制備碳酸二芳基酯的方法與流程
由單酚和光氣制備純碳酸二芳基酯的方法是已知的。碳酸二芳基酯(例如碳酸二苯酯,“DPC”)的制備通常借助連續法——通過制備光氣和隨后使單酚和光氣在惰性溶劑中在堿和氮催化劑存在下在界面中反應來進行。
原則上在文獻中描述了通過例如相界面法制備碳酸二芳基酯,參見例如Chemistry and Physics of Polycarbonates, Polymer Reviews, H. Schnell, 第9卷, John Wiley and Sons, Inc. (1964), 第50/51頁。
在此根據相界面法,溶解在溶劑和水中的反應物互相反應。這些方法的缺點是通過蒸餾從溶劑中分離碳酸二芳基酯并將其重新后處理,以及作為廢產物的含氯化鈉的水相,對此只有有限的使用可能性并且任選需要非常復雜的后處理步驟。
因此,已經開發出單酚的所謂的直接光氣化的方法,其中反應物光氣和單酚不在相界面法中在堿金屬氫氧化物溶液存在下反應,而是在熔體中在催化劑存在下反應,優選不使用另外的溶劑,以形成碳酸二芳基酯和氯化氫而非氯化鈉。
因此,例如,US 2,362,865 (A)描述了通過在170℃至250℃的溫度下使用Al-或Ti-酚鹽的單酚直接光氣化來制備碳酸二芳基酯的方法,但其中沒有描述催化劑的再循環,更沒有描述分離方法。
EP 2 371 806 A和EP 2 371 807 A都同樣描述了通過在20℃至240℃的溫度下使用金屬鹵化物或金屬酚鹽的單酚直接光氣化來制備碳酸二芳基酯的方法。同樣沒有描述催化劑再循環到該方法中。
EP 1 234 845 A同樣描述了單酚在120℃至190℃的溫度下在熔體中與特別純的光氣的反應。使用基于所用單酚計0.1至10摩爾%的量的含氮化合物,例如吡啶作為催化劑。此公開也沒有給出催化劑再循環到該方法中的指示。吡啶與氯化氫形成難揮發的鹽(沸點:222-224℃),其不容易蒸餾出。根據EP 1 234 845 A的教導,因此首先用氫氧化鈉中和反應混合物,以可蒸餾出水、游離吡啶和過量酚的混合物。
WO 2011007001描述了從反應混合物中凝華吡啶鹽酸鹽。為此,在全油泵真空中將二氯硅烷-吡啶加合物加熱到200℃,由此使吡啶鹽酸鹽揮發并作為固體沉淀。
此外,在許多其它專利,如WO 2008/114750 A1、JP 2008-231073 A、JP 2009-114195 A、JP 09-278714 A、JP 09-100256 A、JP 10-245366 A、JP 11-012230 A中描述了單酚在熔體中在均勻可溶的含氮催化劑的存在下與光氣反應以形成碳酸二芳基酯。
JP 10-077250 A、JP 09-24278 A和EP 1 234 845 A提到催化劑的可能的再循環,但沒有提到具體從產物中分離催化劑和著眼于再循環的催化劑后處理方法。此外,提到在反應混合物的中和和洗滌過程中引入水溶液,特別是水和/或氫氧化鈉溶液。
US 5 239 106教導了通過與苯酚的1:1加合物的結晶從含催化劑的反應溶液中分離碳酸二苯酯。但是,在此沒有描述催化劑的分離和再循環。
這些公開無一提供將催化劑,例如吡啶再循環到該方法中的方法的令人滿意的指示。特別地,在使用堿性水溶液的中和步驟后經水相分離出催化劑。
特別地,現有技術沒有給出其中沒有如上文引用的文獻中那樣借助含水添加劑中和鹽酸鹽(這受制于上述缺點)的從含產物的料流中分離催化劑的方法的具體實例。
