通過配合堿金屬化合物,從而調整酰胺化反應速度,可以防止擔心因添加含磷原子化合物而發生的凝膠化。
堿金屬化合物以及前述的含磷原子化合物通常在二羧酸成分與二胺成分反應之前添加到反應體系中。
作為堿金屬化合物,優選堿金屬氫氧化物、堿金屬醋酸鹽、堿金屬碳酸鹽、堿金屬醇鹽等。作為本發明中可以使用的堿金屬化合物的具體例子,可以列舉出氫氧化鋰、氫氧化鈉、氫氧化鉀、氫氧化銣、氫氧化銫、醋酸鋰、醋酸鈉、醋酸鉀、醋酸銣、醋酸銫、甲醇鈉、乙醇鈉、丙醇鈉、丁醇鈉、甲醇鉀、甲醇鋰、碳酸鈉等,可以使用的物質并不限于這些化合物。需要說明的是,所得到的聚酰胺粒料中的含磷原子化合物與堿金屬化合物的比率(摩爾比)從聚合速度控制的觀點、降低黃色指數的觀點出發,優選含磷原子化合物/堿金屬化合物=1.0/0.05~1.0/1.5的范圍、更優選為1.0/0.1~1.0/1.2、更優選為1.0/0.2~1.0/1.1。
此外,反應體系中除了二胺、二羧酸、含磷原子化合物、堿金屬化合物以外還可以添加分子量調節劑等其它的添加劑、后述的其它的單體等。
如上所述,將粒料化了的聚酰胺(縮聚聚酰胺粒料)固相聚合的方法若為使聚酰胺結晶化并且提高分子量(相對粘度)的方法則沒有特別限定。固相聚合例如使用連續式的加熱干燥裝置、被稱為轉鼓式干燥機、圓錐形干燥機、旋轉式干燥機等的旋轉鼓式的加熱裝置以及被稱為諾塔混合機的在內部具備旋轉葉片的圓錐型的加熱裝置等來進行,但不限于它們,可以使用公知的方法、裝置。在這些裝置之中,旋轉鼓式的加熱裝置可以使體系內密閉化并且容易以去除作為著色的原因的氧氣的狀態進行縮聚,故優選使用。
此外,對于固相聚合的條件,若在比所得到的第1聚酰胺粒料的熔點低的條件下進行就沒有限定,例如,優選使反應體系的溫度逐步升溫,在130℃以上并且比所得到的第1聚酰胺粒料的熔點低的溫度下使其反應1~10小時左右、優選反應1.5~6小時左右。此外,在固相聚合中,優選使反應體系處于減壓下,優選在20kPa以下、更優選在10kPa以下左右進行。
如以上所述,在本發明中,通過增大表皮部分中的球晶密度,從而可以使所得到的第1聚酰胺粒料的色相變得良好。因此,將第1聚酰胺粒料作為原料進行成型加工時,可以使成型體的色相變得良好。
<聚酰胺成型體>
對于本發明的第1聚酰胺粒料,在根據需要混合其它的任意成分的基礎上,利用公知的成型方法,可以成型為期望形狀的聚酰胺成型體。對于成型方法,例如,可以列舉出注射成型、吹塑成型、擠出成型、壓縮成型、拉伸、真空成型等。作為聚酰胺成型體,沒有特別限定,可以列舉出薄膜、片材、管、軟管、導管、中空容器、瓶、中空容器或者瓶的預成型坯、纖維、各種形狀的部件等各種成型品。
此外,對于本發明的聚酰胺成型體,可以以在由其它樹脂材料成型的成型體上層疊或粘接等的方式而成型,構成多層結構體、復合纖維、或其它部件等的成型品。作為多層結構體,可以列舉出多層薄膜、多層片材、多層管、多層軟管、多層導管、多層容器、多層瓶、或多層容器或者多層瓶的預成型坯等。
在本發明中,上述的第1聚酰胺粒料由單螺桿擠出成型機等規定的成型機進行成型時,將其截面積設為5~13mm2。第1聚酰胺粒料的截面積不足5mm2時,例如在單螺桿擠出成型機中對粒料進行混煉時,產生粒料纏繞螺桿的不良,即便表皮部分的球晶密度如上述那樣大,也無法使加工穩定性良好。另一方面,截面積大于13mm2時,對用于混煉粒料的混煉裝置的負載變大,例如在單螺桿擠出成型機中產生由該負載引起的振動,從而加工穩定性降低。從這些觀點出發,聚酰胺粒料的截面積優選為6~12mm2、更優選為6~10mm2。
需要說明的是,對于粒料的截面積,在與股線方向(軸向)垂直的截面中,例如用游標尺測定直徑(圓以外的情況下的短徑和長徑),將粒料截面看作圓、或橢圓形,算出面積。需要說明的是,長徑意味著在粒料的上述截面中最長的直徑,短徑是指在該截面中與長徑垂直的直徑的長度。
此外,第1聚酰胺粒料利用單螺桿擠出成型機等規定的成型機進行成型的情況,優選具有上述截面積并且沿該股線方向(軸向)的長度(粒料長度)為1.5~5.0mm。對于第1聚酰胺粒料,通過將粒料長度設為1.5mm以上,從而混煉粒料時,變得容易防止纏繞螺桿。此外,通過設為5.0mm以下,從而變得容易降低對擠出機作用的負載。從這些觀點出發,上述粒料的長度更優選為2.0~4.0mm。
對于具有這樣的特定的粒料尺寸的第1聚酰胺粒料,優選用作用具有高壓縮螺桿的單螺桿擠出成型機進行混煉而成型加工的高壓縮螺桿成型用聚酰胺粒料。第1聚酰胺粒料具有一定的粒料尺寸、并且在表皮部分大量存在微細的球晶,從而防止在混煉時粒料彼此緊密附著、或粒料纏繞螺桿的不良,進而,對單螺桿擠出成型機的負載變小。因此,即便用具有高壓縮螺桿的單螺桿擠出成型機對粒料進行混煉,也可以使加工性良好。
作為具有高壓縮螺桿的單螺桿擠出成型機,可以列舉出料筒中的壓縮比(C/R)為2.0~4.0的單螺桿擠出成型機,可以列舉出后述的單螺桿擠出成型機作為代表性的具體例子。
[聚酰胺成型體的制造方法]
作為使用第1聚酰胺粒料來制造聚酰胺成型體的方法,可以列舉出混煉具有上述特定的粒料尺寸(特定的截面積等)的聚酰胺粒料之后,進行成型加工而得到的聚酰胺成型體的方法。在該方法中,優選利用在內部具有單螺桿的料筒來混煉第1聚酰胺粒料。本發明中,如上所述,將表皮部分的球晶密度設為一定范圍,并且將粒料的截面積設為上述的一定范圍時,即便利用單螺桿進行混煉,也可以使加工穩定性優異。
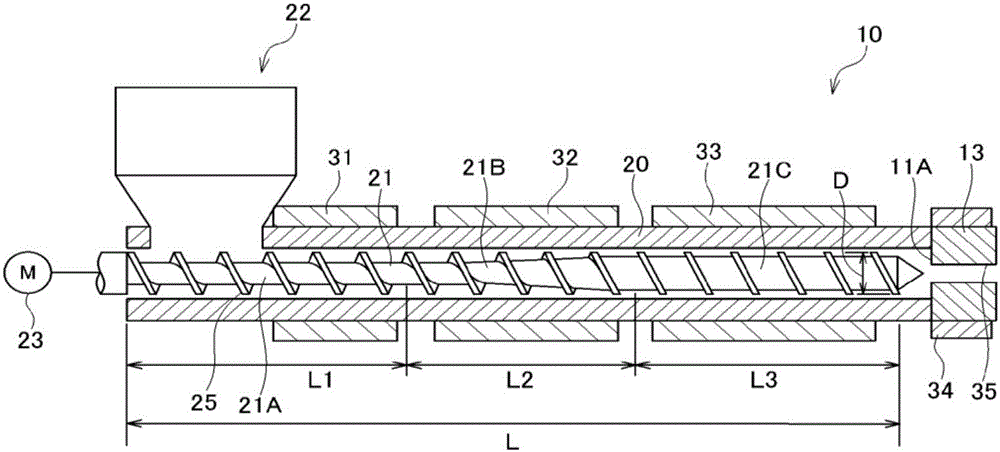
以下,使用圖1對于利用擠出成型機成型聚酰胺成型體的本制造方法的一個實施方式進行說明。圖1為示出在本實施方式中所使用的擠出成型機的概略圖。
如圖1所示,擠出成型機10為單螺桿擠出成型機,具備料筒20、設置于料筒20內部的單螺桿21、口模(未圖示)和用于在料筒20的前端安裝口模的轉接頭13。轉接頭13為用于從料筒20將原料送到口模的連通部。此外,擠出成型機10進一步具備安裝于料筒20的上游側的端部的加料斗22和使螺桿21旋轉的螺桿驅動裝置23。
