用于神經退行性疾病和阿爾茨海默病的治療中的用途的雙重抑制劑化合物的制作方法
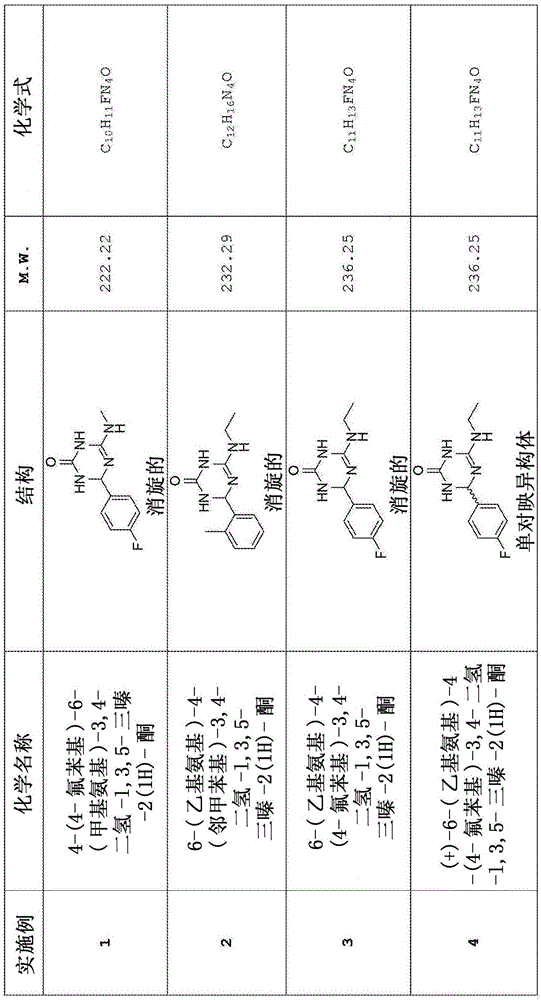
本發明涉及新型三嗪酮衍生物,以及它們作為治療劑(therapeuticagent)來治療多種神經退行性疾病,包括阿爾茨海默病(AD)的用途。
背景技術:
:AD是進行性神經系統疾病,特征在于認知功能惡化、癡呆、記憶喪失和行為改變。AD是在神經學中的主要未滿足的醫療需要,并且據估計,到2050年,全世界可能有超過1億AD患者[Alzheimer'sAssociation,2013Alzheimer'sdiseasefactsandfigures.Alzheimer's&Dementia2013,9,208-45]。AD顯著影響患者及其家屬的生活質量,并且盡管大量投資,但對于AD,有很少的(如果有的話)有效的治療。AD發病機理涉及遺傳和生化因素的復雜的相互作用,包括β-淀粉樣肽的增加的產生(淀粉樣蛋白假說)和微管相關tau蛋白的增加的磷酸化(tau假說)[Querfurth,H.W.andLaFerla,F.M.Mechanismsofdisease:Alzheimer'sdisease.NEnglJMed2010,362,329-44]。通過膜相關的淀粉樣前體蛋白的淀粉樣斷裂(amyloidogeniccleavage)來生成β-淀粉樣肽(Αβ)。兩種主要酶已被確定為負責Αβ形成,即β-分泌酶(BACE-1)和γ-分泌酶。巨大的努力一直致力于任一種這些酶的小分子抑制劑的識別。γ-分泌酶抑制劑已達到III期臨床試驗,但是失敗了,這是由于缺乏功效以及嚴重的副作用(即,皮膚癌)[Blennow,K.;Zetterberg,H.;Haass,C.;Finucane,T.Semagacestat'sfall:wherenextforADtherapies?NatMed2013,19,1214-15]。BACE-1抑制劑仍處于臨床開發,其中最先進的化合物處于III期臨床試驗。BACE-1仍然是少數幾個目標選項之一,其可以用于在AD的淀粉樣蛋白假說(amyloidhypothesis)內的潛在有效的治療[Rafii,M.S.UpdateonAlzheimer'sdiseasetherapeutics.RevRecentClinTrials2013,8,110-18]。至于tau假說,研究活動一直專注于激酶抑制劑的識別,因為幾種激酶已被確定為負責tau磷酸化。在這些酶中,糖原合成酶激酶3β(GSK-3β)已被確定為在此病理級聯(pathologicalcascade)內的關鍵成員之一。GSK-3β負責tau過磷酸化,其引起tau被分離自微管并沉淀為內神經元纏結聚集體[Avila,J.;Wandosell,F.;Hernandez,F.Roleofglycogensynthasekinase-3inAlzheimer'sdiseasepathogenesisandglycogensynthasekinase-3inhibitors.ExpertRevNeurother2010,10,703-10]。此外,GSK-3β已被提議為在β-淀粉樣肽和tau蛋白之間的可能的連接[Hernandez,F.;GomezdeBarreda,E.;Fuster-Matanzo,A.;Lucas,J.J.;Avila,J.GSK3:apossiblelinkbetweenbetaamyloidpeptideandtauprotein.ExpNeurol.2010,223,322-25]。因此,已經長期追求GSK-3β抑制劑[Martinez,A.;Perez,D.I.;Gil,C.Lessonslearntfromglycogensynthasekinase3inhibitorsdevelopmentforAlzheimer'sdisease.CurrTopMedChem2013;13,1808-19]。盡管已經對比地考慮兩種假設,但最近的證據表明,AD是多因素疾病,其中幾種神經退行性途徑可以同時有助于神經元死亡和相關的神經退行性變(神經變性,neurodegeneration)[Ittner,L.M.;Gotz,J.Amyloid-βandtauatoxicpasdedeuxinAlzheimer'sdisease.NatRevNeurosci2011,12,65-72]。在這種情況下,多靶點藥物(MTD),即能擊中影響不同病理途徑的多個目標的小有機分子正在成為有前景的疾病修飾化合物,用于治療復雜的神經系統疾病[Cavalli,A.;Bolognesi,M.L.;Minarini,A.;Rosini,M.;Tumiatti,V.;Recanatini,M.;Melchiorre,C.Multi-target-directedligandstocombatneurodegenerativediseases.JMedChem2008,51,347-72.Bolognesi,M.L.;Simoni,E.;Rosini,M.;Minarini,A.;Tumiatti,V.;Melchiorre,C.Multitarget-directedligands:innovativechemicalprobesandtherapeutictoolsagainstAlzheimer'sdisease.CurrTopMedChem2011,11,2797-806]。借助于單一化合物,同時靶向兩種或更多種蛋白的策略可以提供的治療效果優于選擇性藥物的治療效果。這可以通過,經由使用MTD所提供的,相對于混合劑(混合物,cocktail)或多組分藥物的潛在益處的數目來解釋。MTD的優點可以總結如下:1)在臨床開發中減小的不確定性,因為預測單一化合物的藥物動力學比預測藥物混合劑的藥物動力學要容易得多,從而克服不同的生物利用度、藥物動力學和代謝的問題;2)關于藥效學的確定性;3)改善的功效,這是由于同時抑制多個目標的協同效應;4)改善的安全性,其中通過減少與藥物混合劑的負載相關的副作用(藥物相互作用的降低的風險);這特別與藥物代謝相關,其中不同的藥物對于相同的代謝酶的競爭會影響其毒性。所有這些考慮是特別相關的,因為在藥物開發中,損失率的主要原因之一仍然是候選藥物的藥代動力學分析。另一個重要的優點是簡化的治療方案和改善的依從性(compliance),其對于老年AD患者以及其照顧者是特別重要的[Small,G.;Dubois,B.AreviewofcompliancetotreatmentinAlzheimer'sdisease:potentialbenefitsofatransdermalpatch.CurrMedResOpin2007,23,2705-2713]。在這方面,關鍵問題在于,AD患者易受廣泛的伴隨疾病(共患病,morbidity))的影響,包括高血壓、血管疾病和糖尿病,其往往可以是關聯的。因此,在老年人群中與多重用藥關聯的問題近年來已被確認為關鍵的。這些問題主要包括在此人群中更頻繁地發生的藥物相互作用,這是由于慢性病和受損的器官功能的共存。本身是安全的兩種藥物不能被認為是組合安全的,尤其是在老年患者中。由此得出結論,同時給予的藥物的數目應盡可能減少,因為,對于藥物治療,老年是不可預測的風險因素[Turnheim,K.Whendrugtherapygetsold:pharmacokineticsandpharmacodynamicsintheelderly.ExpGeront2003,38,843-853]。因此,考慮到在老年人中在多重用藥、復合病變、改變的藥效敏感性和藥物動力學的變化之間的相互作用的復雜性,MTD強烈好于聯合治療。MTD的臨床使用還可以簡化治療方案[Youdim,M.B.,andBuccafusco,J.J.CNSTargetsformultifunctionaldrugsinthetreatmentofAlzheimer'sandParkinson'sdiseases.JNeuralTransm2005,112,519-537]。對處方用藥方案的依從性對于有效治療是必不可少的。非依從性是一個普遍的問題,但對于健忘AD患者以及其照顧者是特別具有挑戰性[Small,G.,andDubois,B.AreviewofcompliancetotreatmentinAlzheimer'sdisease:potentialbenefitsofatransdermalpatch.CurrMedResOpin2007,23,2705-2713]。因此,簡化的MTD方案可能增加治療依從性。利用藥物混合劑,不可獲得所有上述優點。多靶配體策略是開發用于治療復雜的神經系統疾病的新型候選藥物的創新方式,尤其是鑒于以下事實:在神經退行性疾病中涉及的主要基本過程具有多因素特性[Cavalli,A.;Bolognesi,M.L.;Minarini,A.;Rosini,M.;Tumiatti,V.;Recanatini,M.;Melchiorre,C.Multi-target-directedligandstocombatneurodegenerativediseases.JMedChem2008,51,347-72]。因此這樣的策略是基于這樣的概念:可以開發單官能團化合物以擊中在構成AD和其它神經退行性疾病的基礎的神經退行性過程中配合的多個靶點,并因此將阻止在相互作用的致病途徑之間的不期望的代償(補償,conpensation)。確實,多靶點化合物可以表示使用藥物組合的實用替代。因為許多神經元病癥共享大部分的神經退行性機制,所以這樣的多靶點化合物還可以用作用于其它疾病的藥劑。與多靶點化合物相關的一個問題在于,就它們的每單位分子量的結合能而言,它們中的許多是低效的。這是因為,它們含有這樣的基團,該基團僅對于靶點之一是重要的,該靶點僅僅由其它基團所容忍。這導致不平衡分布(unbalancedprofile)[Morphy,R.,andRankovic,Z.Fragments,networkbiologyanddesigningmultipleligands.DrugDiscovToday2007,12,156-160;Morphy,R.Theinfluenceoftargetfamilyandfunctionalactivityonthephysicochemicalpropertiesofpre-clinicalcompounds.JMedChem2006,49,2969-2978]。活性的隨后優化不是一項容易的任務。事實上,多靶點化合物被認為是具有其本身的藥理學分布的新化學實體,因此它對靶的功效是不可先驗地預測的,因此必須面對藥物開發的完整的過程。在MTD的上下文中,僅舉幾例,我們可以提及美莫醌(memoquin),幾年前由Cavalli和同事所披露[Cavalli,A.;Bolognesi,M.L.;Capsoni,S.;Andrisano,V.;Bartolini,M.;Margotti,E.;Cattaneo,A.;RecanatiniM.;Melchiorre,C.AsmallmoleculetargetingthemultifactorialnatureofAlzheimer'sdisease.ChemIntEdEngl2007,46,3689-92],和拉多替吉(ladostigil),由Youdim和同事所開發并且目前在II期臨床試驗[Weinreb,0.;Mandel,S.;Bar-Am,0.;Yogev-Falach,M.;Avramovich-Tirosh,Y.;Amit,T.;Youdim,M.B.Multifunctionalneuroprotectivederivativesofrasagilineasanti-Alzheimer'sdiseasedrug.Neurotherapeutics2009,6,163-74]。WO2008/015240披露了N-苯基-普尼拉明(異戊烯胺,prenylamine)衍生物的家族并且聲稱它們用來治療認知、神經退行性或神經元疾病或病癥,如阿爾茨海默病或帕金森病;在體外測定中,它們表現出對酶靶GSK-3β的輕度至中度的抑制作用,并且只有其中的幾個也對BACE表現出輕度至中度抑制作用。EP2138488A1披露了4-(吡啶-4-基)-1H-[1,3,5]-三嗪-2-酮衍生物作為用于治療神經退行性疾病的選擇性GSK-3β抑制劑;除了單靶點抑制劑外,其中描述的化合物是芳族的,并且顯示出三嗪酮核心支架的平面幾何形狀,其中在承載吡啶環的三嗪酮環的位置4處的碳原子是sp2-雜化的,因此不能形成立體異構體。因此,EP2138488的化合物是非手性化合物。多靶點藥物發現中,已報道基于片段的方法發揮關鍵作用。事實上,是片段,而不是類先導(lead-like)化合物,可以具有結合超過單個靶點的能力[Hann,M.M.;Leach,A.R.;Harper,G.Molecularcomplexityanditsimpactontheprobabilityoffindingleadsfordrugdiscovery.JChemInfComputSci2001,41,856-64.Bottegoni,G.;Favia,A.D.;Recanatini,M.;Cavalli,A.Theroleoffragment-basedandcomputationalmethodsinpolypharmacology.DrugDiscovToday2012,17,23-34]。然后,在片段到先導化合物的步驟中,應該細心地保持相對于病理靶點的所期望的生物學分布,同時避免起因于脫靶結合的潛在傾向。在這些前提下,基于片段的方法已用來設計能夠抑制BACE-1和GSK-3β酶(在AD的兩個主要的病理級聯內的兩種證實的靶的小分子)。明顯地,這些酶在祖先上(ancestrally)是相當不同的,具有接近隨機限制的19%的序列同一性。為了設計雙重抑制劑(dualinhibitor),已使用基于配體的方法,其中使負責分別結合于BACE-1和GSK-3β的那些藥效基團特征(feature),如胍基基序和環酰胺基團進行組合,以及,隨后,進行對接仿真(dockingsimulation),其旨在研究進入兩種酶的催化袋的新設計的化合物的相互作用。一系列的4-苯基-6-氨基-3,4-二氫-l,3,5-三嗪-2(lH)-酮發現是具有用于結合于兩種靶所需要的化學特點的框架(支架,scaffold)[PratiF.etal.,Structure-baseddesignandsynthesisofnovelBACE-1/03Κ-3βdualinhibitors.8thAnnualdrugdiscoveryconferenceforneurodegeneration.2月2-4,2014,Miami,FL]。在若干文獻出版物中,描述了在4-苯環處不同取代的4-苯基-6-氨基-3,4-二氫-l,3,5-三嗪-2(1H)-酮的合成、識別(通過物理和光譜數據)和相關的物理化學性能[OstrogovichA.,GazzettaChimicaItaliana1909,39i,540;OstrogovichA.,MedianV.B.,GazzettaChimicaItaliana1929,59,181-198;OstrogovichA.,MedianV.B.,GazzettaChimicaItaliana1929,59,198-200;OstrogovichA.,MedianV.B.,GazzettaChimicaItaliana1934,64,792-800;Wakabayashik.,OkuzuM.,NipponDojoHiryogakuZasshi1970,41(6),237-245;BacalogluR.etal.RevueRoumainedeChimie1972,17(4),747-754;Neamtiuetal.,ZeitschriftfuerPhysikalischeChemie(Leipzig)1976,257,1089-1090;GorbatenkoV.I.,etal.,ZhurnalOrganicheskoiKhimii1976,12(nb.l0),2103-2107]。然而,從神經保護和神經發生的觀點考慮,這些分子沒有被認為是令人滿意的。在本領域中需要發現新的MTD來治療阿爾茨海默病。技術實現要素:因此,本發明的目的是提供新化合物,其具有大范圍的神經保護和抗神經退行性性能并且能夠有效地治療AD。尤其是,本發明的目的之一是提供新化合物,其具有針對BACE-1和GSK-3β的抑制活性,從而表現出增加的神經保護和神經發生活性以治療阿爾茨海默病。根據權利要求1的化合物、根據權利要求7的藥物組合物、根據權利要求10和11的用途已經滿足上述目的。