用于從甘油和酰氯生產氯醇的方法與流程
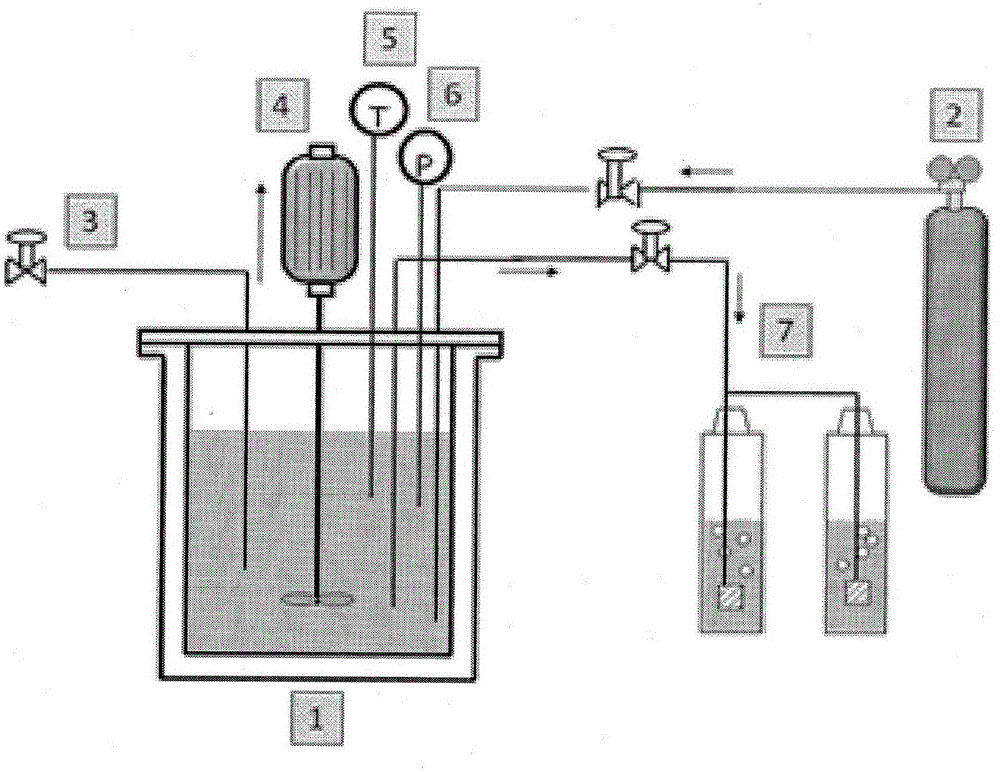
本發明涉及用于從甘油和酰氯直接開始生產氯醇(chlorohydrin)的新方法。酰氯同時發揮反應物和催化劑的作用。一旦以化學計算量(stoichiometricamout)引入酰氯時,其與甘油反應以形成足以用于生產α,γ-二氯丙醇(α,γ-dichlorohydrin)之量的酯和氣態鹽酸。
背景技術:
:甘油用于精細化學領域,并且其用途對于生產α,γ-二氯丙醇是特別重要的,所述α,γ-二氯丙醇構成生產表氯醇(epichlorohydrin)的方法中的中間產物,所述表氯醇進而用于合成環氧樹脂[1-7]。在最近開發的方法中[8-11],為了從甘油開始獲得α,γ-二氯丙醇,在作為催化劑的羧酸的存在下,根據以下反應使甘油與氣態鹽酸反應:正如可注意到的,這些方法產生作為副產物的水;如已經觀察到的,這使反應速率降低。事實上,所述反應經歷甘油的第一次氯化,這導致首先形成α-單氯甘油(α-monochlorohydrin)和水,含有少量的β-單氯甘油(β-monochlorohydrin);接著進行第二次氯化,從其中主要獲得的有期望產物α,γ-二氯丙醇以及非常適中量的α,β-二氯丙醇。因此,更詳細的反應方案如下:追溯到很久之前的使用甘油作為原料的傳統方法考慮在作為催化劑的乙酸的存在下,在約80-100℃的溫度下使甘油與鹽酸在水溶液中反應[12-15]。一些專利描述了使用不互溶的惰性有機溶劑的方法,其中α,γ-二氯丙醇是可溶的,其中反應在反應混合物的沸點下進行,因此,所述沸點可根據所使用的特定溶劑而變化,但是在任何情況下都不超過110℃[16]。另一些專利描述了反應之后進行復雜的中和、萃取和蒸餾,用于回收α,γ-二氯丙醇[17,18]。這些方法都存在相當大的缺點,例如:-由于使用鹽酸水溶液在反應混合物中引入水和未能除去在所述反應后形成的水二者所引起的反應減慢;以及-難以從反應混合物中分離α,γ-二氯丙醇。這些缺點以及作為原料的甘油的高成本在過去阻礙了該方法的發展。目前,最廣泛采用的方法使用丙烯作為原料,其在混合物中得到70%的α,β-二氯丙醇和30%的α,γ-二氯丙醇。該方法同樣存在缺點。事實上,大量的α,β-二氯丙醇帶來了困難,原因是在后續的脫氯化氫生成表氯醇中,該物質比α,γ-二氯丙醇慢得多。這對設備(plant)的大小有影響并導致產率降低,形成副產物[5,6]。在假設甘油可在低成本獲得的條件下,顯然從甘油開始的反應可潛在地更有優勢。在這一點上,應當提到產生約10wt%的作為副產物的甘油之生物柴油的增產通常引起可用性提高,因此甘油的成本逐漸降低。此外,因為甘油生產過程中主要的成本之一是其純化,應指出,可直接對粗制(即,未純化)的甘油進行氯化反應,進一步地降低成本。最近公布了多個考慮從甘油和氣態(無水)鹽酸開始生產二氯丙醇的專利。這些專利都使用羧酸作為催化劑,而且通過所使用的羧酸的類型和/或通過所采用的操作條件彼此區分開。在作為催化劑的羧酸的存在下,從甘油開始生產氯醇比從丙烯開始生產氯醇的優勢之一是:對生產α,γ-二氯丙醇實現的高選擇性,正如已經提到過的,在形成表氯醇的反應中,α,γ-二氯丙醇比α,β-二氯丙醇具有強得多的反應性。因此,可使得脫氯化氫反應器的體積顯著減小,或者假定同樣的體積,生產率提高。在由Solvay提出的專利[8]中,例如建議使用己二酸。由EurochemEngineering提出的專利[9]描述了使用以下羧酸:丙酸、丙二酸、乙酰丙酸、檸檬酸(cytricacid)和琥珀酸,此外,其聲稱通過提餾(stripping)來分離α,γ-二氯丙醇的可能性。在DOWGlobalTechnologies提出的專利[10]中,建議了可能的基于羧酸之催化劑的極長列表。