螺桿21為在螺桿軸的側面形成螺旋狀的螺紋切削部25的構件。螺紋切削部25的外徑D與料筒的內周面的內徑相比稍小、并且設置為固定。
螺桿21自其基端部向前端部具備供給部21A、連接于供給部21A的壓縮部21B和連接于壓縮部21B的計量部21C。供給部21A是指從螺桿的螺紋切削部25所實施的螺紋切削開始的溝的深度(也稱為高度或螺紋深度)為固定的范圍。壓縮部21B是指溝深度緩慢變淺的范圍。計量部21C是指螺桿前端部的溝深度淺至一定的范圍。
對于供給部、壓縮部、計量部的長度L1、L2、L3,將它們的總計設為1時,通常分別為0.30~0.55、0.10~0.35、0.10~0.40左右。此外,供給部的長度L1優選0.40~0.55、壓縮部的長度L2優選0.10~0.30、計量部的長度L3優選0.10~0.40,供給部的長度L1更優選0.45~0.55、壓縮部的長度L2更優選0.10~0.20、計量部的長度L3更優選0.20~0.40。如此,通過使供給部比較長,從而可以在壓縮前以比較長的時間對聚酰胺粒料施加預熱。因此,即便為熔點比較高的含間苯二甲基的聚酰胺,也可以容易地使聚酰胺粒料增塑化。
單螺桿擠出成型機優選所謂高壓縮螺桿型的單螺桿擠出成型機,壓縮比(C/R)優選為較高。具體而言,料筒20中的壓縮比(C/R)優選為2.0~4.0,更優選為2.3~3.5。需要說明的是,壓縮比(C/R)是指,以供給部21A的1螺距部分的樹脂容量(體積)和計量部21C的1螺距部分的樹脂容量(體積)的比表示。螺桿的壓縮比處于上述的范圍,從而以高壓縮對聚酰胺粒料進行剪斷,粒料的增塑化以及混煉變得容易。此外,即便如此以高壓縮進行增塑化,如上所述,聚酰胺粒料的表皮部分的球晶密度高,因此也不易產生粒料彼此緊密附著的不良。
本發明的螺桿有效長度L與螺桿直徑D的比(=L/D)優選為20~35、更優選為23~30。比(L/D)若為20以上,則可以充分地將聚酰胺粒料增塑化,進而進行熔融而混煉。此外,若為35以下,則可以將驅動螺桿的馬達容量抑制在不成為經濟上的問題的程度。
需要說明的是,螺桿有效長度L是指螺桿的螺紋切削部(從螺紋切削開始至螺紋切削最末端)的長度,等于長度L1、L2、L3的總和。
螺桿直徑D若可以混煉本發明的聚酰胺粒料則沒有特別限定,通常為20~120mm左右,優選使用20~90mm左右的螺桿直徑。
螺桿形狀沒有特別限定,從噴出量的觀點出發,優選為全螺紋螺桿,可以為單螺紋也可以為雙螺紋。
在料筒20中自螺桿21的基端部向螺桿前端部,例如,按順序設置加熱器31、32、33。加熱器31、32、33分別對與螺桿的供給部21A、壓縮部21B、計量部21C對應的部分的料筒進行加熱,調整其溫度(料筒溫度)。需要說明的是,加熱器31、32、33可以分別為對與供給部21A、壓縮部21B、計量部21C對應的料筒的整體加熱到規定的溫度,但也可以分別為對與供給部21A、壓縮部21B、計量部21C對應的料筒的大半部分(例如80%以上的部分)加熱到規定的溫度。此外,優選以卷取在轉接頭13上的方式設置加熱器34,轉接頭13(連通部)利用加熱器34而加熱到規定的溫度。進而,優選對未圖示的口模也設置加熱器,對于口模利用其加熱器而加熱到規定的溫度。需要說明的是,在以下,對于分別利用加熱器31、32、33而加熱的螺桿21,將各自的溫度設為溫度C1、C2、C3。此外,將利用設置于轉接頭13的加熱器34而加熱的轉接頭的溫度設為溫度H,并且將利用設置于口模的加熱器而加熱的口模溫度設為溫度D。
料筒20沒有特別限定,利用加熱器31,與供給部21A對應的部分(即、溫度C1)優選加熱至低于聚酰胺粒料的熔點,更優選加熱至低于聚酰胺粒料的熔點并且(聚酰胺粒料的熔點-40℃)以上。此外,與供給部21A相比在下游側的溫度(即、料筒的溫度C2、C3、連通部的溫度H、口模的溫度D)優選調節到聚酰胺粒料的熔點以上,更優選調節到聚酰胺粒料的熔點以上并且(聚酰胺粒料的熔點+40℃)以下。通過進行以上的溫度設定,從而抑制對于聚酰胺粒料的熱歷程、并且在供給部21A中對粒料賦予足夠的余熱,并且在壓縮部21B以及計量部21C中對粒料進行熔融,可以自口模穩定地擠出熔融的聚酰胺。
對于擠出成型機10,利用螺桿21邊使自加料斗22投入到螺桿21的基端側的供給原料移動邊進行增塑化進一步熔融并混煉,將混煉了的原料自處于料筒前端的排出口11A排出。供給原料可以由上述的聚酰胺粒料單體形成,也可以在聚酰胺粒料中混合其它樹脂材料、添加劑等。其它的樹脂材料可以以粒料的形態投入、或也可以以粉體等其它的形態投入。添加劑可以預先配合到由其它的樹脂原料形成的粒料中,也可以以粉體等形態從加料斗22投入。
自螺桿21的排出口11A排出的供給原料介由轉接頭13內的導入路35導入到口模,自口模擠出,成型為規定形狀的成型體。
口模可以使用公知的口模,根據成型體的形狀而適宜選擇。作為具體的口模的例子,可以列舉出平面配合模、直角機頭模(crosshead die)、T口模等。
此外,聚酰胺粒料與其它的樹脂材料一同成型為多層結構體等時,擠出成型機可以具備用于混煉聚酰胺粒料的料筒、以及用于混煉其它樹脂材料的料筒。這些多個料筒為了形成多層結構體而與具備多個流路的口模(例如,多層口模)連接,在該口模中由從各料筒所供給的聚酰胺、其它的樹脂材料成型為多層結構體。
需要說明的是,聚酰胺成型體的制造方法并不限于上述的方法,例如,可以使用具備具有單螺桿的料筒的注射成型機。該情況下,在料筒的前端介由連通部(例如,噴嘴),安裝模腔代替口模。在注射成型機中,在料筒內部所混煉的原料介由連通部而供給到模腔,在模腔中,加工為期望形狀的聚酰胺成型體。對于其它結構,與在擠出成型機中所制造的情況同樣,因此省略其說明。
此外,由注射成型而得到的聚酰胺成型體也可以制成上述的各種成型體,例如,也可以與其它的樹脂材料一同構成多層結構體。作為通過注射成型而成型的多層結構體,可以列舉出代表性的、層疊有由聚酰胺粒料成型的聚酰胺層與由聚對苯二甲酸乙二醇酯等聚酯樹脂成型的聚酯層的多層容器、多層瓶或它們的預成型坯,例如,可以列舉出從內側起包含聚酯層/聚酰胺層/聚酯層的3層結構的容器、瓶、或者它們的預成型坯;從內側起包含聚酯層/聚酰胺層/聚酯層/聚酰胺層/聚酯層的5層結構的容器、瓶、或者它們的預成型坯等。需要說明的是,通過注射成型而成型多層結構體的情況,通常在模腔上連接多個料筒。
