1,3?二取代環丁烷?1,2,3,4?四羧酸及其酸二酐的新型制造方法與流程
本發明涉及用于以高選擇性且高收率獲得1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐的新型制造方法。
背景技術:
1,3-二取代環丁烷-1,2,3,4-四羧酸二酐作為各種工業用途的原料是有用的。另外,成為所述1,3-二取代環丁烷-1,2,3,4-四羧酸二酐的原料的1,3-二取代環丁烷-1,2,3,4-四羧酸也是有用的化合物。
尤其是,式(12a)所示的1,3-二甲基環丁烷-1,2,3,4-四羧酸二酐和成為該酸二酐的原料的式(13a)所示的1,3-二甲基環丁烷-1,2,3,4-四羧酸是作為液晶表示元件、半導體中的保護材料、絕緣材料、濾色器、液晶取向膜、光波導用材料等而廣泛使用的聚酰亞胺的主要原料或合成中間體(參照專利文獻1和非專利文獻1)。
作為式(12a)所示的1,3-二甲基環丁烷-1,2,3,4-四羧酸二酐和式(13a)所示的1,3-二甲基環丁烷-1,2,3,4-四羧酸的結構方面的特征,可列舉出環丁烷環上的兩個甲基位于1位和3位且該甲基的相對構型為反式。
以往,作為1,3-二取代環丁烷-1,2,3,4-四羧酸二酐的制造方法,已知下述制造方法。
根據非專利文獻2,使檸康酸酐和作為光敏劑的二苯甲酮溶解于1,4-二噁烷,對該溶液照射光而進行環化反應,從而合成在環丁烷環上取代有兩個甲基的二甲基環丁烷四羧酸二酐。進而,對所得二甲基環丁烷四羧酸二酐進行水解反應,接著,進行甲酯化反應,從而合成二甲基環丁烷-1,2,3,4-四羧酸四甲酯。但是,尚未確定環丁烷環上的甲基的位置和相對構型。
根據非專利文獻3和非專利文獻4,按照非專利文獻2記載的方法,由檸康酸酐合成二甲基環丁烷-1,2,3,4-四羧酸二酐后,再合成二甲基環丁烷-1,2,3,4-四羧酸四甲酯。進而,記載了有可能生成由環丁烷環上的甲基位置和相對構型帶來的4種二甲基環丁烷-1,2,3,4-四羧酸二酐的非對映體。
根據專利文獻2,使檸康酸酐1000g溶解于醋酸乙酯,對該溶液照射光而進行環化反應,從而以695g的收量合成了檸康酸酐的二聚體。但是,針對所述檸康酸酐的二聚體、針對結構確定和非對映體的生成比沒有任何記載。
根據專利文獻3報告了:通過使乙基馬來酸酐溶解于醋酸乙酯,并對該溶液照射光而進行環化反應,能夠獲得1,3-二乙基環丁烷-1,2,3,4-四羧酸1,2:3,4-二酐與1,2-二乙基環丁烷-1,2,3,4-四羧酸1,4:2,3-二酐的混合物。針對該混合物進行溶劑蒸餾去除、過濾、析晶等精制操作,但尚未實現彼此的分離精制。另外,通過收量和NMR算出的收率為中等程度。
現有技術文獻
專利文獻
專利文獻1:國際公開(WO)2010/092989小冊子
專利文獻2:日本特開平4-106127號公報
專利文獻3:日本特開2006-347931號公報
非專利文獻
非專利文獻1:新訂最新聚酰亞胺基礎和應用(日文:新訂最新ポリイミド基礎と応用)(ISBN 978-4-86043-273-7)、2010年、NTS Co.,Ltd.、344-354頁
非專利文獻2:化學學報(德文:Chemische Berichte)1962年、95卷、1642-1647頁
非專利文獻3:有機化學雜志(Journal of Organic Chemistry)1968年、33卷、920-921頁
非專利文獻4:化學學報(德文:Chemische Berichte)1988年、121卷、295-297頁
技術實現要素:
發明要解決的問題
以往,尚未獲知在構筑環丁烷環的反應時能夠同時控制該環丁烷環上的取代基位置和相對構型的1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐的高選擇性制造方法。因此,在獲得目標化合物時,需要進行精制操作。具體而言,存在如下課題:需要利用升華來精制、使用大量有機溶劑進行析晶、利用懸浮清洗乃至過濾等進行復雜的精制操作,產生大量的廢液、廢棄物,從清潔化學的觀點出發,對環境造成巨大的負擔,生產率差。
本發明的目的在于,提供用于以高選擇性和高收率獲得1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐的新型制造方法,所述1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐作為各種工業用途的原料是有用,作為期望的立體結構,同時滿足:環丁烷環上的兩個取代基位于1位和3位,且該取代基的相對構型為反式。
用于解決問題的方案
本發明人為了解決上述課題而重復進行了深入研究,結果發現:通過生成由乙烯二羧酸衍生物和含氮有機化合物形成的晶體,并對該晶體照射光而進行環化反應,能夠以高選擇性和高收率獲得具有期望立體結構的1,3-二取代環丁烷-1,2,3,4-四羧酸。本發明基于所述見解,具有下述主旨。
〔1〕用式(2)表示的1,3-二取代環丁烷-1,2,3,4-四羧酸的制造方法,其具有下述工序(a)和工序(b):
(R1表示C1~C4烷基、苯基或鹵素原子。)
工序(a):在存在或不存在溶劑的條件下,制造由含氮有機化合物(5)與式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶體(1)的工序。
(R1表示與上述相同的意義。)
工序(b):對通過工序(a)得到的晶體(1)照射光而進行環化反應的工序。
〔2〕根據上述〔1〕所述的制造方法,其中,含氮有機化合物(5)為脂肪族胺、芳香族胺、氧化胺、酰胺、酰亞胺或含氮雜環式化合物。
〔3〕根據上述〔2〕所述的制造方法,其中,含氮有機化合物(5)為含氮雜環式化合物。
〔4〕根據上述〔3〕所述的制造方法,其中,含氮雜環式化合物為煙酰胺或吡啶。
〔5〕根據上述〔1〕~〔4〕中任一項所述的制造方法,其中,在工序(b)中,照射波長為290nm~600nm的光。
〔6〕根據上述〔1〕~〔4〕中任一項所述的制造方法,其中,在工序(b)中,照射波長為300nm~580nm的光。
〔7〕根據上述〔1〕~〔6〕中任一項所述的制造方法,其中,在工序(b)中,在光敏劑的存在下照射光。
〔8〕根據上述〔1〕~〔7〕中任一項所述的制造方法,其中,在式(4C)或式(4M)中,R1表示甲基或乙基。
〔9〕根據上述〔1〕~〔8〕中任一項所述的制造方法,其使用式(4C)所示的化合物。
〔10〕用式(3)表示的1,3-二取代環丁烷-1,2,3,4-四羧酸二酐的制造方法,其通過使利用上述〔1〕所述的制造方法得到的式(2)所示的1,3-二取代環丁烷-1,2,3,4-四羧酸發生脫水縮合反應來制造。
(R1表示C1~C4烷基、苯基或鹵素原子。)
(式中,R1表示與上述相同的意義。)
〔11〕根據上述〔10〕所述的制造方法,其中,在醋酸酐的存在下進行脫水縮合反應。
〔12〕一種晶體,其由吡啶和檸康酸形成,所述晶體在基于Cu-Kα射線的粉末X射線衍射中在衍射角2θ=(12.58±0.2,15.05±0.2,16.08±0.2,17.60±0.2,19.20±0.2,21.57±0.2,23.02±0.2,24.50±0.2,26.45±0.2,27.06±0.2,28.10±0.2,32.49±0.2,35.90±0.2,36.46±0.2和38.43±0.2)處具有峰。
〔13〕根據上述〔12〕所述的晶體,其中,在基于Cu-Kα射線的粉末X射線衍射中在衍射角2θ=(12.58±0.2,15.05±0.2,16.08±0.2,17.60±0.2,18.34±0.2,19.20±0.2,21.57±0.2,23.02±0.2,24.50±0.2,26.45±0.2,27.06±0.2,28.10±0.2,30.39±0.2,32.49±0.2,34.71±0.2,35.90±0.2,36.46±0.2和38.43±0.2)處具有峰。
發明的效果
根據本發明,通過由乙烯二羧酸衍生物和含氮有機化合物制造晶體,接著對該晶體照射光而進行環化反應,能夠以高選擇性和高收率制造具有期望的立體結構的1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐。進而,本發明的制造方法在制造1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐時基本不產生副產物,因此,能夠顯著減輕為了獲得具有期望的立體結構的化合物而進行的精制操作,因此,作為考慮了環境負擔的工業制造方法是有用的。
附圖說明
圖1是實施例中制造的由吡啶和檸康酸形成的晶體(8)的FT-IR譜圖。
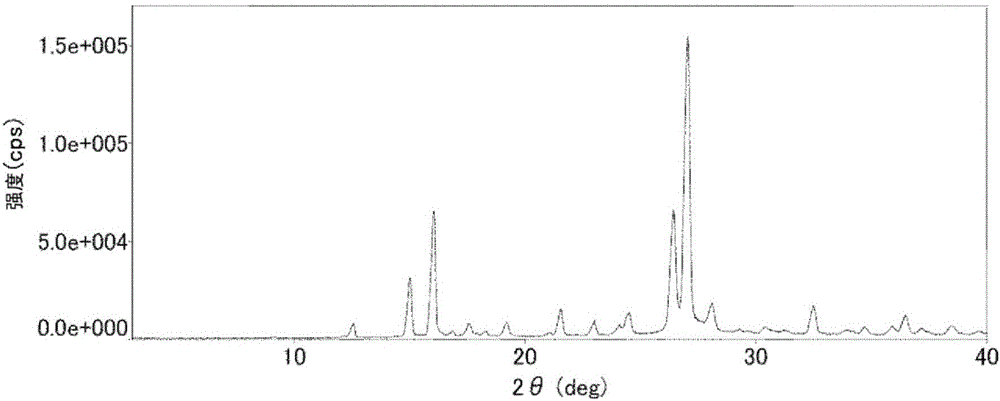
圖2是實施例中制造的由吡啶和檸康酸形成的晶體(8)的粉末X射線衍射譜圖。
圖3是根據實施例中制造的晶體(8)的單晶X射線結構分析結果,利用圓柱模型一并示出吡啶嗡與檸康酸陰離子的堆積結構以及晶胞而得到的圖。
圖4是根據實施例中制造的晶體(8)的單晶X射線結構分析結果,利用ORTEP圖僅示出檸康酸陰離子彼此的構型而得到的圖。
圖5是根據實施例中制造的化合物(13a)的單晶X射線結構分析結果,利用ORTEP圖示出化合物(13a)的分子結構的圖。