但是,考慮到反應速率和產率受到HCl分壓的顯著影響,首要是強調了在壓力下操作的優勢[19,20]。在由CONSERS.p.A.等提出的專利[11]中,建議了這樣的可能性,即依次使用兩種不同的催化劑(首先是蘋果酸,然后是琥珀酸),在低壓(1-2巴)下開始反應,并第二階段中升高壓力(8-10巴)。對羧酸性能進行了深入地研究[19,21,22],并且發現了酸的pKa(其范圍應為4-5)和催化活性之間一定的相關性。例如乙酸、單氯乙酸和三氯乙酸的系列顯示出活性降低,原因是酸性力(acidicforce)隨著含氯量而提高[22]。已經再次證明一些包含羧酸官能團的催化劑在生產單氯甘油中具有選擇性,原因使只有在甘油已被完全轉化成單氯甘油后才可發生后續的氯化[21-23]。已經闡述了能夠描述實驗觀察到的動力學行為的反應機制[23]。然而,這些專利同樣存在缺點,即形成相當大量的副產物水,這造成反應逐漸減慢。最近,一個公開專利中聲稱使用了一類新的催化劑,所述催化劑由酰氯構成,先前從未實驗過[24]。在該專利中,重點在于如何使用酰氯,假定相同的摩爾濃度,與基于羧酸的傳統催化劑相比,其具有更高的活性和選擇性。通過研究這些催化劑的性能,出人意料地發現所述催化劑不必供應鹽酸就可產生期望的反應產物α,γ-二氯丙醇,所述鹽酸在甘油與酰氯反應形成酯的反應后在原位形成。然后鹽酸與酯反應以得到反應產物和作為副產物的羧酸,而非傳統方法中的水。鑒于羧酸是反應的良好催化劑,其進行快速且具有良好的選擇性,而已知水對反應速度有反作用。然后使鹽酸與酯反應以得到以下反應。即,總的來說然后使用1∶2比例的甘油/酰氯,普遍地以高反應速度獲得α,γ-二氯丙醇,其表征在使用酰氯作為催化劑中已經觀察到了催化作用[24]。然后形成高濃度的作為反應的良好催化劑的羧酸而不是水,水對酯的形成有負面作用并因此影響反應速率。技術實現要素:因此,形成本發明的對象是用于從甘油和酰氯生產氯醇的方法。以化學計算量使用的酰氯既用作反應物又用作催化劑。事實上,將通式RCOCl的酰氯與甘油設置為以化學計算量比1∶2接觸,則立即發生以下反應:即,對于形成氯醇的反應所必需的鹽酸與甘油酯在原位同時形成。然后使所形成的鹽酸與酯反應以得到以下反應即,總的來說然后使用1∶2比例的甘油/酰氯,普遍地以高反應速率獲得α,γ-二氯丙醇,這表征在使用酰氯作為催化劑中已經觀察到了催化作用[24]。然而,在這種情況下,形成高濃度的羧酸(而不是水),因為在傳統的方法中反應是由羧酸催化的。羧酸對所述反應具有良好的催化作用;而已知水對酯的形成有負面作用并因此影響反應速率。因此所述反應更快速發生,并且具有良好的選擇性。可使用的酰氯具有以下通式其中R為具有2至10個碳原子的直鏈、支鏈或環狀烷基鏈,其可被一個或更多個R’基團取代,所述R’基團彼此相同或者不同,選自:直鏈、支鏈或環狀脂肪族C1-C10基團;芳香族C1-C10基團;以及一個或多于一個另外的酰氯基團。作為副產物形成的羧酸可經分離、純化并返回到待進行氯化的酰氯的供應體(supplier)中并再次使用,具有明顯的經濟優勢。事實上,以下事實代表了該方法的一個優勢:期望的反應在生產氯醇的位置處無需提供氣態鹽酸即可發生反應。更容易連續操作代表了另一個優勢。本發明的另一個目的是可同樣使用作為生物柴油生產之副產品的粗制甘油(rawglycerol),其將初步處理的操作限于:使用無機酸(優選鹽酸)中和作為催化劑的堿,以及除去殘留的甲醇和任何可能的濕氣。本發明的另一個目的是提供將能使甘油幾乎完全轉化的方法。本發明的另一個目的是提供將在熱力學有利的條件下發生的方法,其具有相應提高的產率和反應速率。