如以上所述,本發明中,增大在表皮部分的球晶密度并且將第1聚酰胺粒料的截面積設為一定的大小,因此例如即便通過具有單螺桿的料筒以高壓縮力進行增塑化、混煉,也可以防止粒料緊密附著、或者纏繞螺桿。因此,無論成型機的種類,可以以高加工穩定性成型成型體。
<第2聚酰胺粒料>
以上說明的第1聚酰胺粒料為在表皮部分觀察到規定的結晶狀態的聚酰胺粒料,在以下說明的第2聚酰胺粒料的特征在于表皮部分以及核部分的指針下降溫度。需要說明的是,如后所述,第2聚酰胺粒料通常處于非晶狀態,通過使第2聚酰胺粒料固相聚合,從而可以得到第1聚酰胺粒料,但第1聚酰胺粒料的制造方法并不限定于該方法。
以下,對于第2聚酰胺粒料詳細地說明。
第2聚酰胺粒料由聚酰胺形成,所述聚酰胺包含二胺單元和二羧酸單元,二胺單元的50摩爾%以上源自間苯二甲胺、二羧酸單元的70摩爾%以上源自己二酸。
第2聚酰胺粒料通常處于非晶狀態。此外,處于非晶狀態的聚酰胺例如通過進行固相聚合,從而進行結晶化,通過固相聚合而發生的結晶化度通常大于25%。
[二胺單元]
在第2聚酰胺粒料中,聚酰胺中的二胺單元包含50摩爾%以上的源自間苯二甲胺的構成單元、優選含有70~100摩爾%、更優選含有80~100摩爾%、進一步優選含有90~100摩爾%。第2聚酰胺粒料中,源自間苯二甲胺的構成單元不足50摩爾%時,難以提高由聚酰胺粒料所得到的成型品的阻隔性能,此外,難以實現期望的強度、彈性模量等各種物性。
在聚酰胺中,作為除間苯二甲胺以外的二胺,可以例示出四亞甲基二胺、五亞甲基二胺、2-甲基-1,5-戊二胺、六亞甲基二胺、七亞甲基二胺、八亞甲基二胺、九亞甲基二胺、十亞甲基二胺、十二亞甲基二胺、2,2,4-三甲基六亞甲基二胺或2,4,4-三甲基六亞甲基二胺等脂肪族二胺;1,3-雙(氨基甲基)環己烷或1,4-雙(氨基甲基)環己烷、1,3-二氨基環己烷或1,4-二氨基環己烷、雙(4-氨基環己基)甲烷、2,2-雙(4-氨基環己基)丙烷、雙(氨基甲基)萘烷、雙(氨基甲基)三環癸烷等脂環族二胺;對苯二甲胺、雙(4-氨基苯基)醚、對苯二胺、雙(氨基甲基)萘等具有芳香環的二胺類等,但不限于它們。
作為除間苯二甲胺以外的二胺,它們之中,優選使用對苯二甲胺。使用對苯二甲胺時,聚酰胺中的二胺單元包含50摩爾%以下源自對苯二甲胺的構成單元,優選含有35摩爾%以下、進一步優選含有10摩爾%以下。
[二羧酸單元]
第2聚酰胺粒料中的聚酰胺中的二羧酸單元包含70摩爾%以上源自己二酸的構成單元、優選含有75~100摩爾%、更優選含有90~100摩爾%。源自己二酸的構成單元不足70摩爾%時,難以提高聚酰胺的阻隔性能,此外,難以實現期望的強度、彈性模量等各種物性。
聚酰胺中的二羧酸單元可以僅包含源自己二酸的構成單元,也可以含有源自除己二酸以外的二羧酸的構成單元。
在聚酰胺中,作為除己二酸以外的二羧酸,可以例示出琥珀酸、戊二酸、庚二酸、辛二酸、壬二酸、癸二酸、1,9-十一烷二酸、1,10-癸烷二羧酸等碳數4~20的α,ω-直鏈脂肪族二羧酸;1,3-環己烷二羧酸、1,4-環己烷二羧酸等脂環族二羧酸;對苯二甲酸、間苯二甲酸、2,6-萘二羧酸等芳香族二羧酸,但不限于它們。它們可以單獨使用或組合使用2種以上。它們之中,對于間苯二甲酸,可以容易地得到阻隔性能優異的聚酰胺而不損害縮聚反應,故優選。
在第2聚酰胺粒料中,作為聚酰胺,最優選全部的二胺單元由源自間苯二甲胺的構成單元組成、全部的二羧酸單元由源自己二酸的構成單元組成的聚己二酰基間苯二甲胺。
[指針下降溫度]
對于第2聚酰胺粒料,粒料的表皮部分的指針下降溫度成為78~92℃,進而,比粒料的核部分的指針下降溫度高,其溫度差為0.1~2.5℃。
即、對于第2聚酰胺粒料,縮小表皮部分和核部分的指針下降溫度的溫度差并且使表皮部分的指針下降溫度高于通常的尼龍MXD6。由此,推斷對于第2聚酰胺粒料,縮小核部分與表皮部分的熱性質之差,并且被表皮部分保護、可抑制氧吸收,可以防止色相的惡化。因此,推定例如通過在減壓、加熱下對第2聚酰胺粒料進行固相聚合而結晶化了的聚酰胺粒料、或加熱成型了的成型品通過降低在粒料內部吸附的氧從而防止氧化劣化,可以良好地保持色相。
需要說明的是,在第2聚酰胺粒料中,表皮部分是指在與粒料的軸向垂直的截面中從粒料外周至粒料直徑的2%的部分,核部分是指距粒料中心為粒料半徑的70%以內的部分。需要說明的是,粒料直徑是指在粒料的上述截面中最長的直徑,粒料半徑是指粒料直徑的1/2的長度。
需要說明的是,指針下降溫度是指如下值:如圖2所示,對于通過粘接劑而固定在支持體110上的粒料112利用切片機與軸向(與粒料化前的股線的流動方向相等)垂直地平滑地切取,對于露出的粒料112的截面的平滑面112A,使用熱探針由局部熱分析進行測定而得到的值。指針下降溫度具體而言是指如下溫度:如圖3所示,與試樣112的平滑面112A接觸的探針114由于試樣112加熱膨脹而上升,由于軟化轉而下降的溫度。需要說明的是,指針下降溫度的詳細測定方法如后所述。
對于第2聚酰胺粒料,表皮部分的指針下降溫度不足78℃時,擔心粒料未被表皮部分充分地保護,不能使由第2聚酰胺粒料而得到的成型體以及固相聚合粒料的色相足夠良好。此外,在上述的聚酰胺的組成中,難以將指針下降溫度提高到92℃以上。從以上的觀點出發,表皮部分的指針下降溫度優選為80~91℃、更優選為83~90℃。
推測指針下降溫度為78℃以上的聚酰胺粒料的表皮部分的形態與以往的情況不同,成為特異的狀態,具有該特異形態的表皮部分保護粒料。具體而言,利用切片機垂直軸向地切取粒料,在規定條件下對使粒料內部的截面露出而得到的樣品的露出截面進行離子銑削處理時,基于形態而形成凹凸,表皮部分的凹凸與核部分的凹凸相比更密實地形成。如此,特異的形態即便成為結晶狀態也可被維持,例如對第2聚酰胺粒料進行固相聚合從而結晶化了的粒料(即,第1聚酰胺粒料)中,表皮部分的球晶密度也大于核部分的球晶密度。