在從屬權利要求內闡述了優選的實施方式。尤其是,本發明人現在已經確定新的系列的4-芳基-6-氨基-3,4-二氫-l,3,5-三嗪-2(lH)-酮,其不同于在現有技術中描述的化合物,能夠抑制BACE-1和GSK-3β酶,具有增加的有效的藥理作用,并具有非平面幾何形狀,其中在三嗪酮環的位置4處的碳原子是sp3-雜化的。這樣的修飾引起較高的構象自由度,包括生成相應的對映異構體的可能性。此外,不同于現有技術的化合物,本發明的化合物的特征在于,在3,4-二氫-l,3,5-三嗪-2(1H)-酮環的位置C6處存在仲或叔氨基、或仲或叔酰胺基團,和相應的已知伯胺比較,其出乎意料地,在星形膠質和小膠質細胞的神經炎癥的藥理學模型中表現出優越的神經保護和神經發生活性。就針對BACE-1和GSK-3β的結構-活性關系(SAR)而言,通過合成和測試多種新的示例性化合物(它們針對兩種酶的抑制活性)已進一步研究了此新系列的化合物。以下段落提供了根據本發明的化合物的各種化學部分的定義,并且旨在統一適用于整個說明書和權利要求,除非另有明確闡述,定義提供了更廣泛的定義。如本文中所使用的術語"烷基",其自身或作為另一取代基的一部分是指脂族烴基。這樣的術語包括直鏈(非支鏈)或支鏈,其可以是完全飽和的、單或多不飽和的。術語"不飽和的"脂族烴基包括烯基和炔基。如在本文中所使用的,術語"烯基"是指烷基,優選具有2至6個碳原子并且含有至少一個碳-碳雙鍵。如在本文中所使用的,術語"炔基"是指烷基,優選具有2至6個碳原子并且含有至少一個碳-碳三鍵。根據本發明的烷基的非限制性實例是,例如,甲基、乙基、丙基、異丙基、正丁基、異丁基、叔丁基、正戊基、異戊基、正己基、乙烯基、1-丙烯基、2-丙烯基、1-或2-丁烯基、乙炔基、1-丙炔基、2-丙炔基、1-或2-丁炔基等。根據本發明的烷基可以是未取代的或者用一個或多個取代基取代的。如在本文中所使用的,術語"環烷基"是指飽和的或部分不飽和的具有單環的碳環基團。它包括環烯基基團。根據本發明的環烷基基團的非限制性實例是,例如環丙烷、環丁烷、環戊烷、環己烷、環庚烷、環戊烯、環己烯、環己二烯等。根據本發明的環烷基可以是未取代的或者用一個或多個取代基取代的。如在本文中所使用的,術語"雜烷基"是指烷基基團,如上所定義的,其通過雜原子,連接于化合物的剩余部分,或者可替換地,是指烷基基團,其中至少一個碳原子被雜原子替換。雜烷基可以是未取代的或者用一個或多個取代基取代的。術語"雜環烷基"基團("非芳族雜環"基團)是指環烷基基團(非芳族基團),其中碳原子的至少之一已被雜原子替換,上述雜原子選自氮、氧和硫。雜環烷基可以是未取代的或取代的。如在本文中所使用的,術語"雜原子"是指不同于碳或氫的原子。雜原子通常意味著包括氧(O)、氮(N)和硫(S)。如在本文中所使用的,雜原子可以可選地被氧化以及氮雜原子可以可選地被季銨化。根據本發明的雜烷基基團的非限制性實例是,例如-CH2-CH2-O-CH3、-CH2-CH2-NH-CH3、-CH2-CH2-N(CH3)-CH3、-CH2-S-CH2-CH3、-CH2-CH2-S(O)-CH3、-CH2-CH2-S(O)2-CH3、-CH=CH-O-CH3、-CH2-CH=NOCH3和-CH=CH-N(CH3)-CH3。多達兩個雜原子可以是連續的,如,例如-CH2-NH-OCH3。雜環烷基的實例包括但不限于內酰胺、內酯、環狀酰亞胺、環狀硫代酰亞胺、環狀氨基甲酸酯、1-(1,2,5,6-四氫吡啶基)、四氫噻喃、4H-吡喃、四氫吡喃、哌啶(2-哌啶基、3-哌啶基)、1,3-二噁英、1,3-二噁烷、1,4-二噁英、1,4-二噁烷、哌嗪、1,3-氧硫雜環己烷(oxathiane)、1,4-氧硫雜環己二烯(oxathiin)、1,4-氧硫雜環己烷、四氫-1,4-噻嗪、2H-1,2-噁嗪、嗎啉(4-嗎啉基、3-嗎啉基)、三噁烷、六氫-1,3,5-三嗪、四氫噻吩、四氫呋喃(四氫呋喃-2-基、四氫呋喃-3-基)、1,2-二硫戊環-3-基、吡咯啉、吡咯烷、吡咯烷酮、吡咯烷二酮(pyrrolidione)、吡唑啉、吡唑烷、咪唑啉、咪唑烷、1,3-間二氧雜環戊烯、1,3-二氧戊環、1,3-二硫雜環戊二烯、1,3-二硫戊環、異噁唑啉、異噁唑烷、噁唑啉、噁唑烷、噁唑烷酮、噻唑啉、噻唑烷和1,3-氧硫雜環戊烷(oxathiolane)。如本文中所使用的,術語"烷氧基"是指這樣的烷基,其通過氧原子,連接于化合物的剩余部分。如在本文中所使用的,術語"鹵素"是指氟、氯、溴和碘。如在本文中所使用的,術語"鹵代烷基"是指單鹵代烷基和多鹵代烷基。例如,術語"鹵代(C1-6)烷基"是指包括但不限于氟C1-6烷基,如三氟甲基、2,2,2-三氟乙基等。如在本文中所使用的,術語"鹵代烷氧基"是指單鹵代烷氧基和多鹵代烷氧基。例如,術語"鹵代(C1-6)烷氧基"是指包括但不限于氟C1-6烷氧基,如三氟甲氧基、2,2-二氟甲氧基等。如在本文中所使用的,術語"芳基"是指由未取代的或取代的單、雙或三碳環系統組成的烴,其中上述環被稠合在一起以及碳環的至少之一是芳族的。術語"芳基"是指,例如環狀芳族,如6元烴環、兩個六元稠合烴環。芳基基團的非限制性實例是,例如苯基、α-或β-萘基、9,10-二氫蒽基、茚滿基、芴基等。根據本發明的芳基可以是未取代的或者用一個或多個取代基取代的。如在本文中所使用的,術語"雜芳基"是指如上所定義的芳基,其中1至4個碳原子獨立地被雜原子替換,上述雜原子選自由氮、氧和硫組成的組。雜芳基基團的非限制性實例是,例如吡咯基、呋喃基、噻吩基、咪唑基、吡唑基、噁唑基、異噁唑基、噻唑基、異噻唑基、吲哚基、苯并呋喃基、苯并噻吩基、苯并咪唑基、苯并吡唑基、苯并[d][1,2,3]三唑-l-基、苯并噁唑基、苯并異噁唑基、苯并噻唑基、苯并異噻唑基、三唑基、噁二唑基、四唑基、吡啶基、吡嗪基、嘧啶基、噠嗪基、喹啉基、異喹啉基、喹唑啉基、喹喔啉基。根據本發明的雜芳基基團可以是未取代的或者用一個或多個取代基取代的。如在本文中所使用的,術語"芳族環"是指,其中取代基碳原子構成不飽和環系統的部分,在環系統中的所有的原子是sp2雜化的以及π電子的總數等于4n+2,其中n是整數。如在本文中所使用的,術語"雜芳族環"是指如上所定義的"芳族環",其中一個或多個碳原子獨立地被雜原子替換,上述雜原子選自由氮、氧和硫組成的組。除非另有說明,如在本文中所使用的,術語"取代的"是指,上述基團的一個或多個氫原子被另一非氫原子或官能團替換,條件是,維持正常價態以及取代導致穩定化合物。尤其是,根據本發明,非氫原子或官能團選自由與以下各項組成的組:烷基、取代的烷基、芳基、取代的芳基、芳烷基、烷氧基、環烷氧基、芳氧基、芳基烷氧基、羥基、雜芳基、雜芳氧基、雜環氧基、三氟甲基、三氟甲氧基、羧基、酰基、芳酰基、雜芳酰基、鹵素、硝基、氰基、烷氧基羰基、芳氧基羰基、芳基烷氧基羰基、環烷氧基羰基、雜芳氧基羰基、酰氧基、烷硫基、芳硫基、烷基亞硫酰基、芳基亞硫酰基、烷基磺酰基、芳基磺酰基、-O-芳酰基、-O-雜芳酰基、氧基(=O)、-C(=O)-NRhRk和-NRpRq,其中每個Rh、Rk、Rp和Rq獨立地表示氫、未取代的或取代的烷基、未取代的或取代的環烷基、未取代的或取代的芳基、未取代的或取代的芳烷基、未取代的或取代的雜芳基、未取代的或取代的雜環基、酰基、芳酰基、雜芳酰基,以及當Rh和Rk、或Rp和Rq連同它們所結合的氮原子一起時,基團-NRhRk或基團NRpRq表示雜環基殘基,并且其中,術語烷基、環烷基、芳基、雜芳基、雜環基是如上所定義的。術語"藥學上可接受的鹽"是指下面確定的式(I)的化合物的鹽,其保留所期望的生物活性并且由管理機構(regulatoryauthority)接受。如在本文中所使用的,術語"鹽"是指根據本發明制備自無機或有機酸或堿的化合物的鹽,以及內部形成的鹽。通常,這樣的鹽具有生理學上可接受的陰離子或陽離子。此外,式(I)的化合物可以形成酸加成鹽或堿鹽,這取決于取代基的種類,以及這些鹽包括在本發明中,只要它們是藥學上可接受的鹽。這樣的鹽的實例包括,但不限于與無機酸(例如,鹽酸、氫溴酸、硫酸、磷酸、硝酸等)形成的酸加成鹽,以及與以下有機酸形成的鹽,如乙酸、三氟乙酸、草酸、酒石酸、琥珀酸、蘋果酸、富馬酸、馬來酸、抗壞血酸、苯甲酸、藻酸、聚谷氨酸和萘磺酸。鹽酸鹽是優選的。生理或藥學上可接受的鹽特別適合于醫療應用,這是由于,相對于母體化合物,它們具有更大的水溶性。使用常規方法,藥學上可接受的鹽還可以制備自其它鹽,包括式(I)的化合物的其它藥學上可接受的鹽。術語"衍生物"是指式(I)的每種化合物,并且是指包括它們的藥學上可接受的水合物、溶劑化物、晶體形式、同位素標記衍生物、互變異構體、幾何或光學異構體、立體異構體、藥物活性衍生物,以及如下文所說明的任何合適的形式。有機化學領域的技術人員將理解,多種有機化合物可與溶劑(在其中它們進行反應或它們從其沉淀或結晶)形成復合物。這些復合物被稱為"溶劑化物"。例如,與水的復合物被稱為"水合物"。本發明的化合物的溶劑化物是在本發明的范圍之內。通過結晶或適當的溶劑的蒸發,可以連同溶劑分子一起容易地分離式(I)的化合物以給出相應的溶劑化物。式(I)的化合物可以是晶體形式(晶型,crystallineform)。在某些實施方式中,式(I)的化合物的晶體形式是多晶型物(polymorph)。本發明還包括同位素標記的化合物,其與在式(I)和下文中敘述的那些化合物相同,但由于以下事實而不同:一個或多個原子被具有不同于通常在自然界中發現的原子質量或質量數的原子質量或質量數的原子替換。可以加入本發明的化合物及其藥學上可接受的鹽的同位素的實例包括氫、碳、氮、氧、磷、硫、氟、碘和氯的同位素,如2H、3H、11C、13C、14C、15N、17O、18O、31P、32P、35S、18F、36Cl、123I、125I。本發明的化合物以及含有上述同位素和/或其它原子的其它同位素的所述化合物的藥學上可接受的鹽是在本發明的范圍內。本發明的同位素標記的化合物,例如其中加入放射性同位素,如3H、14C的那些化合物在藥物和/或底物組織分布檢測中是有用的。氚化,即3H和碳-14,即14C,同位素是特別優選的,因為它們易于制備和檢測。11C和18F同位素在P(T(正電子發射斷層掃描)中是特別有用的,以及125I同位素在SPECT(單光子發射計算機化斷層顯像)中是特別有用的,均可用于腦成像。此外,用較重同位素,如氘,即2H的取代,可以提供某些治療優勢,其來自更大的代謝穩定性,例如增加的體內半衰期或減小的劑量要求,因而在某些情況下可以是優選的。通過執行在方案中和/或在以下實施例中披露的程序,通過用容易獲得的同位素標記的試劑替換非同位素標記的試劑,一般可以制備本發明的式(I)和下文的同位素標記的化合物。包括在本發明中的某些基團/取代基可以存在為異構體,或者具有一種或多種互變異構形式。因此,在某些實施方式中,在一些情況下,式(I)的化合物可以存在為其它互變異構體或幾何異構體,這取決于取代基的種類。在本說明書中,可以僅以上述異構體的一種形式來描述化合物,但本發明包括所有上述異構體、異構體的分離形式或它們的混合物。此外,在一些情況下,式(I)的化合物可以具有不對稱碳原子或軸向不對稱,因此,相應地,它可以存在為旋光異構體如(R)形式、(S)形式等。本發明的范圍包括所有上述異構體,包括外消旋體、對映體和它們的混合物。尤其是,在本發明的范圍內包括所有立體異構形式,包括對映體、非對映異構體和它們的混合物,包括外消旋體,以及一般提及的式(I)的化合物包括所有立體異構形式(除非另有說明)。一般來說,本發明的化合物或鹽應解釋為排除那些(如果有的話),化學上非常不穩定的化合物(本身或在水中),以致它們顯然不適用于制藥用途(通過所有給予途徑,口服、胃腸道外或以其他方式)。對于熟練的化學家來說,上述化合物是已知的。術語"藥物活性衍生物"是指源自式(I)的化合物的任何化合物,其在給予受體以后能夠直接或間接提供本文披露的活性。本發明還包括式(I)的化合物的活性代謝物。具體實施方式根據本發明的第一方面,提供了式(I)的化合物或它們的藥學上可接受的鹽或溶劑化物。在式(I)的化合物中:R1是氫、直鏈或支鏈、未取代的或取代的C1-6烷基;R2選自由以下各項組成的組:直鏈或支鏈、未取代的或取代的C1-6烷基、C2-6烯基、C2-6炔基、未取代的或取代的C3-6環烷基、未取代的或取代的C3-6環烷基C1-6烷基、未取代的或取代的芳基、未取代的或取代的雜芳基、未取代的或取代的芳基C1-6烷基、未取代的或取代的芳氧基C1-6烷基、未取代的或取代的雜芳基C1-6烷基、未取代的或取代的雜芳氧基C1-6烷基、未取代的或取代的雜環烷基C1-6烷基、COR5;R5選自由以下各項組成的組:直鏈或支鏈未取代的或取代的C1-9烷基、未取代的或取代的C3-6環烷基、未取代的或取代的C3-6環烷基C1-6烷基、未取代的或取代的芳基、未取代的或取代的雜芳基、未取代的或取代的芳基C1-6烷基、未取代的或取代的芳氧基C1-6烷基、未取代的或取代的雜芳基C1-6烷基、未取代的或取代的雜芳氧基C1-6烷基、未取代的或取代的雜環烷基C1-6烷基。可替換地,R1和R2連同它們所連接的氮原子一起可以形成4至7元含有多達三個雜原子的氮雜環,其中上述雜原子選自氮和氧。Y1和Y2獨立地選自C或N。R3和R4獨立地選自由以下各項組成的組:氫、鹵素、直鏈或支鏈未取代的或取代的C1-6烷基、未取代的或取代的C1-6烷氧基、未取代的或取代的羥基C1-6烷基、羥基、氰基、硝基、未取代的或取代的氟C1-6烷基、未取代的或取代的氟C1-6烷氧基、氨基、單烷基氨基、二烷基氨基。根據第一實施方式:R1是氫或直鏈C1-6烷基、未取代的或者用一個或多個R6取代基取代的;R2選自由以下各項組成的組:未取代的或者用一個或多個R6取代基取代的直鏈或支鏈C1-6烷基、未取代的或者用一個或多個R6取代基取代的C3-6環烷基、未取代的或者用一個或多個R6取代基取代的C3-6環烷基C1-6烷基、未取代的或者用一個或多個R6取代基取代的芳基、未取代的或者用一個或多個R6取代基取代的雜芳基、未取代的或者用一個或多個R6取代基取代的芳基C1-6烷基、未取代的或者用一個或多個R6取代基取代的芳氧基C1-6烷基、未取代的或者用一個或多個R6取代基取代的雜芳基C1-6烷基、未取代的或者用一個或多個R6取代基取代的雜芳氧基C1-6烷基、未取代的或者用一個或多個R6取代基取代的雜環烷基C1-6烷基或COR5;R5選自由以下各項組成的組:未取代的或者用一個或多個R6取代基取代的直鏈或支鏈C1-9烷基、未取代的或者用一個或多個R6取代基取代的C3-6環烷基、未取代的或者用一個或多個R6取代基取代的C3-6環烷基C1-6烷基、未取代的或者用一個或多個R6取代基取代的芳基、未取代的或者用一個或多個R6取代基取代的雜芳基、未取代的或者用一個或多個R6取代基取代的芳基C1-6烷基、未取代的或者用一個或多個R6取代基取代的芳氧基C1-6烷基、未取代的或者用一個或多個R6取代基取代的雜芳基C1-6烷基、未取代的或者用一個或多個R6取代基取代的雜芳氧基C1-6烷基、未取代的或者用一個或多個R6取代基取代的雜環烷基C1-6烷基;R6選自由鹵素、羥基、烷氧基、氨基、單烷基氨基、二烷基氨基組成的組。可替換地,R1和R2連同它們所連接的氮原子一起可以形成4至7元含有多達兩個氮原子的氮雜環。Y1和Y2獨立地選自C或N;R3和R4獨立地選自由以下各項組成的組:氫、鹵素、直鏈或支鏈未取代的或取代的C1-6烷基、未取代的或取代的C1-6烷氧基、羥基、三氟甲基、氨基、單烷基氨基、二烷基氨基。根據第二實施方式:R1是氫或未取代的或者用一個或多個R6取代基取代的直鏈C1-4烷基;R2選自由以下各項組成的組:未取代的或者用一個或多個R6取代基取代的直鏈或支鏈C1-4烷基、未取代的或者用一個或多個R6取代基取代的C3-6環烷基、未取代的或者用一個或多個R6取代的C3-6環烷基C1-4烷基、未取代的或者用一個或多個R6取代基取代的芳基、未取代的或者用一個或多個R6取代基取代的雜芳基、未取代的或者用一個或多個R6取代基取代的芳基C1-4烷基、未取代的或者用一個或多個R6取代基取代的雜芳基C1-4烷基、未取代的或者用一個或多個R6取代基取代的雜環烷基C1-4烷基、COR5;R5選自由以下各項組成的組:未取代的或者用一個或多個R6取代基取代的直鏈或支鏈C1-7烷基、未取代的或者用一個或多個R6取代基取代的芳基C1-4烷基、未取代的或者用一個或多個R6取代基取代的芳氧基C1-4烷基、未取代的或者用一個或多個R6取代基取代的雜芳基C1-4烷基、未取代的或者用一個或多個R6取代基取代的雜芳氧基C1-4烷基、未取代的或者用一個或多個R6取代基取代的雜環基C1-4烷基;R6選自由鹵素、氨基、單烷基氨基、二烷基氨基組成的組。尤其是,R6可以選自由鹵素、二烷基氨基組成的組。可替換地,R1和R2連同它們所連接的氮原子一起可以形成含有一個氮原子的4至6元氮雜環。