從本發明隨后的描述中將更清楚地體現的上述和其他目的是通過用于生產氯醇并且特別是α,γ-二氯丙醇的方法實現,所述方法包括在甘油和酰氯之間化學計算量反應的步驟。本發明的另一些目的將在隨后本發明的一些實施方案的描述中清楚地體現出。附圖簡述為了能夠更好地理解,將參考附圖對本發明進行描述,所述附圖僅通過非限制性實例舉例說明其一個優選的實施方案。在附圖中:圖1是通過舉例的方式提供的在測試中使用的設備的示意圖,其中:由1表示的是哈氏合金反應器(Hastelloyreactor),容積為300mL;由2表示的是HCl缸(cylinder);由3表示的是用于取出待分析的樣品的閥門;由4表示的是磁攪拌器;由5表示的是溫度讀取器;由6表示的是壓力讀取器;并且由7表示的是用于中和未反應HCl的系統(含有氫氧化鈉溶液的兩個串聯的Drechsel鼓泡器)。圖2是示出了根據實施例1的形式在供應乙酰氯期間和之后反應器中壓力變化的圖;和圖3是示出了根據實施例2的形式在供應乙酰氯期間和之后反應器中壓力變化的圖。發明詳述根據本發明的方法包含引起甘油與酰氯(例如乙酰氯)以化學計算量比1∶2反應的基本操作。甘油與乙酰氯的反應在室溫條件下已經非常快速,但在100-120℃下幾乎是瞬間的。這意味著,如果兩種試劑從一開始就完全接觸,由于迅速產生了HCl,反應器中的壓力顯著提高。因此,將一種試劑逐步添加到另外一種中(優選地,將酰氯添加至甘油)是有利的。以這種方式,保持了4-6巴的適度壓力值,并且可根據調節酰氯流量的方法之期望的持續時間進行調整。因此,根據本發明的方法可在半連續的反應器中進行,隨著酰氯的消耗,向所述反應器中連續地供應酰氯。然而,所述方法也可連續進行,連續供應酰氯和甘油二者。反應溫度可保持在60-250℃,優選100至175℃。可使用具有如上所述通式的酰氯。優選將在先專利中已經使用的酰氯作為催化劑,其可以是單官能團的且包含2至10個碳原子,或者是雙官能團/多官能團的,具有2至10個碳原子。可用于本發明中的酰氯的非限制性實例包括:乙酰氯、丙酰氯、己二酰二氯(hexandioyldichloride)、丙二酰二氯(propandioyldichloride)、丁二酰氯(butandioyldichloride)、苯乙酰氯。在實驗室水平和本發明中提供的實施例中,在先前引入了甘油的反應器中,在攪拌下用泵供應酰氯。在不同的時間從該反應器中采樣以通過化學分析確定所采的不同樣品中產物的分布以追蹤在反應時間中的變化。在下文中提供的實例中,僅通過非限制性舉例說明本發明,詳細地描述了以不同的方式進行的用于生產氯醇(特別是α,γ-二氯丙醇)的測試。圖1示出了用于進行實施例中提供的實驗的裝置的示意圖。反應器1為300-cm3Parr高壓釜,利用經溫度調節的烘箱外部加熱以達到預設的溫度。將甘油以預先確定的量(例如100g)引入到反應器中。通常保持在約1000r.p.m.的磁攪拌器4使反應混合物保持均勻。然后將反應器加熱直到達到反應溫度。然后以預設的恒定流量(1-2cm3/分鐘)引入酰氯,持續供應酰氯直至達到化學計算量值(酰氯摩爾數∶甘油摩爾數=2)。此時,中斷供應酰氯,并讓混合物再反應幾個小時直到發現反應器中HCl的壓力不再降低。在不同的反應時間,通過閥門3取出少量的反應混合物,并進行分析以確定其組成。用讀取器5和6來讀取溫度和壓力。使未反應的HCl通過Drechsel鼓泡器從高壓釜中釋放出來,所述Drechsel鼓泡器中填充有氫氧化鈉飽和溶液并且包含當達到中性時發出信號的指示器。在以下實施例中描述了所得到的結果。實施例1中描述的是通過向圖1中包含100g甘油的反應器以1cm3/分鐘的流量供應乙酰氯持續154分鐘而獲得的系統的行為。乙酰氯的供應量符合乙酰氯/甘油的化學計算量比例2。反應溫度保持在100℃。測試持續共4小時。