此外,由于表皮部分的上述特征,難以切削第2聚酰胺粒料的表面。因此,在粒料移送中不易形成粒料表面被切削并附著于配管的蛇皮(絲絮、フロス)等,可以降低制品上的源自蛇皮的污染,因此在工業上是有利的。
在第2聚酰胺粒料中,核部分的指針下降溫度與表皮部分的指針下降溫度的溫度差如上所述,成為0.1~2.5℃、更優選為0.2~2℃、進一步優選為0.2~1.5℃。溫度差大于2.5℃時,擔心核部分與表皮部分的熱性質之差變大,成型品的物性等變得不穩定。此外,上述溫度差不足0.1℃的粒料難以制造。
[粒料的染色度]
對于第2聚酰胺粒料,觀察利用0.1mol/L碘-碘化鉀溶液染色了的粒料時,優選染色成G≤90(sRGB值)的粒料的比例(染色度)不足50%。如上所述,第2聚酰胺粒料的表皮部分的形態與以往的情況不同,為恰如保護層那樣的狀態,其保護效果可以通過染色度來規定。染色度越小,表示粒料表面的保護效果越大。
第2聚酰胺粒料的染色度不足50%,從而表皮部分作為保護層而充分地起到作用,可以適宜地防粒料的氧吸收。因此,可以使由第2聚酰胺粒料而得到的成型品、通過固相聚合等而結晶化了的粒料等的色相更良好。此外,從這些觀點出發,染色度更優選為20%以下、進一步優選為5%以下。需要說明的是,染色度的具體的測定方法如后所述。
作為聚酰胺的聚合度指標存在多個,通常使用相對粘度。對于第2聚酰胺粒料,其相對粘度優選為1.8~2.4。相對粘度在這樣的范圍的聚酰胺可以通過熔融縮聚法而容易地制造。此外,即便將聚酰胺粒料直接用于成型品的制造,其機械強度以及成型性也比較良好。從這些觀點出發,相對粘度更優選1.9~2.3。
此外,第2聚酰胺粒料優選滿足以下的式(1)的條件。
-110μeq/g≤([COOH]-[NH2])≤110μeq/g (1)
(需要說明的是,式(1)中,[COOH]表示聚酰胺的末端羧基濃度(μeq/g)、[NH2]表示聚酰胺的末端氨基濃度(μeq/g)。)
如此,末端羧基濃度與末端氨基濃度之差小時,耐熱性良好、不易產生色劣化。此外,為了進一步抑制色劣化,([COOH]-[NH2])更優選為-80~80μeq/g。
第2聚酰胺粒料通常處于非晶狀態。另一方面,測定熔點時,具有熔點峰,具有結晶性。因此,對第2聚酰胺粒料進行固相聚合時,成為結晶狀態。第2聚酰胺粒料具有1個熔點峰,其熔點沒有特別限定,優選為200~270℃,更優選為210~260℃。
第2聚酰胺粒料沒有特別限定,通常沿其股線方向(軸向)的長度為1.0~4.0mm左右、優選為2.0~3.5mm。此外,聚酰胺粒料的粒料直徑通常為1.0~4.0mm左右、優選為2.0~3.5mm。粒料的形狀沒有特別限定,通常,如后述那樣以橫截股線的方式切斷而成的形狀,優選為圓柱狀、橢圓柱狀。
<第2聚酰胺粒料的制造方法>
接著,對于第2聚酰胺粒料的制造方法進行說明。第2聚酰胺粒料的制造方法若為可以得到第2聚酰胺粒料的方法則沒有特別限定,例如,可以列舉出以下的方法。
本發明的一實施方式中的第2聚酰胺粒料的制造方法具備如下工序:將上述二胺與二羧酸縮聚而得到的處于熔融狀態的聚酰胺以線料狀送出的工序;和,將以線料狀送出的聚酰胺水冷并切斷而粒料化,之后對粒料化了的聚酰胺進一步水冷4秒以上的工序。
以往,在MXD6尼龍的制造中,熔融縮聚后的溫度并不那樣高的方面、進而裝置的制約出發,通常盡可能縮短水冷時間。另外,已知有將通過縮聚得到的聚酰胺抽出為線料狀,股線直接進行水冷的方法。
與之相對,本制造方法中,以小塊化為粒料狀的狀態對聚酰胺進行水冷,并且,通過將該水冷時間如上所述延長至4秒以上,從而使粒料表面急劇降溫,由此,使表皮部分的形態如上所述那樣成為特異的形態,提高表皮部分的指針下降溫度,并且降低染色度。
另外,聚酰胺粒料的核部分的冷卻速度雖然稍慢于表皮部分,但核部分依然被驟冷,雖然核部分與表皮部分相比在形態上存在少許差別,但指針下降溫度也成為與表皮部分接近的值。因此,雖然核部分的指針下降溫度低于表皮部分的指針下降溫度,但其溫度差如上所述變小。
需要說明的是,在本制造方法中,二胺與二羧酸的縮聚優選通過熔融縮聚法來進行。
對于第2聚酰胺粒料,更詳細而言,省略固相聚合,除此以外可以以與制造第1聚酰胺粒料的方法同樣的方法來制造。即、在上述詳細地說明的第1聚酰胺粒料的制造方法中,至制造縮聚聚酰胺粒料的工序為止來實施,從而可以制造第2聚酰胺粒料。
對于由上述制造方法制造的固相聚合后的第1聚酰胺粒料,表皮部分以及核部分的形態成為特異的形態,因此,表皮部分以及核部分的球晶密度成為特異情況。
同樣地,對于第2聚酰胺粒料,通過由上述制造方法制造,即便在固相聚合前,表皮部分以及核部分的形態也成為特異的形態。因此,在包含二胺單元的50摩爾%以上源自間苯二甲胺、二羧酸單元的70摩爾%以上源自己二酸的聚酰胺的第2聚酰胺粒料中,表皮以及核部分的指針下降溫度如上所述處于一定的范圍。這樣的第2聚酰胺粒料進行固相聚合、或進行成型加工時,可以使該固相聚合后的聚酰胺粒料、成型體的色相良好。
本發明的第2聚酰胺粒料可以根據需要混合其它的任意成分并且通過注射成型、吹塑成型、擠出成型、壓縮成型、拉伸、真空成型等公知的成型方法,成型為期望形狀的成型體。需要說明的是,作為成型體的具體例子,如上所述,可以列舉出與由第1聚酰胺粒料成型的成型體同樣的成型體。
此外,本發明的第2聚酰胺粒料進一步進行固相聚合,從而可以制成高分子量化以及結晶化了的粒料。高分子量化以及結晶化了的粒料也可以與上述同樣地以各種成型方法而成型為各種成型品。
如以上那樣,對于第2聚酰胺粒料,減小表皮部分與核部分的指針下降溫度的溫度差、并且提高表皮部分的指針下降溫度,從而可以減小核部分與表皮部分的熱性質之差,并且將表皮部分的形態制成特異的形態。因此,提供由粒料表面保護粒料即便增加熱歷程色相也不惡化的、非晶狀態的聚酰胺粒料。因此,可以使由本發明的第2聚酰胺粒料得到的固相聚合粒料、成型品的色相良好。