圖6是根據實施例中制造的化合物(12a)的單晶X射線結構分析結果,利用ORTEP圖示出化合物(12a)的分子結構的圖。
圖7是實施例9所使用的流動反應器的示意圖。
圖8是實施例9所使用的流動反應器中的具有雙層管結構的T字型混合器(混合器2)的剖面圖。
具體實施方式
本說明書中,下述符號分別表示下述意義。
“n-”表示正,“s-”表示仲,“t-”表示叔,“o-”表示鄰,“m-”表示間,“p-”表示對。另外,“trans”、“cis”表示環狀化合物的取代基的相對構型,該取代基位于環平面的相反側時示為反式(trans),位于相同側時示為順式(cis)。另外,(E)和(Z)表示下述立體化學:用雙鍵連結的分子的平面部分內鍵合于形成雙鍵的原子上的基團之中,優先級規則的上位的基團存在于相反側時示為(E),存在于同一側時示為(Z)。
“Me”的表述是指甲基。Ca~Cb烷基表示從包含碳原子數a~b個的直鏈狀或支鏈狀的脂肪族烴中喪失1個氫原子而生成的一價基團。作為具體例,可列舉出例如甲基、乙基、正丙基、異丙基、正丁基、異丁基、仲丁基、叔丁基、正戊基、異戊基、新戊基、叔戊基、1,1-二甲基丙基、正己基、異己基等。
作為鹵素原子,可列舉出氟原子、氯原子、溴原子、碘原子等。另外,鹵素的表述也表示這些鹵素原子。
以下,針對本發明中的1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐的制造方法、即工序(a)~工序(c)進行說明。
工序(a):制造由乙烯二羧酸衍生物和含氮有機化合物形成的晶體
本工序中,獲得由含氮有機化合物(5)與式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶體(1)。式中,R1表示C1~C4烷基、苯基或鹵素原子,其中,R1優選為甲基、乙基或苯基的情況,進一步優選為甲基或乙基的情況。
本說明書中的由含氮有機化合物(5)與式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶體(1)在室溫下為固體,具有特征性的粉末X射線衍射峰,是由2種以上的化合物以一定的化學計量比形成的晶體。所述晶體(1)中,作為構成要素的2種以上的化合物在分子或離子層面上具有三維且周期性排列的結構。另外,晶體(1)由2種以上的成分構成,因此也可以說是所謂的多成分晶體。此處,室溫是指1℃~40℃。
另一方面,所述晶體(1)中有時存在下述部分:晶體表面等處的周期性排列發生了崩塌的部分;乙烯二羧酸衍生物和含氮有機化合物在分子或離子層面上發生了聚集而不呈現周期結構的部分。但是,盡管局部具有三維周期性排列缺損的結構但包含上述三維且周期性排列的結構的晶體通過照射光而進行環化反應,從而能夠高選擇性地制造具有期望立體結構的1,3-二取代環丁烷-1,2,3,4-四羧酸時,也包括在本發明的晶體(1)中。
本發明中,工序(a)的由乙烯二羧酸衍生物和含氮有機化合物形成的晶體(1)的結構成為后續工序(b)中的1,3-二取代環丁烷-1,2,3,4-四羧酸、進而工序(c)中的1,3-二取代環丁烷-1,2,3,4-四羧酸二酐的立體選擇性的決定性因素,因此,晶體(1)中的乙烯二羧酸衍生物的分子取向是重要的。本發明中的優選晶體(1)中,兩個乙烯二羧酸衍生物以在后續工序的工序(b)的光環化反應中構筑兩個甲基位于1位和3位且該甲基的相對構型呈現反式的環丁烷環的方式取向。
本發明中使用的屬于乙烯二羧酸衍生物的物質是公知化合物,一部分可以以市售品的形式獲取。例如,檸康酸可以由東京化成工業株式會社、Aldrich公司等獲取。另外,苯基馬來酸可以由HYDRUS CHEMICAL INC.獲取。另外,中康酸可以由東京化成工業株式會社、Aldrich公司等來獲取。
另外,本發明中使用的乙烯二羧酸衍生物的一部分可以按照文獻記載的公知方法來合成。例如,2-異丙基馬來酸可以參照合成通訊(Synthetic Communications)、2013年、43(10)卷、1455-1459頁等來合成,苯基富馬酸可以參照美國化學會志(Journal of the American Chemical Society)、1954年、76卷、1872-1873頁等來合成。
作為本發明中使用的式(4C)所示的乙烯二羧酸衍生物的具體例,可列舉出例如檸康酸、2-乙基馬來酸、2-異丙基馬來酸、2-丙基馬來酸、2-正丁基馬來酸、2-異丁基馬來酸、2-(叔丁基)馬來酸、2-苯基馬來酸、2-氟馬來酸、2-氯馬來酸、2-溴馬來酸、2-碘馬來酸等。
其中,優選為檸康酸、2-乙基馬來酸、2-異丙基馬來酸或2-苯基馬來酸,特別優選為檸康酸或2-乙基馬來酸。
作為本發明中使用的式(4M)所示的乙烯二羧酸衍生物的具體例,可列舉出例如中康酸、2-乙基富馬酸、2-異丙基富馬酸、2-丙基富馬酸、2-正丁基富馬酸、2-異丁基富馬酸、2-(叔丁基)富馬酸、2-苯基富馬酸、2-氟富馬酸、2-氯富馬酸、2-溴富馬酸、2-碘富馬酸等。
其中,優選為中康酸、2-乙基富馬酸、2-苯基富馬酸或2-氟富馬酸,特別優選為中康酸或2-乙基富馬酸。
作為含氮有機化合物(5),可以使用各種化合物。例如可列舉出:在生成上述晶體(1)時,通過在晶體結構中在酸性的式(4C)或式(4M)所示的乙烯二羧酸衍生物與含氮有機化合物(5)之間發生質子移動,從而借助所形成的離子鍵來生成晶體的例子。因此,可以使用能夠與酸性的乙烯二羧酸衍生物發生中和反應那樣的具有堿性的多種含氮有機化合物。
另外,可以使用具有如下取代基、部分骨架的多種含氮有機化合物,所述取代基、部分骨架在生成晶體(1)時,能夠與乙烯二羧酸衍生物形成氫鍵、范德華力等分子間相互作用。作為該取代基,可列舉出氨基、酰胺基、亞氨基、醚基、羥基、羰基、羧基等,作為該部分骨架,可列舉出吡咯系骨架、吡咯啉系骨架、吡咯烷系骨架、吲哚系骨架、吲哚啉系骨架、異吲哚系骨架、咪唑啉系骨架、咪唑烷系骨架、吡啶系骨架、哌啶系骨架、喹啉系骨架、吖啶系骨架、三嗪系骨架等。
本發明中,在后續的工序(b)中,對于由含氮有機化合物(5)與式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶體(1)中的乙烯二羧酸衍生物的雙鍵照射光而使其進行環化反應,因此,作為含氮有機化合物,優選不阻礙光環化反應的推進。因此優選為下述化合物:不與乙烯二羧酸衍生物反應而產生副產物的化合物、具有在生成目標化合物之前不易進行晶體(1)被破壞之類的異構化反應等的結構的化合物、具有耐光照射性的結構的化合物等。作為所述含氮有機化合物的優選例,可列舉出脂肪族胺、芳香族胺、氧化胺、酰胺、酰亞胺、含氮雜環式化合物等。
作為上述脂肪族胺,可列舉出甲胺、乙胺、異丙胺、丁胺、異丁胺、仲丁胺、叔丁胺、戊胺、異戊胺、2-戊胺、叔戊胺、己胺、庚胺、辛胺、2-辛胺、2-乙基己胺、壬胺、癸胺、芐胺、苯乙胺、α-甲基芐胺、墨斯卡靈、多巴胺、二丙胺、二異丙胺、N-甲基乙胺、N-乙基異丁胺、二丁胺、二異丁胺、二芐胺、N,N-二甲基丙胺、三丙胺、N-乙基-N-甲基丁胺、三丁胺、N,N-二甲基芐胺、N,N-二乙基芐胺、三芐胺、環丙胺、環丁胺、環己胺、二環己胺、N,N-二甲基環己胺、環己烷-1β,2β-二胺、(1R,2R)-1,2-環己烷二胺、(1S,2S)-1,2-環己烷二胺等。其中,優選為甲胺、乙胺、環己烷-1β,2β-二胺、(1R,2R)-1,2-環己烷二胺、或者(1S,2S)-1,2-環己烷二胺。
作為上述芳香族胺,可列舉出鄰甲苯胺、間甲苯胺、對甲苯胺、鄰乙基苯胺、間乙基苯胺、對乙基苯胺、對異丙基苯胺、對叔戊基苯胺、二甲苯胺、2,3-二甲苯胺、2,4-二甲苯胺、2,6-二甲苯胺、3,4-二甲苯胺、3,5-二甲苯胺、百里基胺、2,4,5-三甲基苯胺、2,4,6-三甲基苯胺、五甲基苯胺、1-萘胺、2-萘胺、1-蒽基胺、2-蒽基胺、9-蒽基胺、N-丁基苯胺、N-異戊基苯胺、N-芐基苯胺、N-芐基-N-乙基苯胺、N,N-二苯基芐胺、N-甲基鄰甲苯胺、N-甲基對甲苯胺、N,N-二甲基苯胺、N,N-二乙基苯胺、N,N-二丁基苯胺、N,N-二戊基苯胺、N,N-甲基鄰甲苯胺、N,N-甲基間甲苯胺、N,N-甲基對甲苯胺、二苯基胺、二對甲苯胺、N-甲基二苯基胺、N-乙基二苯基胺、三苯基胺、N,N-二芐基苯胺、N-芐基-N-乙基苯胺、鄰苯二胺、間苯二胺、對苯二胺等。其中,優選為鄰甲苯胺、間甲苯胺、對甲苯胺、鄰乙基苯胺、間乙基苯胺或對乙基苯胺。
作為上述氧化胺,可列舉出三甲基氧化胺、吡啶1-氧化物、2,2’-聯吡啶1,1’-二氧化物、4,4’-二甲基-2,2’-聯吡啶1,1’-二氧化物、3,3’-二甲基-2,2’-聯吡啶1,1’-二氧化物等。其中,優選為三甲基氧化胺、吡啶1-氧化物或3,3’-二甲基-2,2’-聯吡啶1,1’-二氧化物。
作為上述酰胺,可列舉出甲酰胺、N,N-二甲基甲酰胺、N,N-二異丙基甲酰胺、乙酰胺、N-乙基乙酰胺、N,N-二甲基乙酰胺、N-氯乙酰胺、N-溴乙酰胺、二乙酰胺、三乙酰胺、丙酰胺、丁酰胺、異丁酰胺、戊酰胺、異戊酰胺、己酰胺、庚酰胺、辛酰胺、丙烯酰胺、氯乙酰胺、二氯乙酰胺、三氯乙酰胺、羥基乙酰胺、乳酰胺、丙酮酰胺、氰基乙酰胺、雷尿酸(fulminuric acid)、草酰胺、丙二酰胺、琥珀酰胺、己二酰胺、L-蘋果酰胺、(R,R)-酒石酰胺等。其中,優選為甲酰胺、N,N-二甲基甲酰胺、N,N-二異丙基甲酰胺或乙酰胺。