實施例2中描述的是通過向圖1中包含100g甘油的反應器以2cm3/分鐘的流量供應乙酰氯持續80分鐘而獲得的系統的行為。乙酰氯的供應量符合乙酰氯/甘油的化學計算量比例2。反應溫度保持在100℃。測試持續其4小時。在實施例3中,將在前面兩個實例中結束時得到的反應混合物進行蒸餾,并按順序分離出:殘留的鹽酸、形成的乙酸以及可能的痕量水。將從蒸餾得到的殘留物進行化學分析,其表明主要形成期望產物α,γ-二氯丙醇。實施例實施例1-通過以1cm3/分鐘的恒定流量供應乙酰氯的甘油與乙酰氯之間的反應測試在圖1的反應器中進行,維持以下條件:起始甘油量100g;乙酰氯的流量1cm3/分鐘(在室溫下供應),供應的持續時間:154分鐘;測試的持續時間:4h;操作溫度100℃。在這些條件中,由于生成了鹽酸而在反應器中設定的壓力是形成酯和隨后形成HCl的反應與消耗HCl形成氯醇的反應之間的動力學平衡的結果。當停止供應乙酰氯,反應再持續86分鐘。反應器中保留的殘余壓力對應于少量未反應的HCl。該HCl通過捕集器(trap)被釋放和減弱,對小樣本分析后得到的液體保留下來用于進行產物的純化(參見實施例3)。圖2中示出了反應器中在供應乙酰氯期間和隨后測試持續期間的壓力圖。正如可從圖2的圖中注意到,形成氯醇的反應極其快速,就約80-100分鐘而言,壓力升高幾乎可以忽略不計,即,所有已經形成的鹽酸都已反應。另一個觀察結果是,在供應乙酰氯結束時,設定為剛好高于7巴值的HCl壓力沒有降低,這是反應結束的標志。表1示出了分別在180分鐘和240分鐘采樣的分析:表1-在不同反應時間時采樣的分析時間αβαγαβ甘油000001001801.88.281.48.602400.55.982.311.30這些分析證實,在達到化學計算量的酰氯量不久之后,可認為反應完成。實施例2-通過以2cm3/分鐘的恒定流量供應乙酰氯的甘油與乙酰氯之間的反應測試再次在圖1所示的反應器中進行,維持以下條件:起始甘油量100g;乙酰氯流量2cm3/分鐘(在室溫下供應),供應的持續時間:80分鐘;測試的總持續時間:4h;操作溫度:100℃。由于生成了鹽酸而在反應器中設定的壓力同樣是形成酯和隨后形成HCl的反應與消耗HCl形成氯醇的反應之間的動力學平衡的結果。在停止供應乙酰氯后,反應再進行160分鐘。同樣地,在這種情況下,反應器中保留的殘余壓力對應于少量未反應的HCl。所述HCl通過捕集器釋放和減弱,并且在對小樣本分析后得到的液體保留下來用于產物的純化(參見實施例3)。圖3示出了在供應乙酰氯期間和整個測試的持續期間反應器中壓力的圖。在這種情形下,乙酰氯的添加非常快,因此,系統中壓力更迅速升高,盡管最終的平臺(plateau)較低,這是消耗了更多鹽酸的標志。另一個觀察是,在供應乙酰氯結束時,設定為剛好高于5巴值的HCl壓力沒有進一步降低,這是反應結束的標志。表2中示出的是分別在180分鐘和240分鐘采樣的分析。表2-在不同反應時間采樣的分析時間αβαγαβ甘油0000010018004.091.94.1024003.792.34.00圖3表明,在130分鐘后,即在達到化學計算量的乙酰氯后,反應器中的HCl壓力保持穩定(settle),因此認為反應終止。此外,該測試還表明可通過適當地調節操作條件(例如甘油的起始量、乙酰氯的添加速率、以及反應溫度)減少反應時間,而反應器中的壓力將根據預先選擇的條件自動調節。此外,以所述的方式操作,即,更快地添加反應性酰氯,提高了對α,γ-二氯丙醇的選擇性。實施例3-實施例1和2中反應產物的純化假設反應進行直到完成,將實施例1和2中得到的產品進行蒸餾。低沸點產物為HCl、乙酸以及可能的痕量水,因此將這些從混合物中除去。將以這種方式得到的殘余物進行驗證分析,在下表3中提供了所考慮的兩種情況的組成。