Y1和Y2獨立地選自C或N;R3和R4獨立地選自由以下各項組成的組:氫、鹵素、直鏈或支鏈未取代的或取代的C1-3烷基、未取代的或取代的C1-3烷氧基、羥基、三氟甲基。根據第三實施方式:R1是氫、未取代或者用一個R6取代基取代的正丙基和乙基、甲基;R2選自由以下各項組成的組:甲基、乙基、正丙基、異丙基、正丁基、異丁基、環丙基、環丙基甲基、苯基、芐基、4-吡啶基甲基、哌啶-l-基丙基、嗎啉-4-基-丙基、4-甲基哌嗪-l-基-丙基或COR5;R5選自由以下各項組成的組:未取代或者用一個R6取代基取代的甲基、乙基、正丙基、異丙基、異丁基、1-丙基丁基、苯氧基甲基;未取代的或者用一個R6取代基取代的雜芳氧基乙基;未取代的或者用一個R6取代基取代的雜環烷基丁基;R6選自由氟和二烷基氨基組成的組。可替換地,R1和R2連同它們所連接的氮原子一起可以形成氮雜環,其選自氮雜環丁烷、吡咯烷或哌啶環系統。Y1和Y2獨立地選自C或N;R3和R4獨立地選自由氫、氟和甲基組成的組。根據本發明的第四實施方式,式(I)的化合物可以選自由下述化合物組成的組:4-(4-氟苯基)-6-(甲基氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(乙基氨基)-4-(鄰甲苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(乙基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(乙基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(乙基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-(丙基氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-(異丙基氨基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-(異丁基氨基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-(苯基氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(二甲基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;6-(二乙基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-(哌啶-l-基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;6-(丁基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(乙基氨基)-4-(吡啶-3-基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-(吡咯烷-l-基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)乙酰胺;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)-2-丙基戊酰胺;5-(1,2-二硫戊環-3-基)-N-(4-(4-氟苯基)-6-氧代-1,4,5,6-四氫-l,3,5-三嗪-2-基)戊酰胺;3-((lH-苯并[d][1,2,3]三唑-l-基)氧基)-N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)丙酰胺;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)-2-苯氧基乙酰胺;2-(4-氟苯氧基)-N-(4-(4-氟苯基)-6-氧代-1,4,5,6-四氫-l,3,5-三嗪-2-基)乙酰胺;6-((3-(二甲基氨基)丙基)氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-((3-(哌啶-l-基)丙基)氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(芐基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-((吡啶-4-基甲基)氨基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;6-(二丙基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。以下化合物可以借助用于上述化合物的相同的方法進行合成:6-(環丙基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;6-((環丙基甲基)氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;6-((3-(二乙基氨基)丙基)氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-((3-嗎啉代丙基)氨基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;4-(4-氟苯基)-6-((3-(4-甲基哌嗪-l-基)丙基)氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮;6-(氮雜環丁烷-l-基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)丙酰胺;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)丁酰胺;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)異丁酰胺;N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)-3-甲基丁酰胺。使用以下一般方法和程序,例如在MichaelSmith,JerryMarch-March'sAdvancedOrganicChemistry:reactionsmechanismsandstructure–第6版,JohnWiley&SonsInc.,2007中舉例說明的,本發明中舉例說明的化合物可以容易制備自可用的起始材料。本領域普通技術人員公知的是,化學官能(chemicalfunction)到另一官能的轉化可能要求在含有此官能團的化合物中的一個或多個反應中心被保護,以避免不希望的副反應。上述反應中心的保護,以及在合成轉化的結束時的隨后的去保護,可以按照,例如,在TheodoraW.GreenandPeterG.M.Wuts-ProtectiveGroupsinOrganicSynthesis,第4版,JohnWiley&SonsInc.,2006中描述的標準程序來完成。將可以理解的是,在給出了典型或優選的實驗條件(即,反應溫度、時間、試劑的摩爾數、溶劑等)的情況下,除非另有說明,也可以使用其它實驗條件。最佳反應條件可隨所用的具體反應物或溶劑而變化,但使用常規優化程序,本領域技術人員可以確定這樣的條件。按照以下描述的合成方法,可以以逐步的方式來進行式(I)的化合物的合成,由此,在進行后續反應以前,通過標準純化技術,如,例如柱層析法,來分離和純化每種中間體。可替換地,可以以所謂的"一鍋法"方法,如本領域中已知的,來進行合成序列的兩個或更多個步驟,由此僅來自兩個或更多個步驟的化合物被分離和純化。可以通過常規技術或方式,例如通過過濾、蒸餾、層析、再結晶和它們的組合,來處理或純化借助于下文描述的方法所制備的式(I)的化合物。可以通過在溶液中使堿性化合物和所期望的酸反應來制備式(I)的化合物的鹽。一般方法在一種實施方式中,可以通過應用在文描述的方案中報道的化學轉化來獲得式(I)的化合物。特別是,當R1是氫、直鏈或支鏈C1-6烷基時,R2是直鏈或支鏈未取代的或取代的C1-6烷基、C2-6烯基、C2-6炔基、未取代的或取代的C3-6環烷基或C3-6環烷基C1-6烷基、未取代的或取代的芳基、雜芳基、芳基C1-6烷基、雜芳基C1-6烷基、雜環基C1-6烷基,或者R2連同R1一起形成C4-7氮雜環,其包括R1和R2所連接的氮原子,以及R3、R4、Y1和Y2是如上文在式(I)中所定義的時,可以按照方案1來合成式(I)的化合物。(P27)(丁醇;步驟;回流;分鐘;)式(I)的化合物可以通過在可商購式(II)的苯甲醛(其中R3、R4、Y1和Y2是如上所定義的)和式(III)的取代的脒基脲(其中R1和R2是如上文在式(I)中所定義的)之間的縮合反應來制備[Ostrogovich,A.,GazzettaChimicaItaliana1909,39,540-549]。作為一般方法,將式(II)和(III)的化合物溶解于濃H2SO,并在室溫下攪拌得到的反應混合物72小時,從而提供式(I)的化合物,作為在三嗪酮環的C4處的外消旋體。如上所定義的式(III)的脒基脲(guanylurea)是可商購的或可以在70%H2SO4中在回流下加熱45分鐘通過化學式(IV)的氰基胍的酸催化水合加以制備,其中R1和R2是如上文在式(I)中所定義的。化學式(IV)的氰基胍是可商購的或可以通過在1-丁醇中,在回流下加熱可商購的化學式(V)的二氰胺鈉和化學式(VI)的伯胺或仲胺鹽酸鹽6-8小時加以制備,其中R1和R2是如上文在式(I)中所定義的。此外,當R1是氫、直鏈或支鏈C1-6烷基,R2是COR5以及R3、R4、R5、Y1和Y2是如上文在式(I)中所定義的時,可以按照方案2來合成式(I)的化合物。式(I)的化合物可以通過化學式(VII)的酰氯或羧酸(其中X分別是氯或/和羥基,以及R5是如在式(I)中所定義的)和化學式(VIII)的中間體,(其中R1、R3、R4、Y1和Y2是如上文在式(I)中所定義的)之間的偶聯反應來制備。作為一般方法,當X是氯時,利用有機溶劑的混合物,例如吡啶/DCM、DMF/DCM、2,6-二甲基吡啶/DMF,在室溫下攪拌3-4小時來進行偶聯反應。當X是羥基基團時,在DMF或DCM中,在活化劑,如EDCI.HC1或EDCI.HCl/HOBt和堿,優選DIPEA或Et3N的存在下,在室溫下攪拌過夜來進行偶聯反應。可以按照先前在方案1中描述的方法來合成如上所定義的化學式(VIII)的化合物。可以借助于通過手性HPLC的分離來獲得式(I)的化合物或式(VIII)的化合物的單對映體(singleenantiomer)。本發明的第二方面涉及藥物組合物,該組合物包含如上文披露的式(I)的化合物及其藥學上可接受的載體、穩定劑、稀釋劑或賦形劑。本領域技術人員知道適用于配制藥物組合物的各種這類載體、稀釋劑或賦形劑化合物。可以將本發明的化合物連同常規使用的佐劑、載體、稀釋劑或賦形劑一起配制成藥物組合物以及其單位劑量的形式,并且這樣的形式可以用作固體,如片劑或填充的膠囊劑,或液體,如溶液、混懸劑、乳劑、酏劑,或填充有藥物組合物的單位劑量的形式的膠囊劑,均用于口服使用,或用于胃腸道外給予(包括皮下和靜脈內使用)的無菌注射溶液的形式。這樣的藥物組合物以及其單位劑型(劑量形式,dosageform)可以包含常規比例的組分,有或沒有另外的活性化合物或成分,并且這樣的單位劑型可以含有任何合適的有效量的活性組分,其相當于待采用的預期日劑量范圍。含有本發明的化合物的藥物組合物可以以在制藥領域中已知的方式進行制備,并且包含至少一種活性化合物。通常,以藥學有效量來給予本發明的化合物。通常由醫生,根據有關情況,包括待治療的病癥、選擇的給予途徑、給予的實際化合物、個體患者的年齡、體重和反應、患者癥狀的嚴重程度等,來確定實際給予的化合物的量。本發明的藥物組合物可以通過各種途徑,包括口服途徑、直腸途徑、皮下途徑、靜脈內途徑、肌內途徑、鼻內途徑和肺途徑給予。用于口服給予的組合物可以采取本體液體溶液(bulkliquidsolution)或混懸劑(懸浮液,suspension)或整裝散劑(散裝粉末,bulkpowder)的形式。然而,更常見地,以單位劑型來呈現組合物以方便精確計量。術語"單位劑型(單位劑量形式,unitdosageform)"是指適合作為用于人主體和其它哺乳動物的單位劑量的物理上分離的單位,每個單位含有經計算會產生所需的治療效果的預定量的活性材料,并聯合與適宜的藥物賦形劑。典型的單位劑型包括液體組合物的預填充的、預先測量的針劑或注射器,或丸劑、片劑、膠囊劑等(在固體組合物的情況下)。適用于口服給予的液體形式可以包括適宜的水性或非水性載體,其含有緩沖劑、懸浮和分散劑、著色劑、調味劑等。固體形式可以包括,例如,任何以下組分或類似特性的化合物:粘合劑,如微晶纖維素、黃芪膠或明膠;賦形劑,如淀粉或乳糖;崩解劑,如藻酸,原生凝膠(羧甲基淀粉鈉,Primogel)或玉米淀粉;潤滑劑,如硬脂酸鎂;助流劑,如膠態二氧化硅;甜味劑,如蔗糖或糖精;或調味劑,如辣薄荷、水楊酸甲酯或橙味味香精(橙味調味劑,orangeflavouring)。可注射組合物通常基于注射無菌鹽水或磷酸鹽緩沖液或在本領域中已知的其它可注射載體。藥物組合物可以以下形式:片劑、丸劑、膠囊劑、溶液、混懸劑、乳劑、散劑(powder,粉末)、栓劑以及作為持續釋放制劑。如果需要,可以通過標準水性或非水性技術來涂布片劑。在某些實施方式中,這樣的組合物和制劑可以含有至少0.1百分比的活性化合物。在這些組合物中活性化合物的百分比當然可以變化,并且可以方便地為約1百分比至約60百分比的單位的重量。在這樣的治療有效組合物中,活性化合物的量是使得達到治療活性劑量的量。還可以以鼻內途徑,作為,例如液滴或噴霧劑來給予活性化合物。片劑、丸劑、膠囊劑等還可以含有粘合劑,如黃芪膠、阿拉伯膠、玉米淀粉或明膠;賦形劑,如磷酸二鈣;崩解劑,如玉米淀粉、土豆淀粉、藻酸;潤滑劑,如硬脂酸鎂;以及甜味劑,如蔗糖、乳糖或糖精。當單位劑型是膠囊劑時,除上述類型的材料之外,它還可以含有液體載體,如脂肪油。各種其它材料可以存在為涂層或用來改變劑量單位的物理形式。例如,片劑可以涂布有蟲膠(shellac)、糖或兩者。除活性組分之外,糖漿劑或酏劑還可以含有作為甜味劑的蔗糖、作為防腐劑的尼泊金甲酯和尼泊金丙酯、染料和調味劑,如櫻桃或橙味香精。為了在通過胃腸道的上部的運輸過程中防止分解,組合物是腸溶衣制劑。用于肺部給予的組合物包括,但不限于干粉組合物,其由式(I)的化合物或其鹽的粉末和適宜的載體和/或潤滑劑的粉末組成。用于肺部給予的組合物可以吸入自本領域技術人員已知的任何適宜的干粉吸入器裝置。根據治療方案且以足以在主體中減少炎癥和疼痛的劑量,來給予組合物。在一些實施方式中,在本發明的藥物組合物中,通常以劑量單位來配制活性成分。對于每日給予,劑量單位可以含有0.1至1000mg的式(I)的化合物/劑量單位。在一些實施方式中,對于具體制劑的有效的量將取決于疾病、病癥或病情的嚴重性、以前的治療、個體的健康狀況和對藥物的反應。在一些實施方式中,按重量計,劑量為制劑的0.001%至約60%。當連同一種或多種其它活性組分一起使用時,可以以比當單獨使用每種時更低的劑量來使用本發明的化合物和其它活性組分。關于制劑,考慮到任何種類的給予途徑、用于藥物的給予的方法和制劑,披露于Remington'sPharmaceuticalSciences,第17版,Gennaroetal.Eds.,MackPublishingCo.,1985,andRemington'sPharmaceuticalSciences,GennaroARed.第20版,2000,Williams&WilkinsPA,USA,andRemington:TheScienceandPracticeofPharmacy,第21版,LippincottWilliams&WilkinsEds.,2005;andinAnsel'sPharmaceuticalDosageFormsandDrugDeliverySystems,第8版,LippincottWilliams&WilkinsEds.,2005。用于口服給予或可注射組合物的上述成分僅僅是代表性的。還可以以持續釋放劑型或從持續釋放藥物遞送系統來給予本發明的化合物。