這些公開無一描述用于制備碳酸二芳基酯的完全無水和無廢水的方法。
現有技術中已知的方法因此不足以在催化劑再循環方面滿足經濟和生態的高要求并另外確保產物(其本身是進一步化學工藝的原材料)的高純度。
但是,工業方法必須考慮經濟方面。在此,催化劑的再循環屬于在這點上評估的重要方面,因為催化劑的高度或甚至完全的排放意味著經濟劣勢并造成不合意的環境污染。形成的廢水必須用極高費用凈化,這對水處理廠帶來巨大的挑戰。在現有技術的方法中,要么需要高技術支出才能實現催化劑的再循環,要么安排催化劑的部分或完全排放。在這兩種情況下都出現額外的廢水流。
在直接光氣化法中,提供催化劑的有效再循環是最重要的。此外,不僅在反應過程中,在后處理中也應該盡可能避免使用水溶液。這是因為含有機物質的廢水首先必須以復雜的方式凈化,然后再棄置。
因此,技術目的是開發省略附加原材料如氫氧化鈉和水的根據單酚在熔體中的直接光氣化法制備碳酸二芳基酯的方法,所述方法通過減少料流排放(清除物(Purge))而經濟地運行并提供最終產物的恒定良好品質。
現在令人驚訝地發現,當使用任選取代的吡啶或其鹽作為催化劑時,形成的鹽酸鹽、氯化氫和碳酸二苯酯可以通過蒸餾將彼此分離并同時可以獲得高純度的碳酸二芳基酯。令人驚訝地,在該蒸餾過程中沒有發生吡啶鹽酸鹽的凝華,因為該鹽保持溶解在分離出的低沸物相中并甚至在室溫下也與酚形成液體混合物。
優選省略反應溶液或母液的中和和/或水的添加。這帶來特別經濟和生態的方法方式。
本發明因此提供一種通過在游離形式和/或其鹽酸鹽形式的至少一種任選取代的吡啶作為催化劑的存在下使至少一種單酚與光氣和/或至少一種氯甲酸芳基酯反應來制備碳酸二芳基酯,優選碳酸二苯酯的方法,其中
a) 所述反應在反應器中在1-50巴(絕對)的壓力下進行,
b) 將反應混合物從反應器轉移到單級或多級蒸餾裝置中,
c) 在至少一個蒸餾塔的頂部分離出含催化劑的餾出物,
d) 將所述含催化劑的餾出物至少部分再循環到步驟a)的反應器中,
e) 經塔的側流分離出碳酸二芳基酯并任選進一步提純。
在步驟a)至e)中都優選不使用水溶液。
步驟a)中的反應優選在高于80℃的溫度下進行,以避免所得碳酸二芳基酯以固體形式沉淀。反應物的反應可以在常壓或略微減小的壓力下,也可以在最多50巴(絕對)的升高的壓力下進行。在此,根據工藝條件,光氣可以存在于凝聚相中或溶解在液相中。通過這種方法制成的碳酸二芳基酯由于它們的高純度而特別適用于根據熔融酯交換法由碳酸二芳基酯和雙酚制備高純度的聚碳酸酯。
可以對該反應中獲得的氯化氫施以一個或多個提純步驟以使其適用于許多其它的用途可能性,特別是用于電化學氧化或熱氧化以形成氯氣。由此獲得的這種氯氣可以用于與一氧化碳制備光氣;所得光氣可用于本發明的方法。
在反應條件下為液體的最終產物在包含步驟b)、c)和e)的多個分離步驟中與副產物和催化劑或其HCl加合物分離。其隨后優選具有大于95%,優選大于99.0%,特別優選大于99.5%的碳酸二芳基酯和任選的酚的含量。該最終產物優選主要包含碳酸二芳基酯。將該反應中所用的催化劑后處理,以使其可至少部分再循環至該反應(步驟d)。
本發明的方法由三個方法區段構成:
I. 包含方法步驟a)的反應,
II. 氯化氫后處理(任選),
III. 通過蒸餾提純產物和分離催化劑,其包含方法步驟b)、c)和e),其中回收的催化劑至少部分再循環至階段I(步驟d))。