表3-在通過蒸餾將低沸點組分純化后反應產物的分析樣品αβαγαβ甘油表105.882.112.10表203.992.23.90參考文獻[1]SolvayInc.Processfortheproductionofalpha-epichlorhydrin.GB799,567(1958).[2]P.Bruin;Epoxyethers,theirpolymersandderivatives.GB814,695(1959).[3]Z.Brojer,P.A.Penczec,;J.H.Sas;Processofmanufacturingepoxideresins.GB1,001,364,(1965)[4]T.Kawabe,M.Kadoma,K.Murakami,H.Itoh;Processforcontinuousproductionofepichlorohydrin;US4113746(1978)[5]S.Carrà,E.Santacesaria,andM.Morbidelli,Ind.Eng.Chem.ProcessDes.Dev.,18(3),424-427,(1979).[6]S.Carrà,E.Santacesaria,andM.Morbidelli,Ind.Eng.Chem.ProcessDes.Dev.,18(3),428-433,(1979)[7]Clarke,H.T.;Hartman,W.W.Epichlorohydrin.InOrganicSyntheses,Coll.;(1941);Vol.1,p233.[8]P.Krafft,P.Gilbeau,B.Gosselin,S.Claessens,(Solvay).Processforproducingdichloropropanolfromglycerol,theglycerolcomingeventuallyfromtheconversionofanimalfatsinthemanufactureofbiodiesel.PCTPatent,WO054167A1,2005.[9]Siano,D.;Santacesaria,E.;Fiandra,V.;Tesser,R.;DiNuzzi,G.;DiSerio,M.;Nastasi,M..(EurochemEngineeringsrl);Processfortheproductionofα,γ-dichlorohydrinfromglycerolandhydrochloricacid.WO111810A2,2006,US2009/0062574,EP1879842B1[10]D.Schreck,W.Kruper,R.D.Varjian,M.E.Jones,R.M.Campbell,K.Kearns,B.D.Hook,,J.R.Briggs,J.G.Hippler,(DowGlobalTech.Inc.).Conversionofmultihydroxylated-aliphatichydrocarbonoresterthereoftoachlorohydrin.PCTPatent,WO2006/020234andEP1771403B1(2007)[11]S.Cassarino,F.Simola;(CONSERSpA)Conversionofglyceroltodichlorohydrinsandepichlorohydrin.WO2009/066327[12]J.B.Conant,O.R.Quayle;Glycerolαγ-dichlorohydrin.InOrganicSyntheses,Coll.;(1941);Vol.1,p292.[13]J.B.Conant,O.R.Quayle;Glycerolα-monochlorohydrin.InOrganicSyntheses,Coll.;(1941);Vol.1,p294.