此外,根據本發明的藥物組合物可以包含第二治療劑,例如神經保護劑或用于阿爾茨海默病治療的已知的試劑,優先選自但不限于(4aS,6R,8aS)-5,6,9,10,11,12-六氫-3-甲氧基-11-甲基-4aH-[1]苯并呋喃并[3a,3,2-ef][2]苯并氮雜-6-醇,還稱為加蘭他敏(galatamine),(S)-3-[1-(二甲基氨基)乙基]苯基N-乙基-N-甲基氨基甲酸酯,還稱為利伐斯的明(卡巴拉汀,rivastigmine),(RS)-2-[(1-芐基-4-哌啶基)甲基]-5,6-二甲氧基-2,3-二氫茚-l-酮,還稱為多奈哌齊,以及3,5-二甲基金剛烷-l-胺,還稱為美金剛(美金胺,memantine)。本發明的第三方面涉及作為藥物的上文披露的式(I)的化合物或其藥物組合物或它們的藥學上可接受的鹽或溶劑化物。事實上,BACE-1和GSK-3β酶的雙重抑制劑可以在各種各樣的中樞神經系統的病理性病癥中找到治療研究。尤其是,這些化合物可用來治療或預防中樞神經系統疾病或病癥,更具體地認知的、神經退行性的或神經元的疾病或病癥。上述疾病或病癥優先選自,但不限于慢性神經退行性病癥,包括癡呆,如阿爾茨海默病、帕金森病、全腦炎帕金森病(panencephaliticparksinsonism)、腦炎后帕金森病、皮克病、皮質基底節變性、亨廷頓病、艾滋病相關性癡呆、癌癥相關性癡呆、2型糖尿病相關認知病癥和2型糖尿病相關性癡呆、肌萎縮側索硬化、多發性硬化和神經損傷疾病,其選自急性卒中和卒中后功能康復、癲癇、情緒病癥,其選自由抑郁癥、精神分裂癥和雙相病癥(bipolardisorders)組成的組、腦出血如由于孤立性腦淀粉樣血管病引起的腦出血、運動神經元疾病、輕度至重度認知病癥、局部缺血、頭部創傷、腦損傷,尤其是創傷性腦損傷、唐氏綜合征、路易體疾病、炎癥和慢性炎性疾病。此外,這些化合物可以在tau蛋白病的領域中找到應用,其中上述tau蛋白病是一類與tau蛋白的病理聚集關聯的神經退行性疾病。tau的過磷酸化可以負責蛋白從神經元微管的分離,從而產生細胞內聚集體,還被稱為神經原纖維纏結(neurofibrillarytangle)(NFT)。阿爾茨海默病本身是tau蛋白病(tau蛋白病理,tau)的實例,然而是過多的神經退行性疾病與更廣泛的tau蛋白的病理聚集關聯。尤其是,本發明的化合物類別可以應用于治療以下疾病:進行性核上麻痹;拳擊員癡呆(慢性創傷腦病);額顳癡呆和帕金森病,其與17號染色體相關;利替可-波帝格(Lytico-Bodig病)(關島帕金森-癡呆綜合征);纏結為主型癡呆,具有類似于阿爾茨海默病的NFT,但沒有斑塊;神經節膠質瘤和神經節細胞瘤;腦膜瘤血管瘤病;亞急性硬化性全腦炎。此外,鑒于本發明的化合物減少淀粉樣蛋白斑塊的形成的能力,它們還可以治療淀粉樣變性(amyloidosis)。淀粉樣變性是指各種病癥,其中通常可溶性蛋白變得不溶,并且沉積在各種器官或組織的細胞外空間中,從而破壞正常功能。在這些病理性病癥中,本發明的化合物可以治療以下疾病:老年性系統性淀粉樣變性;朊病毒蛋白相關疾病,包括克-雅病;腦淀粉樣血管病;家族性角膜淀粉樣變性、具有荷蘭型的淀粉樣變性遺傳性腦出血。尤其是,式(I)的化合物可以用來治療或預防疾病或病癥,其選自:阿爾茨海默病,帕金森病,亨廷頓病,肌萎縮側索硬化,多發性硬化,神經損傷疾病,如急性卒中和卒中后功能康復,癲癇,選自由抑郁癥、精神分裂癥和雙相病癥組成的組的情緒病癥,腦出血,局部缺血,輕度至重度認知病癥,頭部創傷,腦損傷,尤其是創傷性腦損傷,炎癥和慢性炎性疾病,癡呆,其選自由艾滋病相關性癡呆、癌癥相關性癡呆、2型糖尿病相關性癡呆、拳擊員癡呆、額顳癡呆、關島帕金森-癡呆綜合征、纏結為主型癡呆組成的組。在下文中,將通過一些實施例來說明本發明,其不被解釋為限制本發明的范圍。下文在所附的實施例中使用以下縮寫:乙酸(AcOH)、乙腈(MeCN)、乙酰氯(AcCl)、氨(NH3)、乙酸銨(NH4OAc)、氘代氯仿(CDC13)、氘代二甲亞砜(DMSO-d6)、氘代甲醇(CD3OD)、氧化氘(D2O)、二氯甲烷(DCM)、二乙醚(Et2O)、N,N-二異丙基乙胺(DIPEA)、l-(3-二甲基氨基丙基)-3-乙基碳二亞胺鹽酸鹽(EDCI·HCl)、二甲基甲酰胺(DMF)、二甲亞砜(DMSO)、乙醇(EtOH)、乙酸乙酯(EtOAc)、鹽酸(HC1)、羥基苯并三唑水合物(HOBt)、氯化鋰(LiCl)、甲醇(MeOH)、室溫(rt)、碳酸鈉(Na2CO3)、氫氧化鈉(NaOH)、硫酸鈉(Na2SO4)、硫酸(H2SO4)、三乙胺(Et3N)、三氟乙酸(TFA)、水(H2O)。化學品、材料和方法使用購買自Sigma-Aldrich,Fluka(意大利)的所有商業可用的試劑和溶劑,而沒有進一步純化。在快速條件(flashcondition)下,利用SigmaAldrich硅膠級9385,230-400目,進行柱層析純化。用0.20mm硅膠60F254板(Merck,德國)進行薄層色譜法(TLC),其是通過暴露于紫外光(254和366nm)和高錳酸鉀染色,加以可視化。通過利用溴甲酚綠噴顯劑(0.04%,在EtOH中,通過NaOH變成藍色),接著加熱上述板,來可視化涉及胺的產生或消耗的反應。遵循由ChemBioDrawUltra(版本13.0)所用的IUPAC規則來命名化合物。在VarianVXR200或BrukerAvanceIII400(對于1H為200或400MHz;50或對于13C為100MHz)上進行核磁共振(NMR)實驗。利用DMSO-d6、CD3OD、D20或CDC13作為溶劑,在300K下獲得光譜。利用殘留的非氘化溶劑作為內部標準,以百萬分之幾(ppm)為單位,來記錄1H和13C光譜的化學位移。數據報道如下:化學位移(ppm),多重性(表示為:s,單峰;brs,寬單峰;exch,可與D2O交換的質子;d,雙峰;t,三重峰;q,四重峰;m,多重峰以及它們的組合),以赫茲(Hz)為單位的耦合常數(J),以及積分強度。用WatersACQUITYUPLC-MS系統進行UPLC-MS分析,該系統由配備有電霧化電離(ESI)界面的SQD(單四極檢測器)質譜儀(MS)和光電二極管陣列檢測器(PDA)構成。PDA范圍是210-400nm。用ACQUITYUPLCHSST3C18柱(50mmx2.1mm內徑,顆粒尺寸1.8μm)進行分析,該柱具有VanGuardHSST3C18前置柱(預柱,pre-column)(5mmx2.1mm內徑,顆粒尺寸1.8μm)。流動相是在H2O中的10mMNH4OAc,在pH5下,用AcOH調節(A)或在MeCN-H2O(95:5)中的10mMNHOAc,在pH5下(B)。在質量掃描范圍100-500Da內,施加正離子和負離子模式(正和負模式,positiveandnegativemode)的ESI。用WatersAllianceHPLC儀器(其由e2695分離模塊和2998PDA構成)來進行通過手性HPLC的分析。PDA范圍是210-400nm。用DaicelChiralPakAD柱(250mmx4.6mm內徑,顆粒尺寸10μm)進行等度分析。流動相是0.1%TFA庚烷/EtOH(90:10)。用WatersAllianceHPLC儀器(其由1525二元HPLC泵、水分收集器III和2998PDA構成)來進行通過手性HPLC制備(preparativechiralHPLC)的分離。UV檢測是在215nm處。用DaicelChiralPakAD柱(250mmx10mm內徑,顆粒尺寸10μm)進行等度純化。流動相是0.1%TFA庚烷/EtOH(90:10)。用RudolfResearchAnalyticalAutopolII自動旋光儀來測量旋光度,其中使用鈉燈(589nm)作為光源,濃度以g/100mL為單位來表示,使用MeOH作為溶劑和1dm測定池(cell)。通過NMR和UPLC-MS分析,所有最終化合物表現出>95%的純度。為了更好地說明本發明,提供了在圖1中報道的實例化合物的合成。如下所述,合成了在圖1中報道的化合物。制備和實施例制備I(實施例1-15,22-26)式(I)的化合物的一般合成,其中R1是氫、直鏈或支鏈未取代的或取代的C1-6烷基,R2是直鏈或支鏈未取代的或取代的C1-6烷基、未取代的或取代的芳基、未取代的或取代的雜芳基、未取代的或取代的芳基C1-6烷基、未取代的或取代的雜芳基C1-6烷基、未取代的或取代的雜環基C1-6烷基,或R1和R2連同它們所連接的氮原子一起可以形成4至7元含有多達三個雜原子的氮雜環,其中上述雜原子選自氮和氧,以及R3、R4、Y1和Y2是如在式(I)中所定義。一般方法(A):式(IV)的中間體的合成-步驟1,方案1。向雙氰胺鈉(V)(1.2當量)在1-丁醇(1.2M)中的懸浮液,添加適當的胺鹽酸鹽(VI)(1.0當量)。在回流下加熱得到的混合物6-8小時,從而提供白色沉淀物,將其過濾掉。在真空下濃縮濾液以產生粗氰基胍(IV)。當需要時,通過快速色譜法,進行進一步純化。(IVa)N-氰基-N'-甲基胍按照一般方法A,并使用甲胺鹽酸鹽(0.63g,9.30毫摩爾)來獲得標題化合物。借助于Et2O來研制(Triturate)原料,從而提供作為黃色油狀物(yellowoil)的Iva,其用于下一步驟而沒有進一步純化:0.72g(77%)。1H-NMR(CD3OD,400MHz)δ2.74(s,3H)。13C-NMR(CD3OD,100MHz)δ28.4,120.2,163.4。(IVb)N-氰基-N'-乙基胍按照一般方法A,并使用乙胺鹽酸鹽(2.29g,27.07毫摩爾)來獲得標題化合物。借助于DCM/MeOH/33%NH3水性溶液(9:1:0.1)的洗脫提供作為黃色油狀物的IVb:2.5g(73%)。1H-NMR(D20,400MHz)δ1.19(t,J=6.8,3H),3.23(q,J=6.8,2H)。13C-NMR(D20,100MHz)δ13.3,36.5,120.5,161.1。(IVc)N-氰基-N'-丙基胍按照一般方法A并使用丙胺鹽酸鹽(2.30g,24.53毫摩爾)來獲得標題化合物。借助于DCM/MeOH/33%NH3水溶液(9:1:0.1)的洗脫提供作為蠟狀固體的IVc:1.9g(63%)。1H-NMR(CD3OD,400MHz)δ0.90(t,J=7.2,3H),1.49-1.54(m,2H),3.09(t,J=6.8,2H)。13C-NMR(CD3OD,100MHz)δ10.1,22.2,42.8,118.8,161.7。(IVd)N-氰基-N'-異丙基胍按照一般方法A并使用異丙胺鹽酸鹽(0.89g,9.30毫摩爾)來獲得標題化合物。借助于Et2O和DCM來研制原料,從而提供作為白色固體的IVd,其用于下一步驟而沒有進一步純化:0.80g(69%)。1H-NMR(CD3OD,400MHz)(旋轉異構體的4.5:1混合物)δ1.12(d,J=6.0,1.1H,2CH3次要(minor)),1.29(d,J=6.0,4.9H,2CH3主要(major)),3.37-3.43(m,0.8H,CH(major)主要),3.52-3.58(m,0.2,CH次要)。13C-NMR(CD3OD,100MHz)δ19.6(2CH3主要),21.6(2CH3次要),43.3(CH次要),43.9(CH主要),119.1,165.3。(IVe)N-氰基-N'-異丁基胍按照一般方法A,使用異丁胺鹽酸鹽(1.02g,9.30毫摩爾)來獲得標題化合物。借助于Et2O和DCM來研制原料,從而提供作為蠟狀固體的IVe,其用于下一步驟而沒有進一步純化:1.23g(95%)。1H-NMR(CD3OD,400MHz)δ0.88(d,J=6.8,6H),1.75-1.78(m,1H),2.95(d,J=7.2,2H)。13C-NMR(CD3OD,100MHz)δ19.0,28.2,48.4,119.1,161.7。(IVf)N-氰基-N'-苯基胍按照一般方法A,使用苯胺鹽酸鹽(0.65g,5.05毫摩爾)來獲得標題化合物。借助于H2O來研制原料,以提供作為白色固體的IVf,其用于下一步驟而沒有進一步純化:0.80g(定量產率)。1H-NMR(DMSO-d6,400MHz)δ6.98(brs,exch,2H),7.05-7.09(m,1H),7.28-7.35(m,2H),9.03(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ117.6,121.7,124.2,129.2,138.4,159.9。(IVg)N-氰基-N'-二甲基胍按照一般方法A,使用二甲胺鹽酸鹽(1.52g,18.72毫摩爾)來獲得標題化合物。借助于DCM來研制原料,從而提供作為白色固體的IVg,其用于下一步驟而沒有進一步純化:0.40g(18%)。1H-NMR(CD3OD,400MHz)δ2.77(s,6H)。13C-NMR(CD3OD,100MHz)δ36.5,119.0,161.1。(IVh)N-氰基-N',N'-二乙基胍按照一般方法A,使用二乙胺鹽酸鹽(1.02g,9.30毫摩爾)來獲得標題化合物。借助于Et2O來研制原料,從而提供作為黃色固體的IVh,其用于下一步驟而沒有進一步純化:0.95g(73%)。1H-NMR(CD3OD,400MHz)(旋轉異構體的1.5:1混合物)δ1.13(t,J=6.8,3.6H,2CH3主要),1.30(t,J=6.8,2.4H,2CH3次要),3.04(q,J=6.8,1.8H,2CH2次要),3.5(q,J=6.8,2.2H,2CH2主要)。13C-NMR(CD3OD,100MHz)δ10.3(2CH3次要),12.0(2CH3主要),42.2(2CH2次要),42.5(2CH2主要),119.3,159.8。(IVi)N-氰基-l-哌啶甲脒按照一般方法A并使用哌啶鹽酸鹽(1.13g,9.30毫摩爾)來獲得標題化合物。借助于DCM來研制原料,從而提供作為白色固體的IVi,其用于下一步驟而沒有進一步純化:0.46g(33%)。1H-NMR(CD3OD,400MHz)δ1.52-1.57(m,4H),1.62-1.67(m,2H),3.44-3.46(m,4H)。13C-NMR(CD3OD,100MHz)δ23.7,25.2,45.7,119.2,159.9。(IVj)N-氰基-N'-丁基胍按照一般方法A,使用丁胺鹽酸鹽(1.02g,9.30毫摩爾)來獲得標題化合物。借助于DCM來研制原料,從而提供作為膠狀固體(gummysolid)的IVj,其用于下一步驟而沒有進一步純化:1.12g(86%)。1H-NMR(DMSO-d6,400MHz)δ0.83-0.88(m,3H),1.22-1.27(m,2H),1.29-1.39(m,2H),3.00-3.02(m,2H)。13C-NMR(DMSO-d6,100MHz)δ14.0,19.8,31.5,40.11,118.8,161.6。(IVk)N-氰基-l-吡咯烷甲脒按照一般方法A,使用吡咯烷鹽酸鹽(1.00g,9.30毫摩爾)來獲得標題化合物。借助于Εt20來研制原料,從而提供作為淺黃色固體的IVk,其用于下一步驟而沒有進一步純化:0.97g(76%)。1H-NMR(DMSO-d6,400MHz)δ1.76-1.78(m,4H),3.20-3.23(m,4H)。13C-NMR(DMSO-d6,100MHz)δ25.3,47.2,118.8,159.0。(IVI)N-氰基-N'-(二甲基丙烷-3-氨基)胍按照一般方法A,使用N1-N1-二甲基丙烷-l,3-二胺二鹽酸鹽(0.51g,2.90毫摩爾)來獲得標題化合物。用40mL的MeOH/2NHCl水溶液(1:0.5)來吸收(takenup)原料,分為兩部分,加載到兩個2gISOLUTESCX-2柱,然后用MeOH/33%NH3水性溶液(1:0.1)(2x15mL)加以洗脫。在真空下濃縮有機相,從而提供作為膠狀固體的IV1,其用于下一步驟而沒有進一步純化:0.10g(21%)。1H-NMR(CD3OD,400MHz)δ1.68-1.76(m,2H),2.24(s,6H),2.33-2.39(m,2H),3.16-3.22(m,2H)。13C-NMR(CD3OD,100MHz)δ26.8,38.4,44.0,56.1,118.7,160.1。(IVm)N-氰基-N'-(3-(哌啶-l-基)丙烷)胍按照一般方法A,使用3-(哌啶-l-基)丙烷-l-胺二鹽酸鹽(0.51g,2.36毫摩爾)來獲得標題化合物。用20mL的Na2CO3飽和水性溶液來吸收原料并用EtOAc(3x20mL)加以提取。收集有機層,經無水Na2SO4干燥,過濾并真空蒸發。用快速色譜法來純化殘余物,用DCM/MeOH/33%NH3水性溶液(8:2:0.2)加以洗脫。獲得作為膠狀固體的IVm:0.34g(45%)。1H-NMR(CD3OD,400MHz)δ1.44-1.48(m,2H),1.59-1.65(m,4H),1.70-1.77(m,2H),2.44-2.56(m,6H),3.17(t,J=6.4,2H)。