作為上述酰亞胺,可列舉出琥珀酰亞胺、N-溴代琥珀酰亞胺、N-氯代琥珀酰亞胺等。其中,優選為琥珀酰亞胺。
作為上述含氮雜環式化合物,可列舉出吡咯系化合物、吡咯啉系化合物、吡咯烷系化合物、吲哚系化合物、吲哚啉系化合物、異吲哚系化合物、咔唑系化合物、二唑系化合物、咪唑啉系化合物、咪唑烷系化合物、吡啶系化合物、吡啶的取代衍生物、哌啶系化合物、喹啉系化合物、氫喹啉系化合物、異喹啉系化合物、吖啶系化合物、菲啶系化合物、二嗪系化合物、磺胺嘧啶系化合物、氫嘧啶系化合物、哌嗪系化合物、苯并二嗪系化合物、三唑系化合物、苯并三唑系化合物、三嗪系化合物、嘌呤、次黃嘌呤、黃嘌呤、可可堿、茶堿、咖啡因、尿酸、腺嘌呤、鳥嘌呤、3-甲基尿酸、7-甲基尿酸等。
作為上述吡咯系化合物,可列舉出吡咯、甲基吡咯、二甲基吡咯、3-乙基-4甲基吡咯、乙基二甲基吡咯、3-乙基-2,4,5-三甲基吡咯、2,3,4,5-四甲基吡咯、乙酰基吡咯等。作為上述吡咯啉系化合物,可列舉出吡咯啉等。作為上述吡咯烷系化合物,可列舉出吡咯烷等。作為上述吲哚系化合物,可列舉出吲哚、假吲哚、甲基吲哚、2,3-二甲基吲哚、2-苯基吲哚等。作為上述吲哚啉系化合物,可列舉出吲哚啉、靛紅、O-甲基靛紅、N-甲基靛紅、2-氯-3-吲哚酮等。
作為上述異吲哚系化合物,可列舉出異吲哚、異吲哚啉、苯并吡咯酮等。作為上述咔唑系化合物,可列舉出咔唑、靛藍、靛白(leucoindigo)、靛玉紅等。作為上述二唑系化合物,可列舉出吡唑、3,5-二甲基吡唑、2-吡唑啉、吡唑烷、吡唑啉酮、3-甲基-1-苯基-5-吡唑啉酮、2,3-二甲基-1-苯基-5-吡唑啉酮、氨基比林、吲唑等。作為上述咪唑啉系化合物,可列舉出咪唑啉、苦杏素(amarine)、萘甲唑啉等。作為上述咪唑烷系化合物,可列舉出乙烯脲、乙內酰脲、1-甲基乙內酰脲、5-甲基乙內酰脲、二苯基乙內酰脲、肌酸酐等。
作為上述吡啶系化合物,可列舉出吡啶、2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-乙基吡啶、3-乙基吡啶、4-乙基吡啶、2-丙基吡啶、2,3-二甲基吡啶、2,4-二甲基吡啶、2,5-二甲基吡啶、2,6-二甲基吡啶、3,4-二甲基吡啶、3,5-二甲基吡啶、4-乙基-2-甲基吡啶、3-乙基-4-甲基吡啶、5-乙基-2-甲基吡啶、6-乙基-2-甲基吡啶、2,4,6-三甲基吡啶、2,3,4-三甲基吡啶、4-乙基-2,6-二甲基吡啶、2-苯基吡啶、3-苯基吡啶、4-苯基吡啶、2-芐基吡啶、3-芐基吡啶、4-芐基吡啶、2,2’-聯吡啶、3,3’-聯吡啶、4,4’-聯吡啶等。
作為上述吡啶的取代衍生物,可列舉出2-氯吡啶、3-氯吡啶、4-氯吡啶、2-吡啶酮、3-吡啶酚、4-吡啶酮、2-甲氧基吡啶、3-甲氧基吡啶、4-甲氧基吡啶、2-吡啶甲醛、3-吡啶甲醛、4-吡啶甲醛、2-乙酰基吡啶、3-乙酰基吡啶、4-乙酰基吡啶、2-乙氧基吡啶1-氧化物、2-吡啶羧酸、煙酸、異煙酸、煙酰胺、尼可剎米、異煙肼、2-乙基硫異煙胺、2,3-吡啶二羧酸、2,4-吡啶二羧酸、2,5-吡啶二羧酸、2,6-吡啶二羧酸、3,4-吡啶二羧酸、3,5-吡啶二羧酸、3-硝基吡啶、2-吡啶胺、3-吡啶胺、4-吡啶胺、N,N-二甲基-4-吡啶胺等。
作為上述哌啶系化合物,可列舉出哌啶、2-甲基哌啶、毒芹堿(coniine)、3-甲基哌啶、4-甲基哌啶、N-甲基哌啶、1-苯基哌啶、2,6-二甲基哌啶、N-苯甲酰基哌啶、4-哌啶酮等。作為上述喹啉系化合物,可列舉出喹啉、2-甲基喹啉、3-甲基喹啉、4-甲基喹啉、6-甲基喹啉、8-甲基喹啉、2,3-二甲基喹啉、2,4-二甲基喹啉、2,6-二甲基喹啉、2-苯基喹啉、6-苯基喹啉、8-苯基喹啉、2-氯喹啉、2-喹諾酮、4-喹諾酮、5-喹啉醇、6-喹啉醇、7-喹啉醇、8-喹啉醇、α-萘喹啉、β-萘喹啉、2-甲基-4-喹啉醇、4-甲基-2-喹啉醇、6-甲氧基喹啉、2,4-喹啉二醇、2-喹啉羧酸、3-喹啉羧酸、4-喹啉羧酸、5-喹啉羧酸、5-硝基喹啉、6-硝基喹啉、7-硝基喹啉、8-硝基喹啉、2-喹啉胺、3-喹啉胺、4-喹啉胺、黃苯胺、2,2’-二喹啉、3,3’-二喹啉、5,5’-二喹啉、6,6’-二喹啉、2,3’-二喹啉等。
作為上述氫喹啉系化合物,可列舉出1,2,3,4-四氫喹啉、1-甲基-1,2,3,4-四氫喹啉、6-甲氧基-1,2,3,4-四氫喹啉、3,4-二氫-2-喹諾酮、順式-十氫喹啉等。作為上述異喹啉系化合物,可列舉出異喹啉、1-甲基異喹啉、1-異喹諾酮、1,2,3,4-四氫異喹諾酮、1-芐基異喹啉等。作為上述吖啶系化合物,可列舉出吖啶、2-甲基吖啶、3-甲基吖啶、9-甲基吖啶、9-苯基吖啶、3-氨基-9-(對氨基苯基)吖啶、3,6-二氨基-10-甲基氯化吖啶、吖啶滿、吖啶酮、吖啶醇等。
作為上述菲啶系化合物,可列舉出菲啶、苯并[f]喹啉、苯并[g]喹啉、苯并[h]喹啉、苯并[g]異喹啉、1,10-菲咯啉、2,9-二甲基-1,10-菲咯啉、4,7-二苯基-1,10-菲咯啉、2,9-二甲基-4,7-二苯基-1,10-菲咯啉等。作為上述二嗪系化合物,可列舉出噠嗪、嘧啶、吡嗪、2,5-二甲基吡嗪、四苯基吡嗪等。
作為上述磺胺嘧啶系化合物,可列舉出磺胺嘧啶、碘胺二甲氧噠嗪、磺胺苯吡唑、磺胺甲噻二唑、磺胺甲氧基噠嗪、磺胺異噁唑、磺胺索嘧啶等。作為上述氫嘧啶系化合物,可列舉出尿嘧啶、胸腺嘧啶、撲米酮、2-硫尿嘧啶、胞嘧啶、巴比妥酸、5,5-二乙基巴比妥酸、5,5-二丙基巴比妥酸、阿洛巴比妥、5-乙基-5-苯基巴比妥酸、環巴比妥(cyclobarbital)、環己烯巴比妥(hexobarbital)、5-羥基巴比妥酸、5-硝基巴比妥酸、5-氨基巴比妥酸、異戊巴比妥、紫尿酸、阿脲、雙阿脲、紅紫酸、紫螺酸銨、苦杏酸等。作為上述哌嗪系化合物,可列舉出哌嗪、2,5-二甲基哌嗪、2,5-哌嗪二酮等。作為上述苯并二嗪系化合物,可列舉出噌啉、酞嗪、喹唑啉、喹喔啉等。作為上述三唑系化合物,可列舉出1,2,3-三唑、1,2,4-三唑、4-氨基-1,2,4-三唑等。
作為上述苯并三唑系化合物,可列舉出苯并三唑、5-甲基苯并三唑等。作為上述三嗪系化合物,可列舉出1,2,3-三嗪、4-甲基-1,2,3-三嗪、4,6-二甲基-1,2,3-三嗪、4,5,6-三甲基-1,2,3-三嗪、1,2,3-苯并三嗪、4-甲基-1,2,3-苯并三嗪、1,3,5-三嗪、氰尿酰氯、氰尿酸、氰尿酸三甲酯、異氰尿酸甲酯、異氰尿酸乙酯、黑色素、氰尿二酰胺、氰尿酰胺等。
本發明中,作為優選的含氮雜環式化合物,可列舉出吡啶系化合物、吡啶的取代衍生物。作為優選的吡啶系化合物,可列舉出吡啶、2,3-二甲基吡啶、3,5-二甲基吡啶,作為更優選的吡啶系化合物,可列舉出吡啶。作為優選的吡啶的取代衍生物,可列舉出煙酸、異煙酸、煙酰胺、尼可剎米,作為更優選的吡啶的取代衍生物,可列舉出煙酰胺。
上述含氮有機化合物可以單獨使用,也可以組合使用這些之中的2種以上。
本發明中,由含氮有機化合物(5)與式(4C)或式(4M)所示的乙烯二羧酸衍生物形成的晶體(1)可通過將兩者混合而使其充分接觸來制造。關于制造晶體(1)時的含氮有機化合物的用量,相對于乙烯二羧酸衍生物1當量,含氮有機化合物優選為0.1~50當量、更優選為0.2~15當量、特別優選為0.5~3當量。
尤其是,含氮有機化合物為液體時,也可以用作乙烯二羧酸衍生物的溶劑,乙烯二羧酸衍生物為液體時,也可以用作含氮有機化合物的溶劑。
制造晶體(1)時的溫度沒有特別限定,可以在-78℃~反應混合物的回流溫度內任意設定。其中,優選為-30~70℃,更優選為-20~40℃。
制造晶體(1)時的壓力可以為加壓、常壓或減壓中的任一者,優選為0.5~10atm,更優選為0.9~2atm。
制造晶體(1)時的將式(4C)或式(4M)所示的乙烯二羧酸衍生物與含氮有機化合物(5)進行混合的方法可以使用如下方法:將兩者直接混合的方法、使用溶劑進行混合的方法等。
作為將兩者直接混合的方法,可以使用捏合法、混合粉碎法等。捏合法可應用于一者為固體且另一者為液體時的混合,在少量的情況下可以使用乳缽,在大量的情況下可以使用捏合機、有機合成用的反應容器、攪拌機等。
乙烯二羧酸衍生物和含氮有機化合物均為固體時,可以應用混合粉碎法,在少量的情況下可以使用乳缽,在大量的情況下可以使用粉碎機等。利用捏合法或混合粉碎法進行混合時,有時因中和反應等而伴有發熱,因此,也可以一邊對乳缽、反應容器等進行冷卻一邊實施。
作為將乙烯二羧酸衍生物和含氮有機化合物用溶劑進行混合的方法,可以采取使用了溶劑的溫度控制法、溶劑蒸發法、溶劑蒸餾去除法、不良溶劑添加法、溶液-溶液混合法、懸浮液-溶液混合法、懸浮液-懸浮液混合法等。另外,這些混合時,有時因溶解、中和反應等而伴有發熱,因此,也可以一邊對反應容器等進行冷卻一邊實施。