[14]Fauconnier,Sanson,Bull.Soc.Chim.,48,(1887),pp.236-240[15]A.J.Hill,E.J.Fischer,Asynthesisofbeta-chloro-allylchloride;J.Am.Chem.Soc.,(1922)Vol.44,pp.2582-2595[16]E.G.Britton,R.X.Heindel,Preparationofglyceroldichlorohydrin;US2,144,612(1939)[17]E.C.Britton,H.R.Slagh;Glyceroldichlorohydrin;US2,198,600(1940)[18]W.P.Thompson;Recoveryofaliphaticchlorohydrinshavingfrom3to5carbonatomsfromaqueoussolution.GB931,211(1963).[19]R.Tesser,M.DiSerio,R.Vitiello,V.Russo,E.Ranieri,E.Speranza,E.Santacesaria;GlycerolChlorinationinGas_LiquidSemibatchReactor:AnAlternativeRouteforChlorohydrinsProduction;Ind.Eng.Chem.Res.51(2012)8768-8776.[20]B.M.Bell,J.R.Briggs,R.M.Campbell,S.M.Chambers,P.D.Gaarenstroom,J.G.Hippler,B.D.Hook,K.Kearns,J.M.Kenney,W.J.Kruper,D.J.Schreck,C.N.Theriault,C.P.Wolfe,GlycerolasaRenewableFeedstockforEpichlorohydrinProduction.TheGTEProcess,Clean36(2008)657-661[21]E.Santacesaria,R.Tesser,M.DiSerio,L.Casale,D.Verde,NewProcessforProducingEpichlorohydrinviaGlycerolChlorination;Ind.Eng.Chem.Res.49(2010)964-970.[22]R.Tesser,E.Santacesaria,M.DiSerio,G.DiNuzzi,andV.Fiandra;KineticsofGlycerolChlorinationwithHydrochloricAcid:ANewRoutetoαγ-Dichlorohydrin;Ind.Eng.Chem.Res.46(2007)6456-6465.[23]E.Santacesaria,M.DiSerio,R.Tesser,(EurochemEngineeringsrl).Processformonochlorohydrinsproductionfromglycerolandhydrochloricacid.PCTPatentWO132770A1,2008.[24]E.Santacesaria,R.Tesser,R.Vitielio,M.DiSerio;(EurochemEngineeringsrl)Processoperlaproduzionedicloridrine;ItalianPatentRM2013A000488.當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1