13C-NMR(CD3OD,100MHz)δ23.4,24.7,25.5,39.2,53.8,55.5,118.7,161.8。(IVn)N-氰基-N'-芐基胍按照一般方法A,使用芐胺鹽酸鹽(1.34g,9.36毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.1)進行的洗脫提供作為膠狀固體的IVn:0.81g(50%)。1H-NMR(CD3OD,400MHz)4.30(s,2H),7.18-7.29(m,5H)。13C-NMR(CD3OD,100MHz)δ44.7,119.0,127.0,127.2,128.4,138.1,161.7。(IVo)N-氰基-N'-吡啶-4-基甲基胍按照一般方法A,使用吡啶-4-基甲胺二鹽酸鹽(1.69g,9.36毫摩爾)來獲得標題化合物。利用DCM/MeOH/33%NH3水性溶液(8:2:0.2)進行的洗脫,提供作為膠狀固體的IVo:0.93g(57%)。1H-NMR(CD3OD,400MHz)4.40(s,2H),7.25(d,J=5.6,2H),8.39(d,J=5.6,2H)。13C-NMR(CD3OD,100MHz)δ43.4,118.7,122.2,147.6,148.8,161.9。(IVp)N-氰基-N',N'-二丙基胍按照一般方法A,使用二丙胺鹽酸鹽(0.94g,9.30毫摩爾)來獲得標題化合物。用EtOAc來吸收原料并用水洗滌。在真空下濃縮有機層,從而提供作為無色油狀物的IVp,其用于下一步驟而沒有進一步純化:0.76g(50%)。1H-NMR(CD3OD,400MHz)δ0.88(t,J=7.2,6H),1.55-1.60(m,4H),3.24(t,J=7.6,4H)。13C-NMR(CD3OD,100MHz)δ10.2,20.6,49.8,119.4,160.3。一般方法(B):式(III)的中間體的合成-步驟2,方案1。向氰基胍(IV)(1當量)在水(1.2M)中的溶液添加70%含水硫酸(2當量)。在室溫下攪拌得到的混合物15分鐘,然后在回流下加熱1小時。然后將反應混合物冷卻至室溫,并用Na2CO3堿化。在真空中蒸發水相,然后用MeOH來吸收原料。過濾掉殘余物并在真空下濃縮有機溶劑,以提供化合物III,其用于下一步驟而沒有進一步純化。(IIIa)(N'-甲基甲脒基)脲按照一般方法B,使用IVa(0.77g,7.12毫摩爾)來獲得標題化合物。獲得作為黃色油狀物的IIIa:0.66g(83%)。1H-NMR(CD3OD,200MHz)δ2.80(s,3H)。13C-NMR(CD3OD,50MHz)δ26.1,161.2,166.9。(IIIb)(N'-乙基甲脒基)脲按照一般方法B,使用IVb(2.50g,20.46毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIIb:3.90g(定量產率)。1H-NMR(D20,400MHz)δ0.48(t,J=6.8,3H),2.58(q,J=6.8,2H)。13C-NMR(D20,100MHz)δ12.1,35.8,152.8,155.2。(IIIc)(N'-丙基甲脒基)脲按照一般方法B,使用IVc(1.80g,14.26毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIIc:2.40g(定量產率)。1H-NMR(D20,400MHz)δ0.49-0.54(m,3H),1.14-1.25(m,2H),2.71(t,J=6.8,1H),2.83(t,J=6.8,1H)。13C-NMR(D20,100MHz)δ10.1,21.1,42.6,155.7,156.3。(IIId)(N'-異丙基甲脒基)脲按照一般方法B,使用IVd(0.80g,6.39毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIId:0.50g(54%)。1H-NMR(CD3OD,400MHz)(旋轉異構體的1.1:1混合物)δ1.25(d,J=6.8,3.1H,2CH3主要),1.29(d,J=6.8,2.9H,2CH3次要),3.39-3.46(m,0.48H,CH次要),3.81-3.88(m,0.52H,CH主要)。13C-NMR(CD3OD,100MHz)δ19.6(2CH3主要),21.0(2CH3次要),43.9(CH主要),44.0(CH次要),153.3,155.6。(IIIe)(N'-異丁基甲脒基)脲按照一般方法B,使用IVe(1.23g,8.77毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIIe:1.39g(定量產率)。1H-NMR(CD3OD,400MHz)(旋轉異構體的2.3:1混合物)δ0.23(d,J=5.2,4.2H,2CH3主要),0.42(d,J=5.2,1.8H,2CH3次要),0.51-0.53(m,0.3H,CH次要),1.16-1.18(m,0.7H,CH主要),2.39(d,J=6.8,0.6H,CH2次要)2.87(d,J=6.8,1.4H,CH2主要)。13C-NMR(CD3OD,100MHz)δ16.2(2CH3次要),18.0(2CH3要),26.7(CH主要+次要),47.4(CH2次要),47.5(CH2主要)153.6,154.6。(IIIf)(N'-苯基甲脒基)脲按照一般方法B,使用IVf(0.80g,4.90毫摩爾)來獲得標題化合物。獲得作為白色固體的IIIf:0.72g(82%)。1H-NMR(CDC13,400MHz)δ7.01-7.06(m,3H),7.21-7.25(m,2H)。13C-NMR(CDC13,100MHz)δ124.0,125.0,129.5,140.0,155.6,163.6。(IIIg)(N',N'-二甲基甲脒基)脲按照一般方法B,使用IVg(0.72g,5.89毫摩爾)來獲得標題化合物。獲得作為白色固體的IIIg:0.46g(60%)。1H-NMR(DMSO,400MHz)δ2.86(s,6H)。13C-NMR(DMSO,100MHz)δ36.5,160.8,167.0。(IIIh)(N',N'-二乙基甲脒基)脲按照一般方法B,使用IVh(0.95g,6.76毫摩爾)來獲得標題化合物。獲得作為黃色油狀物的IIIh:0.73g(68%)。1H-NMR(CD3OD,400MHz)(旋轉異構體的4.2:1混合物)δ1.11(t,J=6.8,4.8H,2CH3主要),1.18(t,J=6.8,1.2H,2CH3次要),3.36(q,J=6.8,3.2H,2CH2主要),3.46(q,J=6.8,0.8H,2CH2主要)。13C-NMR(CD3OD,100MHz)δ12.0(2CH3次要),12.6(2CH3主要),41.4(2CH2主要),43.1(2CH2次要),158.9,167.1。(IIIi)(哌啶-l-碳酰亞胺基)脲((piperidine-1-carboximidoyl)urea)按照一般方法B,使用IVi(0.46g,3.02毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIIi:0.51g(定量產率)。1H-NMR(CD3OD,400MHz)δ1.65-1.73(m,6H),3.50-3.59(m,4H)。13C-NMR(CD3OD,100MHz)δ23.1,24.9,46.9,153.2,155.3。(IIIj)(N'-丁基甲脒基)脲按照一般方法B,使用IVj(1.07g,7.64毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIIj:1.15g(95%)。1H-NMR(DMSO-d6,400MHz)δ0.85-0.89(m,3H),1.27-1.33(m,2H),1.36-1.43(m,2H),3.04-3.08(m,2H)。13C-NMR(DMSO-d6,100MHz)δ13.7,19.5,31.3,39.6,160.3,166.8。(IIIk)(吡咯烷-l-碳酰亞胺基)脲按照一般方法B,使用IVk(0.97g,7.04毫摩爾)來獲得標題化合物。獲得作為淺黃色固體的IIIk:0.80g(73%)。1H-NMR(DMSO-d6,400MHz)δ1.76-1.79(m,4H),3.25-3.28(m,4H)。13C-NMR(DMSO-d6,100MHz)δ25.1,46.2,155.9,158.1。(III1)(N'-(二甲基丙烷-3-氨基)甲脒基)脲按照一般方法B,使用IV1(0.10g,0.61毫摩爾)來獲得標題化合物。獲得作為黃色膠狀固體的III1:0.11g(95%)。1H-NMR(CD3OD,400MHz)δ1.71-1.76(m,2H),2.25(s,6H),2.37(t,J=7.6,2H),3.20(t,J=7.6,2H)。13C-NMR(CD3OD,100MHz)δ26.7,38.3,43.9,56.3,155.3,156.4。(IIIm)(N'-(3-(哌啶-l-基)丙烷))甲脒基)脲按照一般方法B,使用IVm(0.34g,1.62毫摩爾)來獲得標題化合物。獲得作為黃色膠狀固體的IIIm:0.23g(93%)。1H-NMR(CD3OD,400MHz)δ1.42-1.48(m,2H),1.59-1.63(m,4H),1.76-1.83(m,2H),2.48-2.52(m,6H),3.24-3.31(m,2H)。13C-NMR(CD3OD,100MHz)δ23.6,24.9,25.3,38.7,53.7,54.8,157.4,159.5。(ΙΙΙn)(N'-芐基甲脒基)脲按照一般方法B,使用IVn(0.80g,4.60毫摩爾)來獲得標題化合物。獲得作為白色固體的IIIn:0.82g(93%)。1H-NMR(CD3OD,400MHz)δ4.35(s,2H),7.21-7.30(m,5H)。13C-NMR(CD3OD,100MHz)δ44.1,126.9,127.0,128.1,128.3,160.9,167.5。(IIIo)(N'-吡啶-4-基甲基甲脒基)脲按照一般方法B,使用IVo(0.93g,5.31毫摩爾)來獲得標題化合物。獲得作為膠狀固體的IIIo:0.86g(85%)。1H-NMR(CD3OD,400MHz)δ4.49(s,2H),7.34(d,J=5.6,2H),8.44(d,J=5.6,2H)。13C-NMR(CD3OD,100MHz)δ42.7,122.2,147.6,148.7,159.8,165.6。(IIIp)(N',N'-二丙基甲脒基)脲按照一般方法B,使用IVp(0.77g,4.60毫摩爾)來獲得標題化合物。獲得作為白色固體的IIIp:0.68g(79%)。1H-NMR(CD3OD,400MHz)δ0.79(t,J=7.2,6H),1.42-1.48(m,4H),3.16(t,J=7.6,4H)。13C-NMR(CD3OD,100MHz)δ11.5,21.2,48.2,159.7,167.2。一般方法(C):式(I)的化合物的合成-步驟3,方案1向脒基脲(III)(1當量)在濃H2SO(5.5M)中的溶液添加適當的苯甲醛(II)(1.2當量)。在室溫下攪拌72小時以后,用少量的冷的H2O來稀釋反應混合物。然后用Na2CO3使溶液變成堿性,從而提供沉淀物,其是通過過濾加以收集。而在沒有觀察到沉淀的情況下,則真空濃縮水相。通過借助于有機溶劑的研制(trituration)或通過快速色譜法,來純化粗三嗪酮(I)。實施例14-(4-氟苯基)-6-(甲基氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,并使用4-氟-苯甲醛(IIa)(0.73mL,6.84毫摩爾)和IIIa(0.66g,5.7毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(8.5:1.5:0.1)進行的洗脫提供作為白色固體的實施例1的化合物:0.33g(26%)。1H-NMR(CD3OD,400MHz)δ2.76(s,3H),5.68(s,1H),7.07-7.12(m,2H),7.42-7.45(m,2H)。13C-NMR(CD3OD,100MHz)δ26.5,66.0,115.0(d,J=16.0),128.1(d,J=8.3),138.1,150.7,154.2,162.9(d,J=244)。MS(ESI)m/z:223[M+H]+;MS(ESI)m/z:221[M–Η]-。實施例26-(乙基氨基)-4-(甲苯基)-3,4-二氫-l,3,5-三嗪-2(lH)-酮。按照一般方法C,使用鄰甲苯基-苯甲醛-苯甲醛(lIb)(0.36mL,3.19毫摩爾)和IIIb(0.34g,2.65毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.1)進行的洗脫提供作為白色固體的實施例2的化合物:0.26g(43%)。1H-NMR(DMSO-d6,400MHz)δ1.02(t,J=8.0,3H),2.37(s,3H),3.05(q,J=8.0,2H),5.32(brs,exch,1H),5.77(s,1H),7.14-7.16(m,3H),7.25-7.27(m,1H),7.35(brs,exch,1H),8.48(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ15.4,19.1,35.3,67.1,126.4,126.6,130.7,131.3,135.7,141.9,148.3,153.5.MS(ESI)m/z:233[M+H]+。實施例36-(乙基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.50mL,4.60毫摩爾)和IIIb(0.50g,3.84毫摩爾)來獲得標題化合物。借助于有機溶劑的混合物(DCM/MeOH)的研制提供作為白色固體的實施例3的化合物:0.46g(50%)。1H-NMR(CD3OD,400MHz)δ1.11(t,J=8.0,3H),3.20(q,J=8.0,2H),5.65(s,1H),7.06-7.10(m,2H),7.40-7.43(m,2H)。13C-NMR(CD3OD,100MHz)δ14.9,36.7,67.5,116.3(d,J=22.0),129.5(d,J=8.0),139.6,156.6,158.5,164.2(d,J=244.0)。MS(ESI)m/z:237[M+H]+;MS(ESI)m/z:235[M-H]-。實施例4(+)6-(乙基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。通過實施例3的外消旋化合物的手性HPLC對映體分離來獲得作為白色固體的標題化合物:0.046g(61%)。ee>99.5%(檢測器UV240nm)。關于分析手性HPLC的保留時間:8.475分鐘。1H-NMR(CD3OD,400MHz)δ1.11(t,J=8.0,3H),3.20(q,J=8.0,2H),5.65(s,1H),7.06-7.10(m,2H),7.40-7.43(m,2H)。13C-NMR(CD3OD,100MHz)δ14.9,36.7,67.5,116.3(d,J=22.0),129.5(d,J=8.0),139.6,156.6,158.5,164.2(d,J=244.0).MS(ESI)m/z:237[M+H]+;MS(ESI)m/z:235[M-H]-。[α]20D=+2.1(c0.13,MeOH)[針對相應的三氟乙酸鹽的計算]。實施例5(-)6-(乙基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。通過實施例3的外消旋化合物的手性HPLC對映體分離來獲得作為白色固體的標題化合物:0.043g(57%)。ee>99.4%(檢測器UV240nm)。關于分析手性HPLC的保留時間:16.479分鐘。1H-NMR(CD3OD,400MHz)δ1.11(t,J=8.0,3H),3.20(q,J=8.0,2H),5.65(s,1H),7.06-7.10(m,2H),7.40-7.43(m,2H)。13C-NMR(CD3OD,100MHz)δ14.9,36.7,67.5,116.3(d,J=22.0),129.5(d,J=8.0),139.6,156.6,158.5,164.2(d,J=244.0).MS(ESI)m/z:237[M+H]+;MS(ESI)m/z:235[M-H]-。