通過上述使用了溶劑的溫度控制法來制造晶體(1)的方法是如下方法:首先,使乙烯二羧酸衍生物與含氮有機化合物的反應混合物預先溶解于溶劑,利用高溫與低溫的溶解度差來獲得晶體的方法。
通過上述使用了溶劑的溶劑蒸發法或溶劑蒸餾去除法來制造晶體(1)的方法是首先使乙烯二羧酸衍生物與含氮有機化合物的反應混合物溶解于溶劑,并使溶劑蒸發或蒸餾去除的方法,在使其緩慢蒸發的情況下,大多能夠獲得良好的晶體。蒸餾去除溶劑時,可以使用旋轉蒸發儀等。無法利用1種溶劑良好地進行結晶化時,也可以使用混合溶劑。
通過使用了溶劑的不良溶劑添加法來制造晶體(1)的方法是首先使乙烯二羧酸衍生物與含氮有機化合物的反應混合物溶解于溶劑,并通過添加不良溶劑來獲得晶體的方法。根據所用溶劑的不同,有時乙烯二羧酸衍生物與含氮有機化合物的反應混合物的一部分不會溶解,從而呈現在液體中分散有固體顆粒的漿料。
針對通過使用了溶劑的溶液和懸浮液來制造晶體(1)的方法,以下進行說明。首先,使乙烯二羧酸衍生物溶解于溶劑來制備溶液A。同樣制備含氮有機化合物的溶液B。進而,制備使乙烯二羧酸衍生物懸浮于溶劑的懸浮液A,同樣制備含氮有機化合物的懸浮液B。由上述溶液和懸浮液分別選擇乙烯二羧酸衍生物和含氮有機化合物的溶液或懸浮液并混合,從而得到晶體的方法。如下述那樣,混合有如下方法:同時滴加液體的方法、順序滴加的方法、或者反序添加的方法等。此時使用的溶劑可以僅為1種,也可以利用將多種溶劑混合得到的溶劑。
乙烯二羧酸衍生物的溶液A和懸浮液A的濃度只要不阻礙生成晶體(1)的反應就沒有特別限定,優選為0.01~100mol/L、更優選為0.05~10mol/L、特別優選為0.2~3mol/L。
含氮有機化合物的溶液B和懸浮液B的濃度只要不阻礙生成晶體(1)的反應就沒有特別限定,優選為0.01~100mol/L、更優選為0.05~10mol/L、特別優選為0.2~3mol/L。
溶液-溶液混合法是通過將上述溶液A與溶液B混合而得到晶體(1)的方法。此時的混合中,將同時滴加溶液A和溶液B的方法記作同時滴加,將向溶液B中滴加溶液A的方法記作順序滴加,將向溶液A中滴加溶液B的方法記作反序滴加。
懸浮液-溶液混合法是通過將上述溶液A與懸浮液B混合、或者將懸浮液A與溶液B混合而得到晶體(1)的方法。此時的混合可以從同時滴加、順序滴加或反序滴加中選擇。
懸浮液-懸浮液混合法是通過將上述懸浮液A與懸浮液B混合而得到晶體(1)的方法。此時的混合可以從同時滴加、順序滴加或反序滴加中選擇。
本發明中,也可以按照任意的順序將上述方法組合實施。例如,可以向該不良溶劑添加法中按照任意的順序組合溶劑蒸餾去除法、使用了溶劑的溫度控制法來實施,能夠構筑用于制造晶體(1)的條件。
使用了溶劑的晶體(1)的制造方法中,溶液或懸浮液的攪拌效率高是較好的。以下說明此時能夠使用的攪拌裝置,但不限定于下述記載。作為裝置,可列舉出捏合機、有機合成用的反應容器、攪拌機、超聲波發生器、流動反應器的混合器等。具體而言,可列舉出基于磁力攪拌棒和磁力攪拌器的攪拌、基于攪拌葉片的攪拌、基于非活性氣體鼓泡的攪拌、基于離心式攪拌體的攪拌、能夠配置在反應容器中的擋板等。針對攪拌葉片,優選為螺旋葉片、槳式葉片、MAXBLEND葉片(注冊商標)、盤式渦輪葉片、FULLZONE葉片(注冊商標)等,優選為MAXBLEND葉片(注冊商標)、盤式渦輪葉片或FULLZONE葉片(注冊商標)。
利用工序(a)制造的晶體(1)還因其性狀而異,可以通過過濾等來進行分離,也可以不分離地連續實施后續工序(b)中的照射光的環化反應。本說明書中,將后者稱為工序(a)和工序(b)的連續化。
使用溶劑將上述乙烯二羧酸衍生物與含氮有機化合物進行混合的方法中使用的溶劑只要不阻礙生成晶體(1)的反應就沒有特別限定。作為所述混合時使用的溶劑,可列舉出例如甲苯、鄰二甲苯等芳香族烴系溶劑、己烷、庚烷、石油醚等脂肪族烴系溶劑、環己烷等脂環式烴系溶劑、氯苯、鄰二氯苯等芳香族鹵代烴系溶劑、二氯甲烷、氯仿、四氯化碳、1,2-二氯乙烷、1,1,1-三氯乙烷、三氯乙烯、四氯乙烯等脂肪族鹵代烴系溶劑、二乙基醚、二異丙基醚、1,2-二甲氧基乙烷、四氫呋喃、1,4-二噁烷、環戊基甲基醚等醚系溶劑、三乙基胺、三丁基胺、N,N-二甲基苯胺等胺系溶劑、吡啶、甲基吡啶等吡啶系溶劑、醋酸乙酯、醋酸正丁酯、丙酸乙酯等酯系溶劑、甲醇、乙醇、正丙醇、2-丙醇、乙二醇等醇系溶劑、丙酮、甲基異丁基酮等酮系溶劑、碳酸二甲酯、碳酸二乙酯等碳酸酯系溶劑、乙腈、二甲基亞砜、環丁砜、1,3-二甲基-2-咪唑烷酮、乙二醇二乙酸酯、醋酸、水等。
其中,作為優選的溶劑,可列舉出甲苯、鄰二甲苯、己烷、庚烷、石油醚、鄰二氯苯、二氯甲烷、二乙基醚、四氫呋喃、1,4-二噁烷、吡啶、醋酸乙酯、醋酸正丁酯、甲醇、乙醇、2-丙醇、丙酮、甲基異丁基酮、碳酸二甲酯、碳酸二乙酯、乙腈、乙二醇二乙酸酯或醋酸。作為更優選的溶劑,可列舉出己烷、庚烷、四氫呋喃、吡啶、醋酸乙酯、醋酸正丁酯、甲醇、乙醇、碳酸二甲酯或醋酸。這些溶劑可以單獨使用,或者混合使用2種以上。
作為利用工序(a)制造的晶體(1)的代表例,將通過后述實施例1得到的由吡啶和檸康酸形成的晶體(8)在使用Cu-Kα輻射線的粉末X射線衍射中顯示峰值的衍射角2θ的值(也簡稱為峰值)示于〔表1〕。
〔表1〕
〔表1〕記載的晶體(8)的峰值是根據下述實施例1記載的方法得到的3個批次的晶體(8)的峰值的平均值。
應予說明,粉末X射線衍射的任意峰通常均存在±0.2的誤差。將考慮了該誤差值的晶體(8)的峰值示于〔表2〕。
〔表2〕
接著,將晶體(8)的粉末X射線衍射峰之中的特別的特征性峰值示于〔表3〕。
〔表3〕
將考慮了誤差值的晶體(8)的特征性峰值示于〔表4〕。
〔表4〕
工序(b):對由乙烯二羧酸衍生物和含氮有機化合物形成的晶體(1)照射光來進行環化反應,從而制造1,3-二取代環丁烷-1,2,3,4-四羧酸
本工序(b)中,通過對由乙烯二羧酸衍生物和含氮有機化合物形成的晶體(1)照射光,針對晶體中的乙烯二羧酸衍生物的雙鍵進行環化反應,從而制造式(2)所示的1,3-二取代環丁烷-1,2,3,4-四羧酸。式中的R1如上所示。
本發明的照射光的環化反應的特征在于,其是在晶體中、即固相中的反應,起始原料為固體狀態,因此在原子、分子的移動顯著受限的環境下發生。
作為本發明中的對晶體(1)照射光的環化反應的光源的波長,優選為280~600nm、更優選為290~600nm、進一步優選為300~580nm。
作為光源,可列舉出高壓汞燈、低壓汞燈、超高壓汞燈、氙氣燈、鈉燈、鹵素燈、氣體激光、液體激光、固體激光、太陽光、發光二極管等。其中,優選為高壓汞燈或發光二極管,特別優選為高壓汞燈。
光源為了冷卻而優選容納于夾套,作為此時的夾套材質,可列舉出石英玻璃、Pyrex(注冊商標)、HARIO玻璃、鉬玻璃、鈉玻璃、鉛玻璃、鎢玻璃、VYCOR(注冊商標)、合成石英玻璃(SUPRASIL)等。作為優選的夾套材質,為石英玻璃、Pyrex、HARIO玻璃、鉬玻璃等,作為更優選的夾套材質,為Pyrex。
作為本發明中的對晶體(1)照射光的環化反應中的光源照射方法,可以使用例如將光源設置于反應器內側的內部照射、設置于反應器外側的外部照射等。
作為具體的光源照射方法,有直接對晶體(1)進行光照射的方法、對使晶體(1)分散在溶劑中而得到的漿料照射光的方法等。對漿料照射光時,可以使用將光源設置于反應容器內部的內部照射以及設置于反應容器外側的外部照射中的任一者。
對晶體(1)照射光的環化反應中的氣氛可列舉出大氣、通常的空氣、氮氣、氦氣、氬氣等。其中,優選為大氣、空氣、氮氣或氬氣,特別優選為大氣或氮氣。
對晶體(1)照射光而實施環化反應時的反應溫度沒有特別限定,可以設定為-78℃~反應混合物的回流溫度。其中,優選為-40~90℃,更優選為-20~50℃。
對晶體(1)照射光而實施環化反應時的反應裝置沒有特別限定,從形狀出發,可以使用槽型、管型等,從操作方法出發,可以使用間歇式、連續式、半間歇式等。可列舉出例如間歇反應器、連續槽型反應器、活塞流反應器、流動反應器等。
工序(b)中,對使晶體(1)分散在溶劑中而得到的漿料照射光時,該漿料的攪拌效率優選較高。以下說明此時可以使用的攪拌裝置,但不限定于下述裝置。作為裝置,可列舉出有機合成用的反應容器、攪拌機、超聲波發生器、流動反應器的混合器等。具體而言,可列舉出基于磁力攪拌棒和磁力攪拌器的攪拌、基于攪拌葉片的攪拌、基于非活性氣體鼓泡的攪拌、基于離心式攪拌體的攪拌、能夠配置在反應容器中的擋板等。針對攪拌葉片,優選為螺旋葉片、槳式葉片、MAXBLEND葉片(注冊商標)、盤式渦輪葉片、FULLZONE葉片(注冊商標)等,優選為對于漿料的攪拌效率良好的攪拌葉片,更優選為MAXBLEND葉片(注冊商標)、盤式渦輪葉片或FULLZONE葉片(注冊商標)。
本發明中的對晶體(1)照射光的環化反應也可以在光敏劑的存在下進行。作為光敏劑,可列舉出例如苯、甲苯、丙酮、丁烷-2,3-二酮、杜烯、苯甲腈、苯丁酮、苯丙酮、苯乙酮、氧雜蒽酮、4-甲氧基苯乙酮、4’-乙酰基苯乙酮、蒽酮、苯甲醛、4,4’-二甲氧基二苯甲酮、二苯甲酮、芴、苯并菲、聯苯、硫雜蒽酮、蒽醌、4,4’-雙(二甲氨基)二苯甲酮、菲、萘、4-苯基苯乙酮、4-苯基二苯甲酮、2-碘萘、1,2-二脫氫苊烯(1,2-didehydroacenaphthylene)、2-萘甲腈、1-碘萘、1-萘甲腈、屈、暈苯、苯偶酰、熒蒽、芘、1,2-苯并蒽、吖啶、蒽、苝、萘并萘、2-甲氧基萘、2-乙酰基萘、1,4’-二氰基萘、9-氰基蒽、9,10-二氰基蒽、9,10-二溴蒽、2,6,9,10-四氰基蒽等。