需要說明的是,分別在本發明的第1以及第2聚酰胺粒料中,聚酰胺除二胺單元以及二羧酸單元以外,可以在不損害性能的范圍內含有源自ε-己內酰胺、ω-月桂內酰胺、ω-庚內酰胺等內酰胺類、6-氨基已酸、7-氨基庚酸、11-氨基十一烷酸、12-氨基十二烷酸、9-氨基壬酸、對氨基甲基苯甲酸等的氨基酸等其他單體成分的單元。其中,二胺單元以及二羧酸單元在聚酰胺中為主成分,它們的總量沒有特別限定,通常為總構成單元的80摩爾%以上左右、優選為90摩爾%以上。
此外,本發明的第1以及第2聚酰胺粒料可以分別在不損害其性能的范圍內適宜地含有除聚酰胺以外的其它任意成分。其中,聚酰胺為粒料中的主成分,其含量相對于粒料整體沒有特別限定,通常為80質量%以上左右、優選為90質量%以上。
實施例
以下,利用實施例更詳細地說明本發明,但本發明并不限于它們。需要說明的是,本實施例中,各種測定通過以下的方法來進行。此外,以下示出的壓力只要沒有特別限定則為絕對壓力。
(1)球晶密度
用切片機垂直軸向地切取第1聚酰胺粒料,將使粒料的軸向的中心的截面為平滑面并露出的塊狀的試樣投入到10wt%磷·鎢酸水溶液中,在80℃下進行8小時染色。之后,使用離子銑削裝置(商品名:IM4000、Hitachi High-Technologies Corporation制),在加速電壓2.5kV、放電電壓1.5kV、加工時間6分鐘、照射角度30度、偏心量1.5mm的條件下對平滑面照射離子束,在試樣表面形成源自形態的損傷紋路,使用掃描電子顯微鏡(商品名.SU8020、Hitachi High-Technologies Corporation制)以倍率1000倍進行表面觀察。根據所得到的圖像判斷源自結晶的損傷紋路,各重復3次計數表皮部分、核部分的任意的50μm見方的球晶的數目,測定表皮部分、核部分各自的球晶的密度。需要說明的是,在以上的測定中,將外周被由損傷紋路所形成的條紋圍了70%以上的部分計為1個球晶。
(2)[COOH]-[NH2]
精確稱量聚酰胺粒料0.3~0.5g,在苯甲醇30ml中在氮氣氣流下、160~180℃下進行攪拌溶解。完全溶解之后,在氮氣氣流下冷卻至規定溫度,邊攪拌邊加入10ml甲醇,以0.01摩爾/l氫氧化鈉水溶液進行中和滴定,測定末端羧基濃度([COOH])。需要說明的是,對于第1聚酰胺粒料(即、結晶化了的聚酰胺粒料),上述規定溫度設為80℃,并且對于第2聚酰胺粒料(即、縮聚聚酰胺粒料)將上述規定溫度設為50℃。
精確稱量聚酰胺粒料0.3~0.5g,在室溫下在苯酚/乙醇混合溶液(混合體積比4:1)30ml中進行攪拌溶解。完全溶解之后,邊進行攪拌邊用0.01摩爾/l鹽酸水溶液進行中和滴定,求出末端氨基濃度([NH2])。由測定的這些末端羧基濃度以及末端氨基濃度算出[COOH]-[NH2]。
(3)相對粘度
精確稱量0.2g試樣,在20~30℃下在96質量%硫酸20ml中進行攪拌,使其完全溶解,制備溶液。之后,快速地在坎農芬斯克型粘度計中取該溶液5ml,在25℃的恒溫槽中進行10分鐘放置后,測定下落時間(t)。此外,也同樣測定96質量%硫酸其自身的下落時間(t0)。根據t以及t0利用下式算出相對粘度。
相對粘度=t/t0
(4)熔點(Tm)
使用差示掃描量熱儀(株式會社島津制作所制、商品名:DSC-60),以升溫速度10℃/分鐘在氮氣氣流下進行DSC測定(差示掃描量熱測定),求出熔點(Tm)。
(5)黃色指數YI
使用日本電色工業株式會社制造的ZE-2000,基于JIS K7373在粒料的狀態下進行測定。
(6)結晶化度
利用差示掃描量熱儀(株式會社島津制作所制、商品名:DSC-60),在氮氣氣氛、升溫速度10℃/分鐘的條件下進行測定,根據測定熔解熱量ΔH與聚合物的完全結晶體的熔解熱量ΔHm的比率利用以下的式算出。
結晶化度=ΔH/ΔHm×100[%]
(7)指針下降溫度
作為測定裝置,使用Anasys Instruments Corporation制造的VESTA Nano-TA,作為探針,使用探針前端直徑為30nm的探針,利用熱探針式nano-TA如以下所述進行指針下降溫度的測定。
將第2聚酰胺粒料(縮聚聚酰胺粒料)固定于環氧樹脂塊,利用切片機切取,在粒料的軸向的中心,將相對于軸向(與粒料化前的股線的流動方向相同)垂直的截面設為平滑面而使其露出,制成測定試樣。測定以100℃/分鐘升溫,將使與試樣的平滑面接觸的探針的位移從上升轉向下降的溫度設為指針下降溫度。核部分的指針下降溫度的測定在距粒料的平滑面的中心為粒料半徑70%以內的范圍、表皮部分的指針下降溫度的測定在距粒料外周為粒料直徑的2%以內的范圍,任意地進行6個點,將其的算數平均值作為各自的指針下降溫度。
(8)粒料的染色度
將第2聚酰胺粒料(縮聚聚酰胺粒料)5g浸漬于0.1mol/L碘-碘化鉀溶液(由碘:0.5g、碘化鉀:1.0g、水:100ml制備)中,在23℃下放置12小時,對聚酰胺粒料進行染色,用水洗凈染色了的聚酰胺粒料之后,在室溫(23℃)下使其干燥。在白基準板(以XYZ顏色系統計X=90、Y=94、Z=111)之上排列所得到的染色粒料,在LED光源(CCS制造的PTU-3024)之下,用CCD照相機(Sony制造的XCL-U1000C)拍攝粒料。此時,以粒料不反射光源,進而,拍攝白基準板單體時的sRGB值為140≤R≤150、175≤G≤190、135≤B≤150的方式調節照相機的靈敏度。由以上的條件拍攝染色粒料,求出成為G≤90的粒料的比例。
(9)對于第2聚酰胺粒料的離子銑削觀察條件
用切片機垂直軸向地切取第2聚酰胺粒料(縮聚聚酰胺粒料),將使粒料內部的截面為平滑面并露出的塊狀的試樣投入到10wt%磷·鎢酸水溶液中,在80℃下進行8小時染色。之后,使用離子銑削裝置(商品名:IM4000、Hitachi High-Technologies Corporation制),在加速電壓2.5kV、放電電壓1.5kV、加工時間6分鐘、照射角度30度、偏心量1.5mm的條件下對平滑面照射離子束,在試樣表面形成源自形態的損傷紋路,使用掃描電子顯微鏡(商品名.SU8020、Hitachi High-Technologies Corporation制)以倍率1000倍進行表面觀察。