[α]20D=-0.9(c0.13,MeOH)[針對相應的三氟乙酸鹽的計算]。實施例64-(-氟苯基)-6-(丙基氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.36mL,3.32毫摩爾)和IIIc(0.40g,2.77毫摩爾)來獲得標題化合物。利用DCM/MeOH/33%NH3水性溶液(9:1:0.1)進行的洗脫提供作為白色固體的實施例6的化合物:0.11g(16%)。1H-NMR(DMSO-d6,400MHz)δ0.84(t,J=7.2,3H),1.40-1.49(m,2H),3.03(t,J=6.0,2H),5.59(s,1H),5.75(brs,exch,1H),7.14-7.18(m,2H),7.34-7.38(m,2H),7.55(brs,exch,1H),8.55(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ11.8,22.7,42.3,68.1,115.4(d,J=22.0),128.6(d,J=8.0),141.2,149.0,153.8,161.6(d,J=244.0)。MS(ESI)m/z:251[M+H]+。實施例74-(4-氟苯基)-6-(異丙基氨基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.45mL,4.15毫摩爾)和IIId(0.50g,3.46毫摩爾)來獲得標題化合物。借助于有機溶劑的混合物(DCM/Et2O)的研制提供作為白色固體的實施例7的化合物:0.19g(22%)。1H-NMR(CD3OD,400MHz)δ1.11(m,6H),3.81-3.92(m,1H),5.65(s,1H),7.08-7.12(m,2H),7.41-7.45(m,2H)。13C-NMR(CD3OD,100MHz)δ21.6,21.7,42.1,65.5,114.9(d,J=20.5),128.1(d,J=8.3),138.6,150.5,154.6,164.2(d,J=242.0)。MS(ESI)m/z:251[M+H]+;MS(ESI)m/z:249[M-H]-。實施例84-(4-氟苯基)-6-(異丁基氨基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(1.13mL,10.52毫摩爾)和IIIe(1.34g,8.77毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.05)進行的洗脫提供作為白色固體的實施例8的化合物:0.33g(14%)。1H-NMR(CD3OD,400MHz)δ0.91(d,J=6.8,6H),1.79-1.82(m,1H),3.00-3.05(m,2H),5.69(s,1H),7.08-7.12(m,2H),7.43-7.46(m,2H)。13C-NMR(CD3OD,100MHz)δ19.1,28.1,66.0,115.0(d,J=21.2),128.1(d,J=8.3),138.2,149.2,153.8,162.8(d,J=243.8)。MS(ESI)m/z:265[M+H]+;MS(ESI)m/z:263[M-H]-。實施例94-(4-氟苯基)-6-(苯基氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.30mL,2.70毫摩爾)和IIIf(0.40g,2.24毫摩爾)來獲得標題化合物。用DCM/MeOH(9:1)進行的洗脫提供作為白色固體的實施例9的化合物:0.06g(10%)。1H-NMR(DMSO-d6,400MHz)δ5.78(s,1H),6.89-6.92(m,1H),7.17-7.24(m,4H),7.41-7.45(m,2H),7.52-7.54(m,2H),7.82(brs,exch,1H),8.02(brs,exch,1H),8.52(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ68.3,114.6(d,J=21.0),118.1,121.3,128.2(d,J=9.0),128.6,140.0,145.11,145.6,152.1,162.0(d,J=240.2)。MS(ESI)m/z:285[M+H]+;MS(ESI)m/z:283[M-H]-。實施例106-(二甲基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.45mL,4.25毫摩爾)和IIIg(0.46g,3.54毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(8.5:1.5:0.1)進行的洗脫提供作為白色固體的實施例10的化合物:0.11g(13%)。1H-NMR(CD3OD,400MHz)δ3.02(s,6H),5.67(s,1H),7.11-7.15(m,2H),7.45-7.49(m,2H)。13C-NMR(CD3OD,100MHz)δ36.0,66.2,115.0(d,J=21.2),128.1(d,J=9.1),139.2,153.6,155.6,162.9(d,J=244.0)。MS(ESI)m/z:237[M+H]+;MS(ESI)m/z:235[M–Η]-。實施例116-(二乙基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.59mL,5.52毫摩爾)和IIIh(0.73g,4.60毫摩爾)來獲得標題化合物。借助于有機溶劑的混合物(DCM/Et2O)的研制提供作為白色固體的實施例11的化合物:0.25g(21%)。1H-NMR(CD3OD,400MHz)δ1.12(t,J=7.0,6H),3.42(q,J=7.0,4H),5.66(s,1H),7.07-7.12(m,2H),7.43-7.46(m,2H)。13C-NMR(CD3OD,100MHz)δ12.3,41.5,64.7,115.0(d,J=21.2),128.0(d,J=8.4),137.9,150.8,153.7,162.9(d,J=243.3)。MS(ESI)m/z:265[M+H]+;MS(ESI)m/z:263[M–H]-。實施例124-(4-氟苯基)-6-(哌啶-l-基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.40mL,3.62毫摩爾)和IIIi(0.51g,3.02毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.05)進行的洗脫提供作為白色固體的實施例12的化合物:0.23g(28%)。1H-NMR(CD3OD,400MHz)δ1.55-1.58(m,4H),1.64-1.68(m,2H),3.49-3.52(m,4H),5.67(s,1H),7.10-7.14(m,2H),7.44-7.48(m,2H)。13C-NMR(CD3OD,100MHz)δ23.9,25.4,45.6,64.5,115.0(d,J=22.0),128.1(d,J=8.3),137.5(d,J=3.9),151.8,156.0,164.3(d,J=243.8)。MS(ESI)m/z:277[M+H]+;MS(ESI)m/z:275[M–Η]-。實施例136-(丁基氨基)-4-(-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.94mL,8.76毫摩爾)和IIIj(1.15g,7.30毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.1)進行的洗脫提供作為白色固體的實施例13的化合物:0.71g(39%)。1H-NMR(CD3OD,400MHz)δ0.88-0.92(m,3H),1.32-1.36(m,2H),1.48-1.54(m,2H),3.17-3-18(m,2H),5.70(s,1H),7.06-7.11(m,2H),7.43-7.46(m,2H)。13C-NMR(CD3OD,100MHz)δ14.2,21.0,32.4,41.6,67.1,116.3(d,J=21.9),129.4(d,J=8.4),139.3,151.3,155.2,163.7(d,J=243.8)。MS(ESI)m/z:265[M+H]+;MS(ESI)m/z:263[M–H]-。實施例146-(乙基氨基)-4-(吡啶-3-基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用3-吡啶羧基醛(IIc)(0.43mL,4.61毫摩爾)和IIIb(0.50g,3.84毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(8:2:0.2)進行的洗脫提供作為白色固體的實施例14的化合物:0.08g(10%)。1H-NMR(CD3OD,400MHz)δ1.40(t,J=6.8,3H),3.19(q,J=6.8,2H),5.79(s,1H),7.44-7.47(M,IH),7.89(d,J=8.5,1H),8.50(d,J=5.0,1H),8.58(s,1H)。13C-NMR(CD3OD,100MHz)δ13.4,35.3,65.0,124.0,134.9,138.5,147.2,148.8,151.3,155.2。MS(ESI)m/z:220[M+H]+;MS(ESI)m/z:218[M–H]-。實施例154-(4-氟苯基)-6-(吡咯烷-l-基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.65mL,6.10毫摩爾)和IIIk(0.80g,5.08毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.05)進行的洗脫提供作為白色固體的實施例15的化合物:0.29g(22%)。1H-NMR(CD3OD,400MHz)δ1.91-1.93(m,4H),3.40-3.43(m,4H),5.69(s,1H),7.08-7.13(m,2H),7.46-7.49(m,2H)。13C-NMR(CD3OD,100MHz)δ24.7,46.3,64.8,115.0(d,J=21.2),128.3(d,J=8.3),137.7,155.3,160.4,163.5(d,J=244.4)。MS(ESI)m/z:263[M+H]+。制備II(實施例16-21)式(I)的化合物的一般合成,其中R1是氫、直鏈或支鏈未取代的或取代的C1-6烷基,R2是COR5,以及R3、R4、R5、Y1和Y2是如在式(I)中所定義。一般方法(D):式(VIII)的中間體的合成-步驟1-3,方案1按照在制備I中報道的方法,按照在方案1中的步驟1-3,來獲得式(VIII)的中間體。(VIIIa)6-氨基-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法D,用4-氟-苯甲醛(IIa)(2.5mL,23.80毫摩爾)和可商購的脒基脲硫酸鹽(3.0g,19.80毫摩爾)來獲得標題化合物。借助于有機溶劑的混合物(DCM/MeOH)的研制提供作為白色固體的VIIIa:2.90g(88%)。1H-NMR(DMSO-d6,400MHz)δ5.56(s,1H),5.75(brs,exch,1H)7.15-7.19(m,2H),7.35-7.38(m,2H),7.49(brs,exch,1H),8.51(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ67.5,115.4(d,J=22.0),128.6(d,J=8.0),140.2,150.8,153.5,161.2(d,J=244.0)。MS(ESI)m/z:209[M+H]+。一般方法(E):式(I)的化合物的合成-方案2方法(a):向VIII(l當量)在有機溶劑的混合物,如吡啶/DCM,DMF/DCM和2,6-二甲基吡啶/DMF(0.4M)中的冰冷的溶液/懸浮液滴加適當的酰氯(VII)(1當量)。在0℃下攪拌3-4小時以后,用DCM或EtOAc來稀釋反應混合物并用2NHC1加以洗滌。經無水Na2SO4干燥有機相,并在減壓下蒸發。通過快速色譜法來純化粗三嗪酮(I)。方法(b):用HOBt(1.3當量)來處理適當的羧酸(VII)(1當量)和EDCI.HCl(1.3)在DCM或DMF(0.15M)中的溶液。在室溫下攪拌混合物1小時,然后滴加入VIII(1.1當量)在DCM或DMF(0.12M)中的冰冷懸浮液。在添加Et3N或DIPEA(1.1當量)以后,允許在室溫下攪拌得到的混合物過夜。然后,在反應溶劑分別是DCM或DMF的情況下,反應混合物用DCM加以稀釋并用H2O加以洗滌,或用EtOAc加以稀釋并用5%LiCl水性溶液加以洗滌。經無水Na2SO4干燥有機相并在減壓下蒸發。通過快速色譜法來純化粗三嗪酮(I)。方法(c):在室溫下,攪拌適當的羧酸(VII)(1.2當量)和EDCI.HCl(1.56)在DMF(0.4M)中的溶液1小時,然后滴加入VIII(1當量)在DMF(0.12M)中的冰冷懸浮液。在室溫下攪拌過夜以后,用EtOAc來稀釋反應混合物并用5%LiCl水性溶液加以洗滌。經無水Na2SO4干燥有機相并在減壓下蒸發。然后用有機溶劑來研制殘余物,從而提供感興趣的化合物而沒有進一步純化。實施例16N-(4-(-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)乙酰胺。按照一般方法E,方法(a),自VIIIa(0.08g,0.38毫摩爾)和在2,6-二甲基吡啶/DMF(3:1)中的AcCl(27μL,0.38毫摩爾)來獲得標題化合物。用DCM/MeOH(9.5:0.5)進行的洗脫提供作為白色固體的實施例15的化合物:24mg(25%)。1H-NMR(DMSO-d6,400MHz)δ1.98(s,3H),5.74(s,1H),7.13-7.17(m,2H),7.33-7.36(m,2H),7.96(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ23.8,67.2,114.8(d,J=22.0),127.3(d,J=8.0),139.6,146.9,152.3,161.2(d,J=244.0),174.8.MS(ESI)m/z:251[M+H]+;MS(ESI)m/z:249[M-H]-。實施例17N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)-2-丙基戊酰胺。按照一般方法E,方法(b),從在DMF中的VIIIa(0.15g,0.72毫摩爾)和丙戊酸(104μL,0.65毫摩爾),來獲得標題化合物。借助于DCM/MeOH(9.5:0.5)的洗脫提供作為白色固體的實施例16的化合物:80mg(37%)。1H-NMR(DMSO-d6,400MHz)δ0.81-0.85(m,6H),1.19-1.25(m,4H)1.26-1.35(m,2H),1.45-1.54(m,2H),2.35-2.37(m,1H),5.78(s,1H),7.18-7.20(m,2H),7.35-7-37(m,2H),7.95(brs,exch,1H),9.84(brs,exch,1H),10.76(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ14.0,20.5,34.7,47.3,68.2,115.5(d,J=21.0),127.5(d,J=8.3),139.3,145.6,152.3,162.5(d,J=247.3),180.0。MS(ESI)m/z:335[M+H]+;MS(ESI)m/z:333[M–Η]-。實施例185-(1,2-二硫戊環-3-基)-N-(4-(4-氟苯基)-6-氧代-1,4,5,6-四氫-l,3,5-三嗪-2-基)戊酰胺。按照一般方法E,方法(b),從在DCM中的VIIIa(0.15g,0.72毫摩爾)和(+/-)硫辛酸(0.13g,0.65毫摩爾)來獲得標題化合物。借助于DCM/MeOH(9.5:0.5)的洗脫提供作為白色固體的實施例17的化合物:77mg(31%)。