其中,光敏劑優選為苯、甲苯、丙酮、丁烷-2,3-二酮、苯甲腈、苯丁酮、苯丙酮、苯乙酮、氧雜蒽酮、4-甲氧基苯乙酮、4’-乙酰基苯乙酮、蒽酮、苯甲醛、4,4’-二甲氧基二苯甲酮、二苯甲酮、芴、苯并菲、聯苯、硫雜蒽酮、蒽醌、4,4’-雙(二甲基氨基)二苯甲酮、菲、萘、4-苯基苯乙酮、4-苯基二苯甲酮、1,2-二脫氫苊烯、苯偶酰、芘、吖啶、蒽、苝、2-乙酰基萘或9,10-二溴蒽。
作為本發明中的對晶體(1)照射光的環化反應中的用于將晶體(1)制成漿料的溶劑,可列舉出例如甲苯、鄰二甲苯等芳香族烴系溶劑、己烷、庚烷、2-甲基戊烷、辛烷、石油醚等脂肪族烴系溶劑、環己烷等脂環式烴系溶劑、氯苯、鄰二氯苯等芳香族鹵代烴系溶劑、二氯甲烷、氯仿、四氯化碳、1,2-二氯乙烷、1,1,1-三氯乙烷、三氯乙烯、四氯乙烯等脂肪族鹵代烴系溶劑、二乙基醚、二異丙基醚、1,2-二甲氧基乙烷、四氫呋喃、1,4-二噁烷、環戊基甲基醚等醚系溶劑、三乙基胺、三丁基胺、N,N-二甲基苯胺等胺系溶劑、吡啶、甲基吡啶等吡啶系溶劑、醋酸乙酯、醋酸正丁酯、丙酸乙酯等酯系溶劑、甲醇、乙醇、正丙醇、2-丙醇、正丁醇、2-丁醇、異丁醇、叔丁醇、1-戊醇、2-戊醇、3-戊醇、1-辛醇、2-辛醇、乙二醇等醇系溶劑、丙酮、甲基異丁基酮等酮系溶劑、碳酸二甲酯、碳酸二乙酯等碳酸酯系溶劑、乙腈、二甲基亞砜、環丁砜、1,3-二甲基-2-咪唑烷酮、乙二醇二乙酸酯、醋酸、醋酸酐、水等。
其中,用于將晶體(1)制成漿料的溶劑優選為甲苯、鄰二甲苯、己烷、庚烷、石油醚、鄰二氯苯、二乙基醚、二異丙基醚、吡啶、醋酸乙酯、醋酸正丁酯、甲基異丁基酮或碳酸二甲酯。這些溶劑可以單獨使用,也可以混合使用2種以上。
本發明中,將晶體(1)制成漿料并照射光來進行環化反應的優點可列舉出:能夠對分散在溶液中的晶體高效地照射光、可容易地控制光反應實施中的反應溫度等。此時,作為制備晶體(1)的漿料時的濃度,優選為0.001~100mol/L、更優選為0.01~10mol/L、特別優選為0.05~3mol/L。
工序(c):1,3-二取代環丁烷-1,2,3,4-四羧酸二酐的制造
工序(c)中,以式(2)所示的1,3-二取代環丁烷-1,2,3,4-四羧酸作為起始原料,通過脫水縮合反應來制造式(3)所示的1,3-二取代環丁烷-1,2,3,4-四羧酸二酐。式中的R1如上所示。
所述工序(c)的反應可根據已知的方法、例如合成通訊(Synthetic Communications)、1989年、19(3-4)卷、679-688頁;合成通訊(Synthetic Communications)、1987年、17(3)卷、355-368頁;合成通訊(Synthetic Communications)、2003年、33(8)卷、1275-1283頁等記載的方法來實施。
作為本發明的工序(c)的脫水縮合反應中使用的縮合劑,可列舉出例如醋酸酐、亞硫酰氯、乙酰氯等。另外,縮合劑也可以用作溶劑。
作為工序(c)的脫水縮合反應時使用的溶劑,只要不阻礙反應的進行就沒有特別限定,可列舉出例如甲苯、醋酸乙酯、醋酸酐、亞硫酰氯、乙酰氯、吡啶等。這些溶劑可以單獨使用,也可以混合使用2種以上。另外,也可以在無溶劑的條件下實施該脫水縮合反應。
工序(c)的脫水縮合反應的反應溫度可以設定為-60℃~反應混合物的回流溫度之間的任意溫度。其中,優選為-20~230℃,更優選為0~150℃。
工序(c)在無溶劑的條件下進行的脫水縮合反應的反應溫度可以設定為100℃~起始原料的熱分解溫度之間的任意溫度。反應溫度優選為100~230℃。
實施例
以下,通過本發明的實施例來具體說明,但本發明不限定于它們。實施例中使用的分析裝置、光反應用光源如下所示。
NMR:
ECX 300(JEOL公司制):1H-NMR、13C-NMR
化學位移值通過使用Me4Si(四甲基硅烷)作為內標物質,并利用氘代二甲基亞砜(DMSO-d6)溶劑來測定。
JNM-ECA500(JEOL公司制):定量1H-NMR(13C去耦1H測定)
作為標準物質使用馬來酸,用氘代二甲基亞砜(DMSO-d6)溶劑進行測定。
AVANCE III 500(Bruker公司制):固相13C-NMR
作為標準物質使用金剛烷來進行測定。
氣相色譜法(GC):
GC:6890series GC(Hewlett Packard公司制)
氣相色譜-高分辨率質譜(GC-HRMS):
GC:7890A(Agilent公司制),MS:GCT Premier(Waters公司制)
單晶X射線結構分析:
SMART APEX II ULTRA(Bruker公司制)
粉末X射線衍射:
MiniFlex600(Rigaku公司制)
紅外吸收(IR):
FT-IR ALPHA(Bruker Optics公司制)
光源:作為100W高壓汞燈,使用SEN LIGHTS Co.,Ltd.制造的電源:HB100P-1(5/6)、光源:HL100CH-4,作為400W高壓汞燈,使用SEN LIGHTS Co.,Ltd.制造的電源:HB400P-1、光源:HL400B(L/H/S)-8,作為450W高壓汞燈,使用USHIO INC.制造的UM-452。作為氙氣燈,使用USHIO INC.制造的UV-XEFL(主波長峰為290nm、4.5W〔使用1.5W/cm的長度3cm的量〕)和UV-XEFL(主波長峰為320nm、4.5W〔使用1.5W/cm的長度3cm的量〕)。
超聲波清洗機:
US-18KS(SND Co.,Ltd.制)
卡爾費休水分計:
MKC-510(京都電子工業株式會社制)
液相色譜儀(HPLC):
HPLC:Prominence(島津制作所制)
實施例1-1:由吡啶和檸康酸形成的晶體(8)的制造(其一)
將檸康酸(3.11g,23.90mmol)和吡啶(1.89g,23.89mmol)依次添加至乳缽中,將生成的固體用碾槌碾磨5分鐘并充分混合,從而得到白色固體(4.79g)形式的由吡啶和檸康酸形成的晶體(8)(收率為96%)。
針對由吡啶和檸康酸形成的晶體(8),進行下述分析。
固相13C-NMR:
4mm CP/MAS probe,13C,CP/TOSS法,接觸時間4ms,轉速8kHz,
外部基準試樣adamantane(29.472ppm)
δ172.4,168.4,144.4,144.4,142.5,140.1,134.2,129.8,127.9,25.8ppm
FT-IR:
圖1為晶體(8)的FT-IR譜圖。
粉末X射線衍射:
<粉末X射線衍射的分析條件>
X射線:Cu-Kα
電壓:40kV
電流:15mA
步寬:0.020deg
掃描范圍:2θ=3~40deg
圖2為晶體(8)的粉末X射線衍射譜圖。由所述粉末X射線衍射譜圖讀取的粉末X射線衍射的峰值如下所示。
2θ=12.56,15.03,16.06,17.58,18.29,19.21,21.55,22.99,24.50,26.43,27.04,28.08,30.33,32.48,34.69,35.87,36.44,38.43
實施例1-2:晶體(8)的制造(其二)
將檸康酸(130.1mg,1.00mmol)、吡啶(80.6μL,1.00mmol)和甲醇(2mL)依次添加至螺口瓶(Maruemu Corporation制造的螺紋管)中,混合該反應混合物而使其溶解。接著,將該放有反應混合物的螺口瓶的口用紗布覆蓋,在運轉中的氣流室內以20℃~25℃靜置64小時,使甲醇蒸發,從而得到白色固體(197.2mg)形式的由吡啶和檸康酸形成的晶體(8)(收率為94%)。
針對由吡啶和檸康酸形成的晶體(8),進行下述分析。
粉末X射線衍射:
<粉末X射線衍射的分析條件>與實施例1-1記載的條件相同。
以下示出晶體(8)的粉末X射線衍射的峰值。
2θ=12.56,15.04,16.08,17.58,18.33,19.14,21.55,23.01,24.43,26.42,27.03,28.07,30.40,32.48,34.70,35.88,36.44,38.42
單晶X射線結構分析:
使用Bruker公司制造的單晶X射線衍射計SMART APEX II ULTRA,利用Cu-Kα射線(波長:),冷卻至-50℃并測定。X射線衍射數據的積分處理使用SAINT軟件,空間群確定和晶體結構分析使用SHELXTL-97程序,進行上述作為白色固體的晶體(8)的單晶X射線結構分析。表5示出晶體(8)的晶體數據和結構精密化。
<單晶X射線結構分析的分析條件>
X射線:Cu-Kα
電壓:50kV
電流:24mA
測定溫度:-50℃
<晶體(8)的晶體數據和結構精密化>
〔表5〕
基于晶體(8)的單晶X射線結構分析結果,圖3利用圓柱模型一并示出檸康酸的陰離子與吡啶嗡的堆積結構以及晶胞。
圖3中,碳原子為黑色、氫原子為白色、氮原子和氧原子在圖中用元素符號來表示,圖中的2條點劃線表示最接近的雙鍵彼此。