在方法區段I),即反應中,在上游的方法步驟中將反應物互相混合,以存在光氣在熔融單酚中的基本均勻溶液;這可任選在預定的熔體溫度下通過使用升高的壓力實現。
本發明中制備的碳酸二芳基酯優選是通式(I)的那些
(I)
其中R、R'和R''可以各自彼此獨立地為氫、鹵素或支化或非支化的C1-至C9-烷基或支化或非支化的C1-至C9-烷氧基羰基。R、R'和R''優選在式(I)的兩側上相同。
特別優選的是碳酸二苯酯。
適用于本發明的單酚優選是通式(II)的那些
(II)
其中R、R'和R''可以各自彼此獨立地具有對通式(I)所述的含義。
對本發明而言,“C1-C4-烷基”是例如甲基、乙基、正丙基、異丙基、正丁基、仲丁基、叔丁基;“C1-C6-烷基”另外是例如正戊基、1-甲基丁基、2-甲基丁基、3-甲基丁基、新戊基、1-乙基丙基、環己基、環戊基、正己基、1,1-二甲基丙基、1,2-二甲基丙基、1,2-二甲基丙基、1-甲基戊基、2-甲基戊基、3-甲基戊基、4-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、1,3-二甲基丁基、2,2-二甲基丁基、2,3-二甲基丁基、3,3-二甲基丁基、1-乙基丁基、2-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-1-甲基丙基、1-乙基-2-甲基丙基或1-乙基-2-甲基丙基;“C1-C9-烷基”另外是例如正庚基和正辛基或正壬基。這同樣適用于烷基羰基中的相應烷基。
合適的單酚是例如:苯酚、烷基苯酚如甲酚、對-叔丁基苯酚、對-枯基苯酚、對-正辛基苯酚、對-異辛基苯酚、對-正壬基苯酚和對-異壬基苯酚、鹵代苯酚如對-氯苯酚、2,4-二氯苯酚、對-溴苯酚、2,4,6-三溴苯酚,苯甲醚和水楊酸甲酯或水楊酸苯酯。
特別優選的是苯酚。
所用單酚應具有至少99.90重量%的純度。
反應物優選含有少于300體積ppm的水,因為水的存在促進該裝置材料的腐蝕。
此處所用的單酚可以除了含有從外部引入整個工藝的酚,即來自儲罐的所謂的新鮮酚,還含有來自方法步驟II)和III)的冷凝物料流或來自方法步驟II)的洗滌液體料流的再循環的單酚。這樣再循環的單酚可含有不破壞該反應的來自該方法的副產物,例如殘留量的碳酸二芳基酯、氯化氫或氯碳酸芳基酯。單酚在所用的反應物混合物中優選以大于基于光氣計的化學計算所需量存在。單酚與光氣的摩爾比可以在1.5:1至4:1變化,優選的是2:1至3:1的摩爾比,特別優選的是2.5:1至3:1的摩爾比。
在下文中,術語“氯甲酸芳基酯”用于表示在由單酚和光氣制備碳酸二芳基酯時作為中間產物形成的化合物。
適用于本發明的氯甲酸芳基酯優選是通式(III)的那些
(III)
其中R、R'和R''可以各自彼此獨立地具有對通式(I)所述的含義。
如果通式(III)的氯甲酸芳基酯與通式(II)的單酚反應,R、R'和R''優選各自在式(I)和(II)中具有相同含義。
特別優選的是氯甲酸苯酯。
為避免該制備方法的最終產物中的不合意副產物,所用光氣應具有至少99.80重量%,優選99.96重量%的純度;四氯化碳含量應小于50體積ppm,優選小于15體積ppm。