1H-NMR(DMSO-d6,400MHz)δ1.33-1.39(m,2H),1.49-1.59(m,3H),1.62-1.70(m,1H),1.82-1.90(m,1H),2.31(t,J=4.0,2H),2.36-2.44(m,1H),3.08-3.14(m,1H),3.15-3.21(m,1H),3.58-3.66(m,1H)5.80(s,1H),7.18-7.23(m,2H),7.39-7.42(m,2H)8.00(brs,exch,1H),9.88(brs,exch,1H),10.65(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ24.2,27.9,33.9,36.2,38.0,39.7,55.9,67.5,115.1(d,J=21.0),128.0(d,J=8.3),139.2,145.6,151.2,162.1(d,J=243.0),176.8。MS(ESI)m/z:397[M+H]+;MS(ESI)m/z:395[M–Η]-。實施例193-((lH-苯并[d][1,2,3]三唑-l-基)氧基)-N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)丙酰胺。按照一般方法E,方法(b),從DMF在中的VIIIa(0.10g,0.48毫摩爾)和丙烯酸(33μL,0.48毫摩爾),來獲得標題化合物。在室溫下攪拌過夜以后,觀察到白色固體的沉淀。通過過濾來收集沉淀物并借助于DCM/Et2O的混合物加以研制,從而提供作為白色固體的感興趣的化合物:45mg(25%)。1H-NMR(DMSO-d6,400MHz)δ2.98-3.01(m,2H),4.69-4.71(m,2H),5.79(s,1H),7.19-7.24(m,2H),7.35-7.38(m,2H),7.41-7.45(m,1H),7.66-7.70(m,1H),7.86(d,J=8.8,1H),7.93(d,J=8.4,1H),8.15(br,exch,1H),9.74(br,exch,1H),10.62(br,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ30.5,43.6,67.5,112.5,114.7,115.3(d,J=21.0),124.4,128.1(d,J=9.0),130.3,133.9,134.0,137.8,147.8,150.5,161.8(d,J=246),175.3.MS(ESI)m/z:398[M+H]+;MS(ESI)m/z:396[M-H]-。實施例20N-(4-(4-氟苯基)-6-氧代-l,4,5,6-四氫-l,3,5-三嗪-2-基)-2-苯氧基乙酰胺。按照一般方法E,方法(c),從VIIIa(0.20g,0.96毫摩爾)和苯氧基乙酸(0.18g,1.15毫摩爾)來獲得標題化合物。借助于DCM/Et2O的混合物的研制提供作為白色固體的實施例19的化合物:11mg(3%)。1H-NMR(CD3OD,400MHz)δ4.64(s,2H),5.90(s,1H),6.94-6.96(m,3H),7.13-7.17(m,2H),7.25-7.28(m,2H),7.45-7.48(m,2H)。MS(ESI)m/z:343[M+H]+;MS(ESI)m/z:341[M–Η]-。實施例212-(4-氟苯氧基)-N-(4-(4-氟苯基)-6-氧代-1,4,5,6-四氫-l,3,5-三嗪-2-基)乙酰胺。按照一般方法E,方法(c),從VIIIa(0.16g,0.78毫摩爾)和4-氟-苯氧基乙酸(0.16g,0.94毫摩爾)來獲得標題化合物。借助于DCM/Et2O的混合物的研制提供作為白色固體的實施例20的化合物:34mg(12%)。1H-NMR(DMSO-d6,400MHz)δ4.57(s,2H),5.78(s,1H),6.84-6.87(m,2H),7.04-7.08(m,2H),7.20-7.24(m,2H),7.37-7.40(m,2H),8.42(brs,exch,1H),10.38(brs,exch,1H)。13C-NMR(DMSO-d6,100MHz)δ64.4,69.0,115.3,115.6,115.7,127.8,137.1,147.6,151.0,154.6,155.8(d,J=260),162.0(d,J=244),172.7。MS(ESI)m/z:361[M+H]+;MS(ESI)m/z:359[M–Η]-。實施例226-((3-(二甲基氨基)丙基)氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-F-苯甲醛(IIa)(78μL,0.72毫摩爾)和III1(0.10g,0.60毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(7.5:2.5:0.2.5)進行的洗脫提供作為白色固體的實施例22的化合物:30mg(17%)。1H-NMR(CD3OD,400MHz)51.69-1.73(m,2H),2.22(s,6H),2.38(t,J=7.2,2H),3.21-3.28(m,2H),5.67(s,IH),7.08-7.13(m,2H),7.42-7.46(m,2H)。13C-NMR(CD3OD,100MHz)δ25.8,38.6,46.2,55.8,66.0,115.3(d,J=21.6)128.5(d,J=8.7),139.5,150.6,152.7,162.3(d,J=242.8)。MS(ESI)m/z294[M+H]+;MS(ESI)m/z292[M-H]-。實施例234-(4-氟苯基)-6-((3-(哌啶-1-基)丙基)氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-F-苯甲醛(IIa)(0.20mL,1.80毫摩爾)和IIIm(0.34g,1.50毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(7.5:2.5:0.2.5)進行的洗脫提供作為白色固體的實施例23的化合物:0.12g(24%)。1H-NMR(CD3OD,400MHz)δ1.44-1.46(m,2H),1.56-1.60(m,4H),1.70-1.74(m,2H),2.37-2.44(m,6H),3.20-3.23(m,2H),5.65(s,IH),7.07-7.11(m,2H),7.40-7.44(m,2H)。13C-NMR(CD3OD,100MHz)δ23.6,24.9,25.8,38.6,53.9,55.8,66.0,115.0(d,J=21.2),128.1(d,J=8.4),138.2,150.3,153.2,162.7(d,J=243.8)。MS(ESI)m/z334[M+H]+;MS(ESI)m/z332[M-H]-。實施例246-(芐基氨基)-4-(4-氟苯基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-F-苯甲醛(IIa)(0.56mL,5.20毫摩爾)和IIIn(0.82g,4.30毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(9:1:0.1)進行的洗脫提供作為白色固體的實施例24的化合物:0.12g(36%)。1H-NMR(DMSO-d6,400MHz)δ4.40-4.44(m,2H),5.71(s,1H),7.06-7.11(m,2H),7.26-7.31(m,7H)。13C-NMR(DMSO-d6,100MHz)δ44.5,67.3,114.9(d,J=21.2),126.7,127.0(d,J=8.3),128.06,128.1,138.3,142.7,150.6,152.8,162.5(d,J=242.6)。MS(ESI)m/z299[M+H]+;MS(ESI)m/z297[M-H]-。實施例254-(4-氟苯基)-6-((吡啶-4-基甲基)氨基)-3,4-二氫-l,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-F-苯甲醛(IIa)(0.58mL,5350毫摩爾)和IIIo(0.86g,4.46毫摩爾)來獲得標題化合物。用DCM/MeOH/33%NH3水性溶液(8:2:0.2)進行的洗脫提供作為白色固體的實施例25的化合物:0.18g(14%)。1H-NMR(DMSO-d6,400MHz)δ4.35(s,2H),5.59(s,1H),7.08-7.13(m,2H),7.24-7.26(m,4H),8.47(d,J=4.8,2H)。13C-NMR(DMSO-d6,100MHz)δ42.4,67.1,114.5(d,J=21.3),121.8,127.9(d,J=8.2),139.4,148.6,149.1,152.5,153.2,161.3(d,J=250.0)。MS(ESI)m/z300[M+H]+;MS(ESI)m/z298[M-H]-。實施例266-(二丙基氨基)-4-(4-氟苯基)-3,4-二氫-1,3,5-三嗪-2(1H)-酮。按照一般方法C,使用4-氟-苯甲醛(IIa)(0.47mL,4.38毫摩爾)和IIIp(0.68g,3.64毫摩爾)來獲得標題化合物。借助于有機溶劑的混合物(DCM/Et2O)的研制提供作為白色固體的實施例26的化合物:0.23g(22%)。1H-NMR(CD3OD,400MHz)δ0.88(t,J=7.2,6H),1.55-1.61(m,4H),3.27-3.30(m,2H),3.36-3.41(m,2H),5.65(s,1H),7.10-7.14(m,2H),7.43-7.47(m,2H)。13C-NMR(CD3OD,100MHz)δ9.8,20.8,48.8,64.6,115.0(d,J=22.0),128.0(d,J=9.0),137.9,156.7,161.2,162.9(d,J=243)。MS(ESI)m/z293[M+H]+;MS(ESI)m/z291[M–Η]-。測定本發明的化合物的生化活性的方法BACE-1的抑制通過采用模擬APP序列的肽作為底物(甲氧基香豆素-Ser-Glu-Val-Asn-Leu-Asp-Ala-Glu-Phe-Lys-二硝基苯基,M-2420,Bachem,德國)進行β-分泌酶(BACE-1,Sigma-Aldrich)抑制研究。采用以下方法:在室溫下,用175μL的酶(在20mM乙酸鈉中,其含有3-[(3-膽酰胺丙基)二甲基銨基]-1-丙磺酸酯(CHAPS),0.1%w/v)預溫育5μL的測試化合物(或DMSO,如果制備對照孔)1小時。然后添加底物(3μΜ,最終濃度)并讓其在37℃下反應15分鐘。利用FluoroskanAscent,在λem=405nm(λexc=320nm)處讀取熒光信號。在最終混合物中維持低于5%(v/v)的DMSO濃度保證了酶活性沒有顯著損失。比較有和沒有抑制劑的熒光強度,并計算由于測試化合物的存在導致的百分比抑制。在含有除BACE-1之外的所有試劑的對照孔中測量背景信號并減去。通過以下表達式:100-(IFi/IFox100)來計算起因于存在增加的測試化合物濃度的%抑制,其中IFi和IF0分別是在存在和不存在抑制劑的情況下針對BACE-1獲得的熒光強度。如果可能,通過繪制%抑制與在測定樣品中抑制劑濃度的對數,來獲得抑制曲線。確定線性回歸參數并外推IC50(GraphPadPrism4.0,GraphPadSoftwareInc.)。為了證明BACE-1活性的抑制,將肽模擬物抑制劑(β-分泌酶抑制劑IV,Calbiochem,IC50=20nM)連續稀釋到反應孔。結果示于表1。GSK-3β的抑制人重組GSK-3β購買自Millipore(MilliporeIbericaS.A.U.)。預磷酸化多肽底物購買自Millipore(MilliporeIbericaSAU)。激酶-Glo發光激酶測定獲得自Promega(PromegaBiotechIberica,SL)。ATP和所有其他試劑是來自Sigma-Aldrich。測定緩沖液含有50mM的4-(2-羥基乙基)-1-哌嗪乙磺酸(HEPES)(pH7.5)、1mM乙二胺四乙酸(EDTA)、1mM乙二醇四乙酸(EGTA)和15mM乙酸鎂。采用由Baki開發的方法[Bakietal.AssayandDrugDevelopmentTechnologies2007,5(1),75-83]來分析GSK-3β的抑制。利用白色96孔板,在測定緩沖液中進行激酶-Glo測定。在典型的測定中,將10μL(10μM)的測試化合物(溶解于DMSO,在1mM濃度下并預先在測定緩沖液中稀釋到所期望的濃度)和10μL(20ng)酶加入每個孔,接著加入20μL的含有25μM底物和1μM腺苷三磷酸(ATP)的測定緩沖液。在反應混合物中最終DMSO濃度不超過1%。在30℃下30分鐘溫育以后,用40μL的激酶-Glo試劑來停止酶促反應。在10分鐘以后,利用FluoroskanAscent多模讀取器來記錄輝光型發光(glow-typeluminescence)。活性與總的和消耗的ATP之間的差異成比例。基于在總抑制濃度(5μM)下,分別在不存在抑制劑的情況下以及存在參比化合物抑制劑(SB415826[Coghlanetal.ChemBiol2000,7,793-803],MerckMillipore,IC50=54nM)的情況下測得的最大激酶(AVERAGE陽性)和熒光素酶活性(AVERAGE陰性)來計算抑制活性。確定線性回歸參數并外推IC50(GraphPadPrism4.0,GraphPad軟件公司)。結果示于表1。表1實施例BACE-1IC50(μM)GSK-3βIC50(μM)250.31±6.6140.71±2.70316.05±0.647.11±0.3748.1312.57512.5515.67636.83±9.854.34±0.639n.a.6.93±0.14n.a.:不可獲得測定本發明的化合物的神經保護活性的方法原代細胞培養星形膠質細胞制備自新生(P2)大鼠大腦皮質,如由Luna-Medina等先前描述的[Luna-Medina,R.;Cortes-Canteli,M.;Alonso,M.;Santos,A.;Martinez,A.;Perez-Castillo,A.Regulationofinflammatoryresponseinneuralcellsinvitrobythiadiazolidinonesderivativesthroughperoxisomeproliferator-activatedreceptorgammaactivation.J.Biol.Chem.2005,280,21453-21462]。簡要地說,在腦膜的除去以后,解剖,分離大腦皮質,然后在37℃下用0.25%胰蛋白酶/EDTA加以溫育1小時。在離心以后,用Hanks平衡鹽溶液(HBSS)(Gibco)洗滌沉淀物3次,然后將細胞放入無涂層燒瓶中并保持在含有10%胎牛血清(FBS)的HAMS/Dulbecco改良Eagle培養基(DMEM)(1:1)培養基中。在15天以后,在37℃下,在240rpm的定軌振蕩器上振蕩燒瓶4小時,收集上清液,離心,然后將含有小膠質細胞的細胞沉淀物再懸浮于完全培養基(HAMS/DMEM(1:1),含有10%FBS)并接種在未涂覆的96孔板上。允許細胞黏附2小時,然后除去培養基以消除非黏附少突膠質細胞。添加含有10ng/mL粒細胞巨噬細胞集落刺激因子(GM-CSF)的新的新鮮培養基。然后胰蛋白酶消化黏附在燒瓶上的剩余的星形膠質細胞,收集,離心,并平板接種在含有完全培養基的96孔板上。通過此方法獲得的培養物的純度是>98%,如通過免疫熒光,并借助于OX42(小膠質細胞標記)和膠質細胞原纖維酸性蛋白(GFAP,星形膠質細胞標志物)抗體所確定的。亞硝酸鹽測量在不同的神經退行性疾病中,GSK-3β的過表達和/或過活性導致增加水平的小膠質細胞活化。此外,GSK-3β被證明會調節在星形膠質細胞中的炎性耐受性[Beurel,E.;Jope,R.S.Glycogensynthasekinase-3regulatesinflammatorytoleranceinastrocytes.Neuroscience2011,169,1063-1070]。因此,我們探索了在基于細胞的測定中這些新的ΒΑ0Ε-1/03Κ-3β雙重抑制劑是否可以發揮抗炎作用。通過評估來自原代培養的膠質細胞的產生的亞硝酸鹽來測試選擇的化合物的潛在的抗炎活性。連同選擇的化合物(10uM)一起溫育星形膠質細胞和小膠質細胞的原代培養物1小時,然后連同有效的抗炎劑,如脂多糖(LPS)(10g/mL)一起來培養細胞另外24小時。LPS能夠誘導培養基中的亞硝酸鹽產生和積累,其是通過標準Griess反應加以測定。