實施例1
使用具備可調溫的流通油的分凝器、全凝器、氮氣導入管、用流通油的夾套覆蓋反應槽整面、用于二胺滴加的罐以及泵的、500升不銹鋼制間歇式反應裝置,如下所述合成聚酰胺。
投入己二酸(純度99.85wt%)150.0kg(1024.9mol),進行充分氮氣置換之后,在壓力0.1MPa下邊攪拌邊將己二酸加熱至190℃。到達溫度后,添加次磷酸鈉一水合物8.6g(以聚酰胺中的磷原子濃度計為5ppm),邊將反應裝置內的壓力維持在0.1MPa邊經110分鐘滴加間苯二甲胺(純度99.99wt%)138.8kg(1018.7mol)。以二胺的滴加終止時的溫度為240℃的方式調整加熱,將分凝器出口側蒸汽溫度控制在101~104℃,餾出的蒸汽通過全凝器而凝縮,放出到體系外。二胺滴加終止后,邊攪拌邊以壓力0.1MPa保持20分鐘之后,進而減壓至80kPa,進一步保持20分鐘攪拌。自二胺滴加終止至減壓終止為止,將反應液溫升溫至256℃。
反應終止后,停止攪拌,用氮氣將反應裝置內加壓至0.30MPa(表壓力),從裝置底部的線料模頭(口模直徑:8mm)以口模閥開度60%在256℃下將聚合物抽出為線料狀。使抽出的股線在水溫25℃的水槽中著水,自著水開始0.8秒后,在水槽中利用水道(water slide、ウォータースライダー)型的造粒機進行切斷而粒料化。之后,將粒料化了的聚酰胺邊在水槽中持續冷卻邊輸送到水槽中,自切斷開始在5.9秒后使其離水,得到非晶狀態的粒料化了的聚酰胺。所得到的粒料的長度3mm、粒料直徑3mm、冷卻終止后的粒料的溫度為40℃。此外,造粒機中的刀具的牽引速度為200m/分鐘。
需要說明的是,自水槽取出的粒料立即暫時保管于保管容器中,在該保管容器內的大量粒料中插入鎧裝熱電偶并測定的溫度,將其作為水冷終止后的粒料溫度。水冷終止后的粒料溫度的測定方法在以下的實施例、比較例中也是同樣的。
所得到的非晶狀態的粒料化了的聚酰胺(縮聚聚酰胺粒料)的結晶化度18%、YI=-3、([COOH]-[NH2])=46μeq/g、相對粘度2.1、熔點(Tm)239℃、表皮部分的指針下降溫度85.5℃、核部分的指針下降溫度84.8℃,用0.1mol/L碘-碘化鉀溶液進行染色時,染色為G≤90的粒料的比例(染色度)為1%。
此外,對于所得到的縮聚聚酰胺粒料進行離子銑削后進行觀察時,從粒料外周表面至約30μm的位置為止密集地形成凹凸,并且比該位置靠中心側(核部分)稀疏地形成凹凸,聚酰胺粒料的表皮部分的形態為特異的狀態。
接著,在常溫(23℃)空氣中將縮聚聚酰胺粒料放置6小時進行空氣冷卻之后,實施固相聚合。固相聚合通過如下方式進行:在250L的不銹鋼制轉鼓中投入150kg非晶狀態的粒料化了的聚酰胺,將原料投入后的轉鼓內保持在1.0kPa以下,并且以3小時升溫至130℃,進而,需要3小時升溫至195℃之后進行冷卻。在表1中示出由固相聚合而得到的聚酰胺粒料(第1聚酰胺粒料)的評價結果。
在圖4、5中示出根據上述的球晶密度的測定方法,對聚酰胺粒料的薄片試樣照射離子束之后的觀察圖像。圖4為表皮部分的照片、圖5為核部分的照片。
實施例2
使用具備可調溫的流通油的分凝器、全凝器、氮氣導入管、反應槽整面被流通油的夾套覆蓋、用于二胺滴加的罐以及泵的、500升不銹鋼制間歇式反應裝置,如下所述合成聚酰胺。
投入己二酸(純度99.85wt%)150.0kg(1024.9mol),進行充分氮氣置換之后,在壓力0.4MPa下邊攪拌邊將己二酸加熱至190℃。溫度到達后,添加次磷酸鈉一水合物8.6g,邊將反應裝置內的壓力維持在0.4MPa邊經過110分鐘滴加間苯二甲胺(純度99.99wt%)138.8kg(1018.8mol)。以二胺的滴加終止時的溫度為240℃分鐘方式調整加熱,將分凝器出口側蒸汽溫度控制在101~104℃,餾出的蒸汽通過全凝器而凝縮,放出到體系外。在二胺滴加終止后,邊進行攪拌邊在壓力0.4MPa下保持20分鐘,然后,以0.01MPa/分鐘的速度經過30分鐘降壓至常壓,進而,減壓至80kPa,進而保持20分鐘攪拌。自二胺滴加終止至減壓終止為止,將反應液溫升溫至256℃。
反應終止后,停止攪拌,用氮氣將反應裝置內加壓至0.30MPa(表壓力),將聚合物從裝置底部的線料模頭(口模直徑:8mm)以口模閥開度60%在256℃下抽出為線料狀。使抽出的股線在水溫25℃的水槽中著水,自著水開始0.7秒后,在水槽中利用水道型的造粒機進行切斷而粒料化。之后,粒料化了的聚酰胺邊在水槽中持續冷卻邊在水槽中輸送,自切斷開始5.0秒后使其離水,得到非晶狀態的粒料化了的聚酰胺。所得到的粒料的長度3mm、粒料直徑3mm、水冷終止后的粒料的溫度為45℃。此外,造粒機中的刀具的牽引速度為200m/分鐘。
所得到的非晶狀態的粒料化了的聚酰胺(縮聚聚酰胺粒料)的結晶化度19%、YI=-2、([COOH]-[NH2])=49μeq/g、相對粘度2.1、熔點(Tm)239℃、表皮部分的指針下降始溫度為86.3℃、核部分的指針下降溫度為85.2℃,用0.1mol/L碘-碘化鉀溶液進行染色時,染色為G≤90的粒料的比例(染色度)為15%。
此外,對于所得到的縮聚聚酰胺粒料進行離子銑削觀察時,自粒料表面至30μm的位置密集地形成凹凸、并且比該位置靠中心側稀疏地形成凹凸,聚酰胺粒料的表皮部分的形態成為特異的狀態。
在常溫(23℃)空氣中將所得到的縮聚聚酰胺粒料放置6小時,進行空氣冷卻之后,在與實施例1同樣的條件下實施固相聚合。在表1中示出由固相聚合而得到的聚酰胺粒料的評價結果。
實施例3
使用具備可調溫的流通油的分凝器、全凝器、氮氣導入管、用流通油的夾套覆蓋反應槽整面、用于二胺滴加的罐以及泵的、500升不銹鋼制間歇式反應裝置,如下所述合成聚酰胺。