根據本發明,使用取代或未取代的吡啶作為催化劑。其可以以游離堿的形式或完全或部分以其鹽酸鹽的形式存在。“以游離堿的形式”或“以游離形式”對本發明而言是指該吡啶環的氮不以質子化形式存在。
優選最多10摩爾%,特別優選最多1摩爾%的任選取代的吡啶以游離形式存在。其余部分以鹽酸鹽形式存在。
根據本發明充當催化劑的吡啶優選是通式(IV)的那些
(IV)
其中R1和R2可以各自彼此獨立地為H、支化或非支化的C1-至C9-烷基、C5-或C6-環烷基、OH、OR3、NHR3或NR3R4,其中R3和R4各自彼此獨立地為C1-至C4-烷基。特別優選的是R1和R2為H。
合適的吡啶是例如吡啶、2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-乙基吡啶、3-乙基吡啶、4-乙基吡啶、2-異丙基吡啶、3-異丙基吡啶、4-異丙基吡啶、2-丁基吡啶、4-叔丁基吡啶、2,3-二甲基吡啶、2,4-二甲基吡啶、2,5-二甲基吡啶、2,6-二甲基吡啶、3,4-二甲基吡啶、3,5-二甲基吡啶、3,4-二乙基吡啶、3,5-二乙基吡啶、3-乙基-4-甲基吡啶、2-(3-戊基)吡啶、4-(3-戊基)吡啶、2-二甲基氨基吡啶、4-二甲基氨基吡啶、2-甲氧基吡啶、2,6-二甲氧基吡啶、4-環己基吡啶、4-(5-壬基)吡啶、4-苯基丙基吡啶和2-羥基吡啶。
特別優選的是吡啶。
在一個特別優選的實施方案中,該催化劑是吡啶鹽酸鹽。
根據本發明使用的催化劑可以以基于存在的單酚計0.001摩爾%至10摩爾%的量,優選0.01摩爾%至5摩爾%的量使用。
該催化劑以在單酚熔體中的溶液的形式使用。這種溶液根據本發明含有從方法區段III)經過或未經過單獨的催化劑后處理而再循環到作為方法區段I)的反應中的催化劑的至少部分量。對于將催化劑再循環到方法區段I)中,催化劑后處理因此不是絕對必要的,而是完全可能的。
最早在反應物完全充分混合后,優選在反應器中進行催化劑的添加,以避免反應物在混合過程中過早反應和因此在不合適的方法區段中過早生成氯化氫。
來自方法區段III)的催化劑的再循環可以任意頻繁地進行;在連續方法中,可優選連續再循環部分量的催化劑,而任選從工藝循環中排出部分量,以防止催化劑污染或任選慮及催化劑的失活。在需要時,可以將新鮮催化劑添加到再循環量的催化劑中。在一個優選實施方案中,再循環至少25重量%,特別優選至少50重量%,非常特別優選至少75重量%,特別是非常特別優選至少85重量%的催化劑。但是,在一個優選實施方案中,再循環最多99重量%,優選最多95重量%的催化劑。
反應物單酚和光氣以上示摩爾比或以上述優選摩爾比互相混合,其中單酚始終作為熔體存在,且光氣根據主導壓力為氣體或液體。在常壓和高于60℃的溫度下,主要存在雙相的氣液混合物,因為光氣在單酚中的溶解度也如同在碳酸二芳基酯中一樣隨溫度提高而降低。
因此,熔融單酚和光氣的混合物必須在反應相中非常劇烈混合并再分散,以借助相界面的令人滿意的補充確保反應物的充分反應。或者,在凝聚均相中可以顯著提高光氣與酚的反應(由于與氣態光氣和液態酚的兩相混合物相比,光氣在酚中的濃度提高)。提高溫度也對反應速率具有加速作用,因此100℃至250℃,優選110℃至220℃的升高的溫度可能是有利的。但是,由于如上文提到的這樣的溫度不利于光氣在酚中的溶解度,在加壓下在升高的溫度下進行反應是特別有利的。因此,將反應物在升高的溫度在常壓下,優選在最多50巴(絕對)的升高的壓力下,特別優選在最多30巴(絕對)的升高的壓力下,非常特別優選在4至25巴(絕對)的壓力下互相混合并反應。混合區中的溫度應至少為單酚的熔點,但100℃至250℃的反應溫度是有利的。