進而,基于晶體(8)的單晶X射線結構分析結果,圖4以ORTEP圖的形式僅示出晶體(8)中的檸康酸的陰離子彼此的構型。
根據上述單晶X射線結構分析的結果,晶體(8)是僅由摩爾比為1:1的檸康酸的陰離子與吡啶嗡這兩種構成要素組成的晶體。
進而,根據圖4的結果,在晶體(8)的晶體結構中,最接近的雙鍵彼此平行地存在,用下述式(8A)表示的平行雙鍵間的距離L1為
實施例1-3:晶體(8)的制造(其三)
向反應容器中添加庚烷(28mL)和吡啶(1.87mL,23.25mmol),將所得溶液保持在20℃~25℃。接著,耗費10分鐘向該溶液中滴加溶解于醋酸乙酯(28mL)的檸康酸(2.75g,21.14mmol),以20℃~25℃攪拌20分鐘,制備由吡啶和檸康酸形成的晶體(8)的漿料。停止攪拌后,將過濾出的固體用庚烷(15mL)與醋酸乙酯(15mL)的混合溶劑進行清洗,接著,用己烷(30mL)清洗,并進行真空干燥。由此,以白色固體(4.14g)的形式得到由吡啶和檸康酸形成的晶體(8)(收率為94%)。
針對由吡啶和檸康酸形成的晶體(8),進行下述分析。
粉末X射線衍射:
<粉末X射線衍射的分析條件>與實施例1-1記載的條件相同。
以下示出晶體(8)的粉末X射線衍射的峰值。
2θ=12.61,15.08,16.11,17.64,18.39,19.26,21.61,23.05,24.57,26.51,27.12,28.15,30.43,32.53,34.74,35.94,36.50,38.44
根據上述粉末X射線衍射的結果,通過實施例1-1~1-3得到的晶體可視為相同的物質。此外,上述式(8)表示由吡啶和檸康酸形成時的吡啶/檸康酸(1:1)的晶體。
實施例2-1:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其一)
將由吡啶和檸康酸形成的晶體(8)(100.0mg、0.478mmol)鋪展在玻璃制培養皿上,蓋上培養皿的蓋子。將該培養皿置于設定為25℃的冷板(cool plate)上,用100W的高壓汞燈進行50小時的光照射來進行環化反應。培養皿與光源的距離設為1cm。停止反應后,以淡黃色固體(80.2mg)的形式回收反應混合物。根據該反應混合物的分析,目標化合物(13a)的收率為83%、轉化率為94%、選擇性>99%。收率通過使用馬來酸作為標準品的定量1H-NMR來確定。另外,轉化率和選擇性根據GC分析中的目標化合物、無用的非對映體、源自原料的峰的相對面積比來算出。
用于求出上述環化反應中的轉化率和選擇性的GC樣品的制備的相關概要示于(式A)。
為了求出轉化率和選擇性,在使用晶體(8)的光環化反應后,采取反應混合物的一部分,進行后述GC樣品制備法B,由目標化合物(13a)及其非對映體衍生成化合物(14a)及其非對映體后,進行GC分析和GC-HRMS分析。
接著,用GC分析來追蹤與目標化合物(13a)的非對映體相當的副產物,因此,通過與使用晶體(8)的光環化反應不同的方法,進行以檸康酸酐(11)作為起始物質的溶液中的光環化反應來作為工序(z),從而合成化合物(12a)及其非對映體。進行后述的GC樣品制備法A,針對化合物(12a)及其非對映體進行水解反應、接著進行甲酯化反應,從而經由目標化合物(13a)及其非對映體而衍生成化合物(14a)及其非對映體,然后進行GC分析和GC-HRMS分析。應予說明,檸康酸酐溶液中的照射光的環化反應的有關詳情在后述的參考例中進行敘述。
記載GC分析條件和GC-HRMS分析條件。
<GC分析條件>
柱:TC-5(0.53mm×30m、膜厚1.5μm)
載氣:氦氣
流量:3.3mL/min(恒定流量)
狹縫比:1/10、試樣注入量:3μL
柱溫:80℃(保持2分鐘),升溫速度:10℃/分鐘、250℃(保持11分鐘)
注入口溫度:280℃
檢測器溫度:280℃
<GC-HRMS分析條件>
柱:DB-5(0.25mm×30m、膜厚0.25μm)
載氣:氦氣
流量:1mL/min(恒定流量)
狹縫比:1/50、試樣注入量:0.2μL
柱溫:80℃(保持3分鐘),升溫速度:25℃/分鐘、250℃(保持7.2分鐘)
注入口溫度:280℃
離子化方法:EI、CI+
記載GC樣品制備法和分析結果。
<GC樣品制備法A>
該制備法是(式A)記載的制備工序I、后續的制備工序II。照射光的環化反應后,將蒸餾去除了溶劑的反應混合物(20mg)采取至螺口瓶中,添加甲醇(3mL)和0.05mol/L的氫氧化鈉水溶液(2mL)后,將該溶液以20℃~25℃攪拌20分鐘。將該反應混合物(1mL)的一部分取出并置于螺紋管中,添加甲苯(0.2mL)后,添加己烷溶液的三甲基甲硅烷基重氮甲烷(0.2mL、約0.6mol/L、東京化成工業株式會社制造的市售品),以20℃~25℃攪拌20分鐘。將該反應混合物的有機層(0.4mL)取出一部分,用甲醇(1mL)稀釋,將如此得到的溶液作為GC分析樣品。
如(式A)所示那樣,化合物(12a)通過GC樣品制備法A而衍生成化合物(14a)。記載其分析結果。
GC分析:保留時間=18.62分鐘
GC-HRMS分析:m/z calcd for C16H25O8[M+C2H5]+:345.1549,found345.1577
化合物(12a)的非對映體是化合物(12b)、化合物(12c)或化合物(12d)之一,利用GC樣品制備法A衍生成化合物(14b)、化合物(14c)或化合物(14d)之一。記載其分析結果。
GC分析:保留時間=18.97分鐘
GC-HRMS分析:m/z calcd for C16H25O8[M+C2H5]+:345.1549,found345.1561
根據由GC-HRMS測得的質量數結果可以確認到:與化合物(14a)的分子量相同但GC的保留時間不同的該非對映體。
<GC樣品制備法B>
該制備法是(式A)記載的制備工序II。進行照射光的環化反應后,采取經過濾操作等而取出的反應混合物(25mg),添加甲醇(0.5mL)和甲苯(0.5mL)來制備溶液。接著,向該溶液中添加己烷溶液的三甲基甲硅烷基重氮甲烷(0.8mL、約0.6mol/L、東京化成工業株式會社制造的市售品),以20℃~25℃攪拌20分鐘。將該反應混合物(0.12mL)取出一部分,用甲醇(1.5mL)稀釋,將如此得到的溶液作為GC分析樣品。
化合物(13a)通過GC樣品制備法B而衍生成化合物(14a)。記載其分析結果。
GC分析:保留時間=18.62分鐘
GC-HRMS分析:m/z calcd for C16H25O8[M+C2H5]+:345.1549,found345.1542
未反應的檸康酸通過GC樣品制備法B而衍生成(Z)-2-甲基-2-丁烯二酸二甲酯。記載其分析結果。
GC分析:保留時間=9.68分鐘
GC-HRMS分析:m/z calcd for C9H15O4[M+C2H5]+:187.0970,found187.0972
轉化率以基于GC分析結果的(Z)-2-甲基-2-丁烯二酸二甲酯、化合物(14a)及其非對映體的相對面積比來算出。
選擇性以基于GC分析結果的化合物(14a)及其非對映體的相對面積比來算出。
實施例2-2:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其二)
將由吡啶和檸康酸形成的晶體(8)(5.00g、23.90mmol)以及作為溶劑的己烷(100mL)添加至能夠用磁力攪拌器攪拌的光化學反應實驗裝置(SEN LIGHTS Co.,Ltd.制)中,將光源設置于內部的中央。將反應溫度設定為20℃~25℃并攪拌漿料,用100W的高壓汞燈進行20小時的照射光的環化反應。停止反應后進行過濾,從而回收白色固體的反應混合物(4.33g)。根據該反應混合物的分析,目標化合物(13a)的收率為94%、轉化率為95%、選擇性>99%。收率通過使用馬來酸作為標準品的定量1H-NMR來確定。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比算出。
實施例2-3:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其三)
將由吡啶和檸康酸形成的晶體(8)(25.00g、119.50mmol)、庚烷(200mL)和醋酸正丁酯(200mL)添加至能夠用磁力攪拌器攪拌的光化學反應實驗裝置(SEN LIGHTS Co.,Ltd.制)中,將光源設置于內部的中央。將反應溫度設定為20℃~25℃并攪拌漿料,用400W的高壓汞燈進行12小時的照射光的環化反應。停止反應后進行過濾,從而回收白色固體的反應混合物(19.64g)。根據該反應混合物的分析,目標化合物(13a)的收率為93%、轉化率為95%、選擇性>99%。收率通過該反應混合物的收量和1H-NMR來算出。轉化率和選擇性根據實施例2-1記載的方法來分析,由GC分析的各種峰的相對面積比來算出。
對上述光反應后得到的白色固體的反應混合物進行分離精制。關于精制,從該反應混合物取出10.00g,溶解至甲醇(70mL)中,進行過濾后,蒸餾去除溶劑。接著,溶解至醋酸(45mL)后,將該溶液以60℃攪拌2小時30分鐘,然后冷卻至20℃~25℃,并進行過濾。其后,蒸餾去除所得固體中包含的溶劑,使用真空泵進行真空干燥,從而得到白色固體形式的目標化合物(6.12g)。通過1H-NMR而算出的收率為83%。