投入己二酸(純度99.85wt%)150.0kg(1024.9mol),進行充分氮氣置換之后,在壓力0.4MPa下邊攪拌邊將己二酸加熱至190℃。溫度到達后,添加次磷酸鈉一水合物8.6g,邊將反應裝置內的壓力維持在0.4MPa邊經過110分鐘滴加間苯二甲胺/對苯二甲胺混合物(摩爾比:80/20)(純度99.99wt%)138.8kg(1018.7mol)。以二胺的滴加終止時的溫度為258℃的方式調整加熱,將分凝器出口側蒸汽溫度控制在143~147℃,餾出的蒸汽通過全凝器而凝縮,放出到體系外。在二胺滴加終止后,邊進行攪拌邊在壓力0.4MPa下保持20分鐘,然后,以0.01MPa/分鐘的速度經過30分鐘降壓至常壓,進而減壓至80kPa,進而保持20分鐘攪拌。自二胺滴加終止至減壓終止為止,將反應液溫升溫至260℃。
反應終止后,停止攪拌,用氮氣將反應裝置內加壓至0.30MPa(表壓力),將聚合物從裝置底部的線料模頭(口模直徑:8mm)以口模閥開度60%在260℃下抽出為線料狀。使抽出的股線在水溫25℃的水槽中著水,自著水開始0.8秒后,在水槽中利用水道型的造粒機進行切斷而粒料化。之后,將粒料化了的聚酰胺邊在水槽中持續冷卻邊在水槽中輸送,自切斷開始5.9秒后使其離水,得到非晶狀態的粒料化了的聚酰胺。所得到的粒料的長度3mm、粒料直徑3m、水冷終止后的粒料的溫度為50℃。此外,造粒機中的刀具的牽引速度為200m/分鐘。
所得到的非晶狀態的粒料化了的聚酰胺(縮聚聚酰胺粒料)的結晶化度20%、YI=-1、([COOH]-[NH2])=43μeq/g、相對粘度2.1、熔點(Tm)253℃、表皮部分的指針下降溫度87.2℃、核部分的指針下降溫度85.8℃,用0.1mol/L碘-碘化鉀溶液進行染色時,染色為G≤90的粒料的比例(染色度)為45%。
此外,對于所得到的縮聚聚酰胺粒料進行離子銑削觀察時,自粒料表面至30μm的位置密集地形成凹凸、并且比該位置靠中心側稀疏地形成凹凸,聚酰胺粒料的表皮部分的形態成為特異的狀態。
將所得到的縮聚聚酰胺粒料在常溫(23℃)空氣中放置6小時,進行冷卻之后,在與實施例1同樣的條件下實施固相聚合。在表1中示出由固相聚合而得到的聚酰胺粒料的評價結果。
比較例1
在與實施例1同樣的條件下使其反應而合成聚酰胺。
反應終止后,停止攪拌,用氮氣將反應裝置內加壓至0.30MPa(表壓力),將聚合物從裝置底部的線料模頭(口模直徑:8mm)以口模閥開度60%在256℃下抽出為線料狀。使抽出的股線在水溫25℃的水槽中著水,自著水開始0.8秒后,在水槽中利用水道型的造粒機進行切斷而粒料化。之后,將粒料化了的聚酰胺邊在水槽中持續冷卻邊在水槽中輸送,自切斷開始2.8秒后使其離水,得到非晶狀態的粒料化了的聚酰胺。所得到的粒料的長度3mm、粒料直徑3mm、水冷終止后的粒料的溫度為70℃。此外,造粒機中的刀具的牽引速度為200m/分鐘。
所得到的非晶狀態的粒料化了的聚酰胺(縮聚聚酰胺粒料)的結晶化度18%、YI=-2、([COOH]-[NH2])=45μeq/g、相對粘度2.1、熔點(Tm)239℃、表皮部分的指針下降溫度76.8℃、核部分的指針下降溫度73.8℃,用0.1mol/L碘-碘化鉀溶液進行染色時,染色為G≤90的粒料的比例(染色度)為60%。
此外,對于所得到的縮聚聚酰胺粒料進行離子銑削觀察時,在粒料表面附近凹凸勉強密集地形成,呈現凹凸密集的部分距表面為10μm左右,在其它部分凹凸稀疏地形成。
接著,在常溫(23℃)空氣中將縮聚聚酰胺粒料放置6小時,進行冷卻之后,在與實施例1同樣的條件下實施固相聚合。在表1中示出由固相聚合而得到的聚酰胺粒料的評價結果。
在圖6中示出根據上述的球晶密度的測定方法,對聚酰胺粒料的薄片試樣照射離子束之后的表皮部分的觀察圖像。
比較例2
在與實施例3同樣的條件下反應而合成聚酰胺。
反應終止后,停止攪拌,用氮氣將反應裝置內加壓至0.30MPa(表壓力),將聚合物從裝置底部的線料模頭(口模直徑:8mm)以口模閥開度60%在260℃下抽出為線料狀。使抽出的股線在水溫25℃的水槽中著水,自著水開始0.8秒后,在水槽中利用水道型的造粒機進行切斷而粒料化。之后,將粒料化了的聚酰胺邊在水槽中持續冷卻邊在水槽中輸送,自切斷開始2.8秒后使其離水,得到非晶狀態的粒料化了的聚酰胺。所得到的粒料的長度3mm、粒料直徑3mm、水冷終止后的粒料的溫度為75℃。造粒機中的刀具的牽引速度為200m/分鐘。
所得到的非晶狀態的粒料化了的聚酰胺(縮聚聚酰胺粒料)的結晶化度17%、YI=-1,([COOH]-[NH2])=44μeq/g、相對粘度2.1、熔點(Tm)253℃、表皮部分的指針下降溫度77.8℃、核部分的指針下降溫度75.3℃,用0.1mol/L碘-碘化鉀溶液進行染色時,染色為G≤90的粒料的比例(染色度)為70%。
此外,對于所得到的縮聚聚酰胺粒料進行離子銑削觀察時,在粒料表面附近凹凸勉強密集地形成,呈現凹凸密集的部分距表面為10μm左右,在其它部分凹凸稀疏地形成。
在常溫(23℃)空氣中下將所得到的非晶狀態的粒料化了的聚酰胺放置6小時,進行冷卻之后,在與實施例1同樣的條件下實施固相聚合。在表1中示出通過固相聚合而得到的聚酰胺粒料的各種性狀。
[表1]
表1
※ΔYI為由固相聚合后的YI減去固相聚合前的YI的值。
實施例1~3中,表皮部分的球晶密度大、球晶密集地存在,因此可以用表皮部分保護粒料,使得所得到的聚酰胺粒料的黃色指數YI良好。另一方面,比較例1、2中,在離子銑削后觀察時,在表皮部分以及核部分的任一者中,源于球晶的紋路不明確,難以測定球晶密度。因此,認為未明確地形成表皮部分的球晶,此外,即便存在,也未密集地存在,粒料未被表皮部分充分地保護。因此,所得到的聚酰胺粒料的黃色指數YI與實施例相比惡化。
此外,實施例1~3中,縮小固相聚合前的縮聚聚酰胺粒料的表皮部分與核部分的指針下降溫度之差,并且提高表皮部分的指針下降溫度。因此,固相聚合后的聚酰胺粒料的黃色指數YI也是良好的。另一方面,比較例1、2中,粒料的表皮部分的指針下降溫度低,因此固相聚合后的粒料的黃色指數YI與實施例相比變差。
實施例4
[基于雙螺桿擠出成型機的聚酰胺成型體的制造]