在反應物基本完成混合后,優選將上述催化劑之一優選以所述優選量作為在單酚中的溶液添加到該混合物中。由于單酚與光氣形成氯碳酸芳基酯作為中間體的催化反應在上述溫度和壓力下極快進行并消去氣態氯化氫,該反應優選通過多級方式進行。該反應可以在絕熱條件下進行,因為其只有輕微的放熱(W?rmet?nung)。在第一階段,即所謂的主反應器中,除已進一步反應的碳酸二芳基酯外主要形成氯碳酸芳基酯,特別是在升高的壓力下和優選在120℃至230℃的溫度下,特別優選在130℃至210℃的溫度下,對于碳酸二苯酯的制備,非常特別優選在170℃至200℃的溫度下,并且在15至120分鐘,優選45至90分鐘的反應器液體停留時間下。在第二階段中,在所謂的后反應器中在優選170℃至250℃,特別優選190℃至230℃,非常特別優選200℃至210℃的略更高的溫度下在15至120分鐘,優選45至90分鐘的反應器停留時間下,氯碳酸芳基酯與仍存在的單酚反應以形成碳酸二芳基酯。在此,也可以將第二階段中在所謂的后反應器中的壓力降低到2至20巴。壓力的這種降低可以有利地在所謂的閃蒸階段(Flash-Stufe)中進行,其中由于壓力降低而可以特別好地從反應熔體中分離出在主反應器中形成的氯化氫氣體。在后反應器中的第二反應階段下游也可任選存在用于分離出殘留量的氯化氫的閃蒸階段。其也可任選集成到后續方法區段III)(通過蒸餾的產物提純和催化劑分離)的第一蒸餾塔中,并在此加速氣相和液相的分離。
連續反應器優選很好地適合作為用于反應物在所示反應條件下的反應的反應器,但也可以使用攪拌釜作為分批式反應器。特別好合適的連續反應器是例如攪拌釜級聯、泡罩塔、板式塔、填料塔或具有用于充分混合反應介質的固定內裝件的塔或反應蒸餾塔。
這樣的塔也可互相組合,例如泡罩塔與上疊的精餾塔,在這種情況下不同于反應物的上述混合,可以在塔組合的不同位置處分開引入反應物。因此,例如,在上述塔組合的情況下,可以將光氣引入下方的泡罩塔并將單酚與催化劑一起引入具有大約10個理論塔板的上方的精餾塔。從泡罩塔中取出形成的碳酸二芳基酯。
反應物的相應的分開計量加入也可以在反應蒸餾塔中如下進行,即通過在塔中部引入光氣并在塔頂部與催化劑一起引入單酚。從塔底取出反應混合物。這樣的塔可具有至少5個,優選大約20個塔板。
在反應器的另一任選實施方案中,反應物可以在主反應器中在1至25巴(絕對)的壓力下在足夠高的任選更長的停留時間下,但在反應器下部的優選120℃至190℃,特別優選160℃至180℃的較低溫度下完全反應。在反應器上部必須另外加熱,以在此實現最多250℃,優選最多230℃的略更高的溫度。隨后可以借助閃蒸或另一脫氣技術進行反應混合物的基本脫氣和低沸物的分離。
特別優選的是泡罩塔,如上所述的反應物混合物自下向上流經其中。在此,在泡罩塔的塔頂部取出氣態氯化氫并在該塔軸(Kolonnen-Shaft)的上端取出反應混合物。將其經該塔的底部供入充當后反應器的下一泡罩塔。在停留反應器(Verweil-Reaktor)末端從最后一個泡罩塔中取出完全反應的反應混合物,并供入后續的方法區段III)(通過蒸餾的產物提純和催化劑分離)。在每種情況下在泡罩塔的頂部取出的氯化氫氣體在后續的方法區段II)(氯化氫后處理)中提純。在各個階段之間也可通過在閃蒸中的減壓和隨后的增壓而另外分離氯化氫。