采取該所得目標化合物的一部分,制備飽和溶解在加熱至70℃的乙腈中的溶液。接著,將該溶液冷卻至20℃~25℃,進行靜置而重結晶,進行所得單晶的單晶X射線結構分析。
化合物(13a)的分析
1H NMR(DMSO-d6):
δ12.46(br),3.30(s,2H),1.42(s,6H)ppm
13C-NMR(DMSO-d6):
δ175.9,175.9,171.5,171.5,51.7,51.7,44.2,44.2,20.7,20.7ppm
單晶X射線結構分析:
基于化合物(13a)的單晶X射線結構分析結果,圖5以ORTEP圖的形式示出化合物(13a)的分子結構。
實施例2-4:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其四)
向能夠用磁力攪拌器攪拌的光化學反應實驗裝置(SEN LIGHTS Co.,Ltd.制)中添加醋酸乙酯(110mL)和吡啶(7.49mL,93.01mmol),冷卻至5℃。接著,耗費30分鐘向該反應混合物中滴加溶解于醋酸乙酯(110mL)的檸康酸(11.00g,84.55mmol)。其后,將該反應混合物以5℃攪拌20分鐘,制備由吡啶和檸康酸形成的晶體(8)的漿料。向該反應容器中插入100W的高壓汞燈,以5℃一邊攪拌一邊進行22小時的照射光的環化反應。其后,利用桐山漏斗濾取固體,用醋酸乙酯(30mL)清洗并真空干燥,從而得到白色固體的反應混合物(12.82g)。根據該反應混合物的分析,目標化合物(13a)的收率為93%、轉化率為95%、選擇性>99%。收率通過該反應混合物的收量和1H-NMR來算出。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比算出。
實施例3-1:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造
將由吡啶和檸康酸形成的晶體(8)(200.0mg、0.956mmol)以及作為溶劑的醋酸乙酯(4mL)添加至螺口瓶(Maruemu Corporation制造的螺紋管)中,從而制備漿料。其后,以能夠用磁力攪拌器進行攪拌的方式進行操作,將光源設置于螺口瓶的外部。螺口瓶與光源的距離設為4.5cm。用100W的高壓汞燈以20℃~25℃進行20小時的照射光的環化反應。停止反應后進行過濾,得到白色固體的反應混合物(116.0mg)。根據該反應混合物的分析,目標化合物(13a)的收率為69%、轉化率為97%、選擇性>99%。收率通過該反應混合物的收量和1H-NMR來算出。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比來算出。
除了將溶劑醋酸乙酯變更成表6記載的溶劑之外,利用與實施例3-1記載的反應相同的條件來實施反應。將所用的溶劑、轉化率、選擇性、收率和回收時的外觀的有關實驗結果作為實施例3-2~3-8來示于表6。
表6中,AcOn-Bu表示醋酸正丁酯,hexane/AcOn-Bu(v/v=1/1)表示己烷(2mL)與醋酸正丁酯(2mL)的混合溶劑,heptane/AcOn-Bu(v/v=1/1)表示庚烷(2mL)與醋酸正丁酯(2mL)的混合溶劑。
〔表6〕
實施例4:(3aR,3bR,6aS,6bS)-3a,6a-二甲基環丁烷[1,2-c:3,4-c’]二呋喃-1,3,4,6(3aH,3bH,6aH,6bH)-四酮(12a)的制造
[工序(a)和工序(b)的連續化]
作為工序(a),將吡啶(60mL)和檸康酸(15.55g、119.52mmol)依次添加至能夠用磁力攪拌器攪拌的光化學反應實驗裝置(SEN LIGHTS Co.,Ltd.制)中,以20℃~25℃攪拌10分鐘。攪拌結束后,添加作為溶劑的庚烷(175mL)和醋酸正丁酯(175mL),攪拌30分鐘,制備由吡啶和檸康酸形成的晶體(8)的漿料。接著,將光源設置于該反應容器的內部中央,作為工序(b),將通過工序(a)得到的晶體(8)的漿料以20℃~25℃用400W的高壓汞燈進行8小時的光照射并攪拌。通過停止光照射來終止反應后,對所得漿料進行過濾,從而得到白色固體的反應混合物。根據該反應混合物的分析,目標化合物(13a)的轉化率為97%、選擇性>99%。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比來算出。
為了對光反應后得到的粗產化合物(13a)進行精制,使該反應混合物溶解于四氫呋喃(250mL)并過濾后,蒸餾去除溶劑。接著,使用醋酸(40mL)和醋酸正丁酯(40mL)的混合溶劑進行懸浮清洗,再次進行過濾。其后,使用真空泵進行真空干燥,從而得到精制的化合物(13a)。
[工序(c)]
將上述精制后的化合物(13a)、甲苯(60mL)和醋酸酐(31.8mL、336.4mmol)添加至反應容器中,以100℃攪拌3小時。攪拌結束后,通過將反應容器冷卻至20℃~25℃而終止反應。對所得反應混合物進行過濾后,使用醚溶劑來清洗所得固體,使用真空泵進行真空干燥,從而得到白色固體形式的目標化合物(12a)(11.36g)。通過1H-NMR算出的源自起始原料檸康酸的總收率為85%。采取該所得目標化合物的一部分,進行單晶X射線結構分析。
化合物(12a)的分析結果如下所示。
1H NMR(DMSO-d6):
δ3.88(s,2H),1.38(s,6H)ppm
13C-NMR(DMSO-d6):
δ173.5,173.5,168.1,168.1,49.0,49.0,44.1,44.1,15.7,15.7ppm
單晶X射線結構分析:
基于化合物(12a)的單晶X射線結構分析結果,圖6以ORTEP圖的形式示出化合物(12a)的分子結構。
實施例5:使用了光敏劑的(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造
將作為光敏劑的二苯甲酮(21.78mg、0.12mmol)和作為溶劑的庚烷(10mL)添加至螺口瓶(Maruemu Corporation制造的螺紋管)中。確認二苯甲酮溶解后,向螺口瓶中添加由吡啶和檸康酸形成的晶體(8)(500.0mg、2.39mmol)而制備漿料。制備結束后,以能夠用磁力攪拌器攪拌的方式進行操作,將光源設置于螺口瓶的外部。螺口瓶與光源的距離設為4.5cm。用100W的高壓汞燈以20℃~25℃進行5小時30分鐘的照射光的環化反應。通過停止光照射來終止反應后,過濾所得漿料,從而得到白色固體的反應混合物(471.9mg)。根據該反應混合物的分析,目標化合物(13a)的轉化率為34%、選擇性>99%。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比來算出。
實施例6:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造
工序(a)
將中康酸(6M)(130.1mg、1.00mmol)、甲醇(2mL)和煙酰胺(9)(122.1mg、1.00mmol)依次添加至螺口瓶(Maruemu Corporation制造的螺紋管)中,混合該反應混合物而使其溶解。接著,將該放有反應混合物的螺口瓶的口用紗布覆蓋,在運轉中的氣流室內以20℃~25℃靜置64小時,使甲醇蒸發,從而得到白色固體形式的由煙酰胺和中康酸形成的晶體(10)。上述式(10)表示由煙酰胺和中康酸形成的煙酰胺/中康酸的晶體。
工序(b)
將由煙酰胺和中康酸形成的晶體(10)(100.0mg、0.396mmol)鋪展在玻璃制培養皿上。蓋上培養皿的蓋子,并置于設定為25℃的冷板上,將培養皿與光源的距離設定為3cm后,用100W的高壓汞燈進行20小時的光照射。通過停止光照射而終止反應后,回收白色固體形式的反應混合物。根據該反應混合物的分析,目標化合物(13a)的轉化率為23%、選擇性>99%。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比來算出。原料使用中康酸,GC樣品制備中,未反應的中康酸衍生成(E)-2-甲基-2-丁烯二酸二甲酯。因此,(E)-2-甲基-2-丁烯二酸二甲至的峰視作源自原料的峰。
實施例7-1:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其一)
[工序(a)和工序(b)的連續化]
作為攪拌裝置,使用帶磁力攪拌器的光化學反應實驗裝置(SEN LIGHTS Co.,Ltd.制)。向該裝置的反應容器中依次添加碳酸二甲酯(110mL)和吡啶(7.49mL、93.01mmol),冷卻至10℃。冷卻結束后,耗費30分鐘向該反應溶液中滴加溶解于碳酸二甲酯(110mL)的檸康酸(11.00g、84.55mmol)。滴加結束后,將該反應混合物以10℃攪拌20分鐘。攪拌結束后,得到由吡啶和檸康酸形成的晶體(8)的漿料。所得晶體(8)不進行分離/精制地直接用于后續工序。將該漿料以10℃進行攪拌,用100W的高壓汞燈進行17小時的光照射。反應結束后,用漏斗濾取反應混合物中的固體后,用碳酸二甲酯(30mL)進行清洗。通過對所得固體進行真空干燥,從而得到白色固體形式的目標物14.23g。根據該白色固體的分析,目標化合物(13a)的收率為88%、轉化率為94%、選擇性>99%。收率通過該反應混合物的收量和1H-NMR分析來算出。轉化率和選擇性按照實施例2-1記載的方法,由GC分析的各種峰的相對面積比來算出。