準備包括螺桿直徑為30mm的雙螺桿擠出機,在前端安裝T口模。在該雙螺桿擠出機成型機中將各溫度C1/C2/C3/C4/H/D設定為230℃/270℃/270℃/270℃/270℃/270℃。需要說明的是,C1~C4從供給部側依次示出料筒的溫度。在該雙螺桿擠出機的加料斗中投入實施例1的由固相聚合得到的聚酰胺粒料,以螺桿轉速100rpm在料筒內將聚酰胺粒料增塑化混煉,通過T口模用60分鐘擠出厚度0.05mm、寬度250mm的薄膜形狀的聚酰胺成型體,測定此時的馬達負載振動幅度,從而評價加工性。
[基于第1單螺桿擠出成型機的聚酰胺成型體的制造]
準備具有包含螺桿直徑D為25mm的全螺紋螺桿的螺桿,L/D為24、壓縮比(C/R)為3.0、L1/L2/L3為0.50/0.12/0.38且在前端安裝T口模的單螺桿擠出成型機(第1單螺桿擠出成型機),在該單螺桿擠出成型機中,將各溫度C1/C2/C3/H/D設定為230℃/260℃/265℃/265℃/260℃。在該單螺桿擠出成型機的加料斗中投入實施例1的由固相聚合而得到的聚酰胺粒料(第1聚酰胺粒料),以螺桿轉速50rpm在料筒內對聚酰胺粒料進行增塑化混煉,通過T口模用60分鐘擠出厚度0.05mm、寬度200mm的薄膜形狀的聚酰胺成型體,測定此時的馬達負載振動幅度,從而評價加工性。
[基于第2單螺桿擠出成型機的聚酰胺成型體的制造]
準備具有包含螺桿直徑D為40mm的全螺紋螺桿的螺桿,L/D為26、壓縮比(C/R)為3.2、L1/L2/L3為0.50/0.12/0.38且在前端安裝T口模的單螺桿擠出成型機(第2單螺桿擠出成型機),在該單螺桿擠出成型機中,將各溫度C1/C2/C3/H/D設定為250℃/260℃/265℃/265℃/260℃。在該單螺桿擠出成型機的加料斗中投入實施例1的由固相聚合而得到的聚酰胺粒料(第1聚酰胺粒料),以螺桿轉速60rpm在料筒內對聚酰胺粒料進行增塑化混煉,通過T口模用60分鐘擠出厚度0.1mm、寬度400mm的薄膜形狀的聚酰胺成型體,測定此時的馬達負載振動幅度,從而評價加工性。
實施例5
制造聚酰胺粒料時,將聚合物抽出時的氮氣加壓設為0.35MPa(表壓力),將口模閥開度設為70%,除此以外與實施例1同樣地制造聚酰胺。使用所得到的固相聚合后的聚酰胺粒料,與實施例4同樣地利用雙螺桿擠出成型機、第1單螺桿擠出成型機、以及第2單螺桿擠出成型機制造聚酰胺成型體,評價其加工性。
實施例6
制造聚酰胺粒料時,將聚合物抽出時的氮氣加壓設為0.40MPa(表壓力),將口模閥開度設為80%,除此以外與實施例1同樣地制造聚酰胺。使用所得到的固相聚合后的聚酰胺粒料,與實施例4同樣地利用雙螺桿擠出成型機、第1單螺桿擠出成型機、以及第2單螺桿擠出成型機制造聚酰胺成型體,評價其加工性。
實施例7
使用實施例2的由固相聚合而得到的聚酰胺粒料,與實施例4同樣地利用雙螺桿擠出成型機、第1單螺桿擠出成型機、以及第2單螺桿擠出成型機而制造聚酰胺成型體,評價其加工性。
實施例8
使用實施例3的由固相聚合而得到的聚酰胺粒料,將第1單螺桿擠出成型機的溫度C1/C2/C3/H/D設定為230℃/275℃/280℃/280℃/275℃,將第2單螺桿擠出成型機的溫度C1/C2/C3/H/D設定為250℃/275℃/280℃/280℃/275℃,除此以外與實施例4同樣地使用雙螺桿擠出成型機、第1單螺桿擠出成型機、以及第2單螺桿擠出成型機,制造聚酰胺成型體,評價其加工性。
實施例9
制造聚酰胺粒料時,將聚合物抽出時的氮氣加壓設為0.25MPa(表壓力),將口模閥開度設為30%,除此以外與實施例1同樣地制造聚酰胺粒料。使用所得到的固相聚合后的聚酰胺粒料,利用雙螺桿擠出成型機、第1單螺桿擠出成型機以及第2單螺桿擠出成型機制造聚酰胺成型體,評價其加工性。
實施例10
制造聚酰胺粒料時,將聚合物抽出時的氮氣加壓設為0.50MPa(表壓力),將口模閥開度設為100%,除此以外與實施例1同樣地制造聚酰胺粒料。使用所得到的固相聚合后的聚酰胺粒料,利用雙螺桿擠出成型機、第1單螺桿擠出成型機以及第2單螺桿擠出成型機制造聚酰胺成型體,評價其加工性。
比較例3
使用比較例1的由固相聚合得到的聚酰胺粒料,與實施例4同樣地利用雙螺桿擠出成型機、第1單螺桿擠出成型機、以及第2單螺桿擠出成型機制造聚酰胺成型體。
比較例4
使用比較例2的由固相聚合得到的聚酰胺粒料,與實施例8同樣地利用雙螺桿擠出成型機、第1單螺桿擠出成型機、以及第2單螺桿擠出成型機制造聚酰胺成型體。
[表2]
MXDA:間苯二甲胺 PXDA:對苯二甲胺
根據以上的結果明確,實施例4~10中,表皮部分的球晶密度大,球晶密集地存在,因此可以用表皮部分保護粒料,使所得到的聚酰胺粒料的黃色指數YI良好。進而,實施例4~10中,通過雙螺桿擠出成型機而成型時,加工性良好。
此外,實施例4~8中,使用使表皮部分的球晶密集地存在并且使粒料的截面積成為一定的大小的聚酰胺粒料,從而利用單螺桿擠出成型機而成型為成型體的情況下,也可以實現低馬達負載且穩定的擠出成型。需要說明的是,實施例4~8中,使用尺寸大的第2單螺桿擠出成型機時雖然馬達負載變大,但對于該尺寸的成型機,為在實用上不存在問題的水平。
與之相對,實施例9中,在表皮部分中,球晶密集地存在,但粒料截面積小,因此使用單螺桿擠出成型機時,粒料纏繞螺桿,不能進行擠出成型。此外,實施例10中,在表皮部分中,球晶密集地存在,但粒料的截面積大,因此使用單螺桿擠出成型機時,馬達負載變大,不能實現穩定的擠出成型。
進而,比較例3、4中,通過離子銑削進行觀察,表皮部分以及核部分的任一者不能充分地確認到源于球晶的紋路,不能測定球晶密度,這樣的聚酰胺粒料未明確地形成球晶,此外即便存在也不是密集地存在。比較例3、4中,如此球晶未密集地存在,因此未充分地保護粒料,使用單螺桿擠出成型機時,粒料彼此緊密附著而不能擠出成型。進而,比較例3、4中,不能使所得到的聚酰胺粒料的黃色指數YI良好。
附圖標記說明
10 擠出成型機
13 轉接頭
20 料筒
21 螺桿
21A 供給部
21B 壓縮部
21C 計量部
D 螺桿外徑
L 螺桿有效長度
L1 供給部的長度
L2 壓縮部的長度
L3 計量部的長度
- 還沒有人留言評論。精彩留言會獲得點贊!