自培養基收集上清液并混合與等容積的Griess試劑(Sigma)。然后在室溫下溫育樣品15分鐘并利用板讀數器在492/540nm處讀取吸光度。當用LPS刺激原代星形膠質細胞和小膠質細胞時,我們觀測到亞硝酸鹽生產的顯著誘導,其通過通過三嗪酮處理來減小(圖2A,2B)。特別是,實施例3、6和9的6-(烷基氨基)和6-(苯基氨基)化合物通常表現出比用作參比化合物(在以下表2中的化合物A-C)的6-氨基三嗪酮更高的效力。更有趣的是,表現出平衡的GSK-33/BACE-1抑制譜(inhibitoryprofile)的實施例3和6的化合物在測試的衍生物中結果是最有效的,并且在星形膠質細胞中它們能夠將產生的亞硝酸鹽恢復到基礎水平(圖2A)。測定本發明的化合物的神經原性活性的方法神經球培養物神經球(NS)培養物源自成年大鼠的海馬,并且使用已建立的傳代方法,神經球被誘導用于增殖,以根據公布的方法實現最佳細胞擴增[Ferron,S.R.;Andreu-Agullo,C.;Mira,H.;Sanchez,P.;Marques-Torreon,M.A.;Farinas,I.Acombinedex/invivoassaytodetecteffectsofexogenouslyaddedfactorsinneuralstemcells.NatureProtoc.2007,2,849-859]。大鼠被斷頭,去除大腦,并解剖海馬,如所描述的[Morales-Garcia,J.A.;Luna-Medina,R.;Alfaro-Cervello,C.;Cortes-Canteli,M.;Santos,A.;Garcia-Verdugo,J.M.;Perez-Castillo,A.Peroxisomeproliferator-activatedreceptorgammaligandsregulateneuralstemcellproliferationanddifferentiationinvitroandinvivo.Glia2011,59,293-307]。簡要地說,將細胞接種到12孔培養皿并在含有10ng/mL表皮生長因子(EGF,Peprotech,London,UK)、10ng/mL成纖維細胞生長因子(FGF,Peprotech)和B27培養基(Gibco)的DMEM/F12(1:1,Invitrogen)中培養。在培養3天以后,在一周期間,在存在或不存在指定化合物(10μM)的情況下,培養NS。之后,在不存在外源性生長因子的情況下,在100g/mL聚-L-賴氨酸包被的玻片上平板接種來自10天的培養物的NS,時間為72小時。免疫細胞化學對于新的AD修飾藥物而言,神經發生是關鍵特性,因為它在受損腦中賦予增加內源性再生的潛力作為修復機制,并且減少神經元損失和變性。考慮到,GSK-3β抑制被認為會調節和增加神經發生[Lange,C.;Mix,E.;Frahm,J.;Glass,A.;Muller,J.;Schmitt,0.;Schmole,A.C.;Klemm,K.;Ortinau,S.;Hubner,R.;Freeh,M.J.;Wree,A.;Rolfs,A.SmallmoleculeGSK-3inhibitorsincreaseneurogenesisofhumanneuralprogenitorcells.Neurosci.Lett.2011,488,36-40;Morales-Garcia,J.A.;Luna-Medina,R.;Alonso-Gil,S.;Sanz-SanCristobal,M.;Palomo,V.;Gil,C.;Santos,A.;Martinez,A.;Perez-Castillo,A.GlycogenSynthaseKinase3InhibitionPromotesAdultHippocampalNeurogenesisinVitroandinVivo.ACSChem.Neurosci.2012,3,963-971],所以感興趣的是,證實將實施例3和6的化合物加入原代大鼠神經干細胞的NS培養物是否可以調節分化向著神經元表型進行。因此,對細胞進行免疫細胞化學處理以檢測神經標記,如β-微管蛋白和微管相關蛋白(MAP2),其表明本發明的化合物的潛在神經源性作用。對細胞進行免疫細胞化學處理,如先前描述的[Luna-Medina,R.;Cortes-Canteli,M.;Alonso,M.;Santos,A.;Martinez,A.;Perez-Castillo,A.Regulationofinflammatoryresponseinneuralcellsinvitrobythiadiazolidinonesderivetivesthroughperoxisomeproliferator-activatedreceptorgammaactivation.J.Biol.Chem.2005,280,21453-21462]。簡要地說,在處理期結束時,在24孔細胞培養板中的玻璃蓋玻片上培養NS培養物。然后用磷酸鹽緩沖鹽水(PBS)來洗滌培養物,在25℃下用4%多聚甲醛固定30分鐘,并在37℃下用0.1%TritonX-100透性化處理30分鐘。在用所選擇的一抗(多克隆抗-β-微管蛋白(克隆Tuj1;Abcam)和小鼠單克隆抗MAP2(Sigma))溫育1小時以后,用磷酸鹽緩沖鹽水洗滌細胞并在37℃下用相應的Alexa標記的二抗(Alexa-488和Alex-647以分別揭示β-微管蛋白和MAP2,MolecularProbes,Leiden,荷蘭)加以溫育45分鐘。隨后,利用TCSSP5激光掃描共聚焦顯微鏡(LeicaMicrosystems)來獲得圖像。調節共聚焦顯微鏡設置以產生最佳信噪比。Dapi染色用作核標記。如圖3所示,當用三嗪酮處理神經球時,觀測到一些神經源性效應。特別是,當與對照比較時,在用實施例3的化合物處理的培養物中,β-微管蛋白陽性細胞(未成熟神經元標記)的數目顯著增加。顯著地,實施例6的化合物顯著擴增MAP-2陽性細胞(成熟神經元標記)的數目,而在用參比化合物A處理的細胞中只可察覺到輕微的增加。用來測定本發明的化合物的神經毒性作用和膠質調節活性的方法原代細胞培養物混合膠質細胞培養物制備自新生Wistar大鼠(Rattusnorvegicus)的大腦皮質,如先前描述的[Monti,B.;D'Alessandro,C.;Farini,V.;Bolognesi,A.;Polazzi,E.;Contestabile,A.;Stirpe,F.;Battelli,M.G.Invitroandinvivotoxicityoftype2ribosome-inactivatingproteinslanceolinandstenodactylinonglialandneuronalcells.Neurotoxicology2007,28,637-644]。簡要地說,腦組織清除腦膜,在37℃下胰蛋白酶消化15分鐘,然后,在機械解離以后,洗滌細胞懸液并平板接種在聚-L-賴氨酸(Sigma-Aldrich,St.Louis,MO,USA,10μg/mL)涂布的燒瓶(75cm2)上。在補充有100mL/L熱滅活FBS(Lifetechnologies)、2毫摩爾/L谷氨酰胺(Sigma-Aldrich)和100μmοl/L硫酸慶大霉素(Sigma-Aldrich)的Eagle基本培養基(BME,LifetechnologiesLtd,Paisley,UK)中培養混合膠質細胞10-13天。通過機械振動,小膠質細胞收獲自混合膠質細胞培養物,再懸浮于無血清的新鮮培養基,然后以1.5x106個細胞/1.5mL培養基/孔的密度平板接種在未涂覆直徑為35mm的平皿上,用于蛋白質印跡分析,或以1x105個細胞/0.2mL培養基/孔的密度平板接種在96孔上,用于MTT測定。使細胞附著30分鐘,然后洗滌以除去未貼壁細胞。這些原代培養物是純小膠質細胞,其中大于99%的貼壁細胞對于同工凝集素(isolectin)B4為陽性以及對于星形膠質細胞和少突膠質細胞標記為陰性。為了制備純化的星形膠質細胞培養物,劇烈振蕩10天齡原代混合膠質細胞培養物發分離小膠質細胞和少突膠質細胞(生長在星形膠質細胞層的頂部)。用胰蛋白酶(0.25%)/EDTA(Lifetechnologies)來分離剩余的貼壁細胞,然后在室溫下將獲得的細胞懸液放置在未涂覆燒瓶中,以允許小膠質細胞附著于塑料表面。在20-30分鐘以后,在燒瓶的輕微搖動以后,收集非粘附或松散貼壁細胞,然后再次進行粘附步驟。收集含有非貼壁細胞的上清液并離心,將細胞再懸浮于無血清的新鮮BME培養基(Lifetechnologies),并以1.5x106個細胞/1.5mL培養基/孔的密度再接種在聚-L-賴氨酸涂布的(Sigma-Aldrich)直徑為35mm的平皿上,用于蛋白質印跡分析,或以1x105個細胞/0.2mL培養基/孔的密度再接種在96孔上,用于MTT測定。之后,進行蛋白質印跡分析。小腦粒狀神經元(CGN)的原代培養物制備自Wistar品系的7日齡大鼠,如先前描述的[Monti,B.;D'Alessandro,C.;Farini,V.;Bolognesi,A.;Polazzi,E.;Contestabile,A.;Stirpe,F.;Battelli,M.G.Invitroandinvivotoxicityoftype2ribosome-inactivatingproteinslanceolinandstenodactylinonglialandneuronalcells.Neurotoxicology2007,28,637-644]。簡要地說,細胞分離自小腦并以在補充有100mL/L熱滅活FBS(Lifetechnologies)、2毫摩爾/L谷氨酰胺、100μmοl/L硫酸慶大霉素和25毫摩爾/LKCl(均來自Sigma-Aldrich)的BME中的2x105個細胞/cm2的密度平板接種在直徑為350mm的平皿上或在24孔板中,其預先涂布有10μg/mL聚-L-賴氨酸。16小時以后,添加10μΜ胞嘧啶阿拉伯呋喃糖苷(Sigma-Aldrich)以避免膠質細胞增生。在體外7天以后(7DIV),將分化的神經元轉移到含有25毫摩爾/LKCl的無血清BME培養基并用于MTT測定。MTT測定通過MTT測定來評估在暴露于增加濃度的實施例3和6的化合物(0、10、20和50μM)24小時的原代培養物中不同的腦細胞的生存力[Monti,B.;D'Alessandro,C.;Farini,V.;Bolognesi,A.;Polazzi,E.;Contestabile,A.;Stirpe,F.;Battelli,M.G.invitroandinvivotoxicityoftype2ribosome-inactivatingproteinslanceolinandstenodactylinonglialandneuronalcells.Neurotoxicology2007,28,637-44]。簡要地說,以0.1mg/mL的最終濃度,將噻唑藍(thiazolylblue)加入培養基。在37℃下,在黑暗中,在對CGN溫育20分鐘和對膠質細胞溫育2小時以后,將MTT沉淀物溶解于含有5%TritonX-100的0.1MTris-HCl緩沖液(均來自Sigma-Aldrich),并用多片分光光度讀數器(Bio-Rad)讀取在570nm處的吸光度。重要的是,在神經膠質細胞和神經元細胞中,高達50μΜ,實施例3和6的化合物并不表現出任何毒性(圖4a,4b和4c)。蛋白質印跡分析在AD的進展中,從神經保護M2到細胞毒性Ml膠質細胞的轉化被認為是關鍵步驟[Boche,D.;Perry,V.H.;Nicoll,J.A.Review:activationpatternsofmicrogliaandtheiridentificationinthehumanbrain.Neuropathol.Appl.Neurobiol.2013,39,3-18]。這種M1/M2表型分類是基于促或抗炎細胞因子和受體的特定模式,除了別的以外,通過GSK-3β活性,來調節其釋放和表達[Goldmann,T.;Prinz,M.RoleofmicrogliainCNSautoimmunity.Clin.Dev.Immunol.2013,2013,208093]。在這方面,已報道GSK-3β激活會培育并保持促炎狀態。在此基礎上,我們評估了在膠質細胞上,實施例3和6的化合物調節誘導的一氧化氮合酶(iNOS)作為M1標記物,以及表達在髓樣細胞2(TREM2)上的觸發受體(triggeringreceptro)作為M2標記物的表達水平的能力。在存在或不存在不同濃度(0、5、10和20μM)的實施例3和6的化合物的情況下,將暴露于LPS(100ng/mL)24小時的小膠質細胞和星形膠質細胞是直接置于冰冷的裂解緩沖液(Tris50mM,SDS1%,蛋白酶抑制劑混合劑0.05%)中,然后通過使用Lowry方法來確定蛋白質含量[Lowry,0.H.;Rosebrough,N.J.;Farr,A.L.;Randall,R.J.ProteinmeasurementwiththeFolinphenolreagent.J.Biol.Chem.1951,193,265-275]。將20μg蛋白提取物再懸浮于20μL加樣緩沖液(0.05MTris-HClpH6.8;40g/L的十二烷基硫酸鈉;20mL/L甘油;2g/L溴酚藍和0.02M二硫蘇糖醇;所有化學品均來自Sigma-Aldrich)并加載到10%的十二烷基硫酸鈉-聚丙烯酰胺凝膠(SDS-PAGE;Bio-RadLaboratoriesSrL,Segrate,MI,IT)上,在電泳并轉移到硝酸纖維素膜(GEHealthcareEuropeGmbH,Milano,MI,IT)上以后,用在磷酸鹽緩沖鹽水(PBS,Sigma-Aldrich),pH7.4中的5%非脂乳(Bio-Rad)/0.1%吐溫-20來封閉膜1小時,然后在4℃下用在0.1%吐溫-20/PBS中的一抗(兔多克隆抗iNOS或抗TREM21:1000,兩者均來自SantaCruzBiotechnologyInc.,SantaCruz,CA,USA,或小鼠單克隆抗β肌動蛋白,1:2000,Sigma-Aldrich)加以溫育過夜。然后在室溫下在0.1%吐溫-20/PBS中用與辣根過氧化物酶(1:2000;SantaCruz)結合的抗兔或抗小鼠二抗來溫育膜90分鐘。通過使用增強化學發光方法(ECL;Bio-RAD)來檢測標記蛋白。通過利用來自NIH的ScionImage軟件來進行光密度分析。如圖5所示,當用LPS刺激原代大鼠膠質細胞的培養物時,我們觀測到預期的iNOS誘導,通過用實施例3和6的化合物的24小時共處理,iNOS誘導以劑量依賴的方式減小(圖5a和b)。此外,沿著相同的方法,在用LPS處理的小膠質細胞中,我們觀測到TREM2表達的減小,通過用實施例3和6的化合物24小時共處理,TREM2表達得到恢復(圖5c)。表2當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- SeZn7MT3的組成、制備方法及其在防治阿爾茨海默癥中的應用與流程
- 纖維支架材料、其制備方法、及亞甲藍負載量的調節方法與流程
- 大駁骨在制備預防和/或治療神經退行性疾病的藥物中的應用及藥物的制造方法與工藝
- 花葉假杜鵑在制備預防和/或治療神經退行性疾病的藥物中的應用及藥物的制造方法與工藝
- 大腦內穩態調節蛋白的組成、制備方法及其在防治阿爾茨海默癥中的應用與流程
- 一種藥物組合物及其制備方法和在制備防治神經退行性疾病的藥物中的應用與流程
- 片層碳?四氧化三鐵復合體在抑制Aβ聚集中的應用的制造方法與工藝
- 一種干細胞球切割收集裝置及干細胞球的切割分離傳代方法與流程
- 一種具有改善記憶功效的多肽及其分離制備方法和應用與制造工藝
- 治療或預防神經退行性病癥的組合物和方法與制造工藝
- SeZn7MT3的組成、制備方法及其在防治阿爾茨海默癥中的應用與流程
- 纖維支架材料、其制備方法、及亞甲藍負載量的調節方法與流程
- 大駁骨在制備預防和/或治療神經退行性疾病的藥物中的應用及藥物的制造方法與工藝
- 花葉假杜鵑在制備預防和/或治療神經退行性疾病的藥物中的應用及藥物的制造方法與工藝
- 大腦內穩態調節蛋白的組成、制備方法及其在防治阿爾茨海默癥中的應用與流程
- 一種藥物組合物及其制備方法和在制備防治神經退行性疾病的藥物中的應用與流程
- 片層碳?四氧化三鐵復合體在抑制Aβ聚集中的應用的制造方法與工藝
- 一種干細胞球切割收集裝置及干細胞球的切割分離傳代方法與流程
- 一類抗氧化超短肽及其應用的制造方法與工藝
- 一種具有改善記憶功效的多肽及其分離制備方法和應用與制造工藝