實施例7-2:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其二)
[工序(a)和工序(b)的連續化]
作為攪拌裝置,使用帶磁力攪拌器的光化學反應實驗裝置(SEN LIGHTS Co.,Ltd.制)。向該裝置的反應容器中添加醋酸乙酯(110mL)和吡啶(14.98mL、186.02mmol),冷卻至5℃。冷卻結束后,耗費30分鐘向該反應溶液中滴加溶解于醋酸乙酯(110mL)的檸康酸(22.00g、169.10mmol)。滴加結束后,將該反應混合物以5℃攪拌20分鐘。攪拌結束后,得到由吡啶和檸康酸形成的晶體(8)的漿料。所得晶體(8)不進行分離/精制地直接用于后續工序。將該漿料以5℃進行攪拌,用100W的高壓汞燈進行33小時的光照射。光照射后的反應混合物中的水分量利用卡爾費休水分計(MKC-510、京都電子工業株式會社制)進行測定,結果為1570ppm。反應結束后,用漏斗濾取反應混合物中的固體后,用醋酸乙酯(40mL)進行清洗。通過將所得固體進行真空干燥,從而得到白色固體形式的目標物26.03g。根據該白色固體的分析,目標化合物(13a)的收率為93%、轉化率為94%、選擇性>99%。收率根據該反應混合物的收量和1H-NMR分析來算出。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比來算出。
實施例8-1:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其一)
將由吡啶和檸康酸形成的晶體(8)(100.0mg、0.478mmol)和醋酸乙酯(2mL)添加至螺口瓶(Maruemu Corporation制造的螺紋管)中,制備漿料。制備結束后,將該放有漿料的螺口瓶以其與光源的距離達到5cm的方式進行設置。設置結束后,作為光源而使用氙氣燈(主波長峰為290nm、4.5W),進行2小時的光照射,使得漿料的溫度保持在20℃~25℃。應予說明,攪拌使用磁力攪拌器。根據攪拌結束后的該漿料的HPLC分析,目標化合物(13a)的轉化率為3%。
<HPLC分析條件>
檢測器:差示折射率檢測器
柱:Develosil C30-UG5(內徑4.6mm、長度150mm、粒徑5μm)
洗脫液:重量濃度為0.2%的三氟醋酸水溶液:乙腈=95:5(體積比)
流速:1.5mL/分鐘、
柱溫:35℃
保留時間:3.06分鐘〔目標產物(13a)〕、3.39分鐘〔檸康酸(6C)〕
實施例8-2:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造(其二)
將由吡啶和檸康酸形成的晶體(8)(300.0mg、1.434mmol)和醋酸乙酯(6mL)添加至螺口瓶(Maruemu Corporation制造的螺紋管)中,制備漿料。制備結束后,將該放有漿料的螺口瓶以其與光源的距離達到5cm的方式進行設置。設置結束后,作為光源而使用氙氣燈(主波長峰為320nm、4.5W),進行77小時的光照射,使得漿料的溫度保持在20℃~25℃。應予說明,攪拌使用磁力攪拌器。根據攪拌結束后的該漿料的HPLC分析(分析條件與實施例8-1相同),目標化合物(13a)的轉化率為74%。
實施例9:(1R,2R,3S,4S)-1,3-二甲基環丁烷-1,2,3,4-四羧酸(13a)的制造
使用流動反應器來進行工序(a)和工序(b)。將流動反應器的概略圖示于圖7,將流動反應器中的具備雙層管結構的T字型混合器(混合器2)的剖面圖示于圖8。流動反應器使用將內徑2mm、外徑3mm和長度10m的FEP管(由四氟乙烯/六氟丙烯共聚物形成的氟樹脂制的管)纏繞于光源的燈夾套而得到的裝置,將該裝置設置于超聲波清洗機。
工序(a):
制備濃度為0.34mol/L的檸康酸(437.9mg、3.366mmol)的醋酸乙酯溶液。此外,制備濃度為0.34mol/L的吡啶(266.3mg、3.366mmol)的醋酸乙酯溶液。將檸康酸的醋酸乙酯溶液和吡啶的醋酸乙酯溶液分別用注射泵以0.9mL/min的速度進行送液,在混合器1(圖7中的21)中進行混合。其后,利用混合器2(圖7中的32)來混合氮氣,從而形成由晶體(8)的漿料和氮氣得到的段塞流(漿料與氮氣交替排列的流體)。應予說明,混合器2為了避免管的閉塞而具有圖8所示的雙層管結構,并設置在50℃的恒溫槽中。
工序(b):
在基于450W的高壓汞燈的光照射下,進一步在超聲波照射下進行環化反應。利用質量流量控制器調整段塞流,使得其在11分鐘內通過光和超聲波的照射部分,來進行反應。根據所回收的流出液的HPLC分析,目標化合物(13a)的轉化率為34%。
參考例1
按照前述非專利文獻2,進行檸康酸酐(11)溶液中的照射光的環化反應。
將檸康酸酐(11)(1.38g、12.31mmol)、作為溶劑的1,4-二噁烷(10mL)和作為光敏劑的二苯甲酮(93.0mg、0.51mmol)添加至螺口瓶(Maruemu Corporation制造的螺紋管),以能夠用磁力攪拌器進行攪拌的方式進行操作,將光源設置在螺口瓶的外部。將螺口瓶與光源的距離設為4.5cm。用100W的高壓汞燈以20℃~25℃進行18小時的照射光的環化反應。停止反應后,根據反應混合物的分析,目標化合物(12a)的轉化率為68%、選擇率為50%。轉化率和選擇性由GC分析中的各種峰的相對面積比來算出。原料使用檸康酸酐,GC分析中,檸康酸酐的峰示作原料的峰。
GC分析和GC-HRMS分析用的樣品的制備方法和分析結果如下所述。從懸浮的反應混合物中采取懸浮液(100μL),用二甲基亞砜(1.5mL)稀釋,作為分析樣品。
目標化合物(12a)的分析結果如下所示。
GC分析:保留時間=15.60分鐘
GC-HRMS分析:m/z calcd for C10H9O6[M+H]+:225.0399,found 225.0386
目標化合物(12a)的非對映體的分析結果如下所示。
GC分析:保留時間=15.81分鐘
GC-HRMS分析:m/z calcd for C10H9O6[M+H]+:225.0399,found 225.0403
根據由GC-HRMS測定的質量數結果可以確認到:與化合物(12a)的分子量相同但GC的保留時間不同的該非對映體。然而,無法確定化合物(12a)的非對映體的立體結構。推測目標化合物(12a)的非對映體為化合物(12b)、化合物(12c)或化合物(12d)中的一者。
參考例2
按照前述專利文獻2記載的方法,進行檸康酸酐(11)溶液中的照射光的環化反應。
將檸康酸酐(11)(1.42g、12.67mmol)和作為溶劑的醋酸乙酯(10mL)添加至螺口瓶(Maruemu Corporation制造的螺紋管)中,以能夠用磁力攪拌器進行攪拌的方式進行操作,將光源設置于螺口瓶的外部。未添加光敏劑,將螺口瓶與光源的距離設為4.5cm。用100W的高壓汞燈以20℃~25℃進行60小時的照射光的環化反應。停止反應后,蒸餾去除反應混合物的溶劑,使用真空泵進行真空干燥,從而得到白色固體的反應混合物(1.36g)。根據該反應混合物的分析,目標化合物(12a)的轉化率為88%、選擇率為41%。GC樣品的制備法使用前述GC樣品制備法A。轉化率和選擇性按照實施例2-1記載的方法進行分析,并由GC分析的各種峰的相對面積比來算出。
前述專利文獻2和非專利文獻2中,針對剛剛進行照射光的環化反應之后、即進行精制操作之前的目標化合物和無用的非對映體的選擇性沒有記載。根據上述參考例的結果可確認:相對于目標化合物1,3-二甲基環丁烷-1,2,3,4-四羧酸二酐,無用的非對映體生成了1倍~1.4倍左右。因而,以往的制造方法中,由于選擇性如此低,因此需要復雜的精制操作,推測其在生產效率的方面會造成不良影響。
產業上的可利用性
通過本發明得到的滿足環丁烷環上的兩個取代基位于1位和3位且該取代基的相對構型為反式的1,3-二取代環丁烷-1,2,3,4-四羧酸及其酸二酐作為聚酰亞胺等各種工業用的原料、合成中間體而在廣泛的領域中得以應用,是有用的化合物。
應予說明,2014年5月9日申請的日本專利申請2014-098037號的說明書、權利要求書、附圖和摘要的全部內容援引至此,作為本發明說明書的公開內容并入。
附圖標記說明
11:放有化合物(6C)的醋酸乙酯溶液的注射泵
12:放有化合物(7)的醋酸乙酯溶液的注射泵
21:T字型混合器(混合器1)
31:恒溫槽
32:具有雙層管結構的T字型混合器(混合器2)
33:質量流量控制器
34:氮氣瓶
41:超聲波清洗機
42:燈夾套
43:光源
44:回收目標生成物的容器
- 還沒有人留言評論。精彩留言會獲得點贊!