裝置材料必須滿足在高溫下對氯化氫和光氣的耐受性的高要求,并優選選自材料:黑鋼、不銹鋼、鋼合金、鎳基合金(例如Hastelloy C)、陶瓷、石墨、搪瓷涂覆的材料、PTFE包覆的材料。
任選的方法區段II)(氯化氫后處理)的目標是分離和提純副產物氯化氫。為此,收集反應A)中形成的氣相并將氯化氫氣體與可任選再循環以進一步反應形成碳酸二芳基酯的其它組分分離。可以蒸餾副產物氯化氫以提高純度。此外,可以混入來自方法區段III)的氣態子流。
在這一方法區段II)中,合并來自方法區段I)的含HCl的料流并共同提純。優選在此不中和氯化氫。在低沸點組分中的主要產物是94重量%或更多的氯化氫氣體;副產物是大于3重量%的過量使用的單酚以及痕量的氯碳酸芳基酯、碳酸二芳基酯和光氣和作為來自光氣的副產物的痕量的一氧化碳和四氯化碳。可以借助各種步驟將副產物與主要產物氯化氫基本分離,以獲得具有大于99.0體積%,優選大于99.8體積%的純度的氯化氫氣體和小于1000體積ppm,優選小于500體積ppm的殘留含量的光氣和/或氯碳酸酯。氯化氫中的有機化合物含量同樣應小于1000體積ppm,優選小于50體積ppm;特別地,含氯烴的含量應小于50體積ppm。
通過一個或多個下述步驟實現這一目的。優選通過多級法實現這一目的。優選通過蒸餾分離出氯化氫。
在所謂的第一冷凝階段中,在合適的溫度下部分冷凝出具有比氯化氫更高的沸點的副產物。在此,特別是從氯化氫氣體中基本除去以較高濃度存在的較高沸點組分,例如單酚和碳酸二芳基酯,并再循環至該反應。當除了較低溫外還任選使用升高的壓力時,這種分離特別成功。第一冷凝階段中的優選溫度為至少80℃,并且對于碳酸二苯酯的制備而言特別優選90℃。優選將壓力設定為8至25巴(絕對),用于制備碳酸二苯酯的特別優選的壓力為12巴(絕對)。也可任選通過多級方式在各種溫度和/或壓力下從氯化氫氣體流中冷凝副產物。
如果足夠低的溫度或足夠高的壓力在技術上不可實現或難以實現,也可以跳過這種第一冷凝階段,以在后續的所謂的HCl洗滌階段中在合適的裝置中使用熔融碳酸二芳基酯從氯化氫料流中洗除出副產物。如果這一HCl洗滌階段是避開第一冷凝階段的氯化氫的第一提純階段,這一HCl洗滌階段也可本身通過多級進行并在各種降低的溫度水平下運行,以提高該洗滌的效率。在此,單酚特別地極易溶于碳酸二芳基酯。當例如在后續的方法區段C)(碳酸二芳基酯后處理)中的合適位置取出用于該洗滌的碳酸二芳基酯時,痕量的氯碳酸酯和光氣也可在這一方法步驟中反應形成碳酸二芳基酯。原則上,從這一方法區段到經蒸餾的碳酸二芳基酯的各個碳酸二芳基酯料流都適用于該HCl洗滌階段,并且對上述有機氯化合物的反應有利的是從方法區段III)中取出用于HCl洗滌階段的含催化劑和酚的碳酸二芳基酯料流,以能使仍存在于氯化氫氣體中的有機氯化合物在短時間內反應。
這樣的合適碳酸二芳基酯是離開方法區段I)(反應)并為進一步后處理而供入方法區段III)的第一階段(碳酸二芳基酯后處理)的粗制碳酸二芳基酯。在這種碳酸二芳基酯中存在足夠量的催化劑和單酚。或者,經蒸餾的碳酸二芳基酯可以以任意方式用于HCl洗滌階段,因為要洗出的副產物在DPC中的物理溶解度足夠高。但是,經蒸餾的純碳酸二芳基酯優選用于HCl洗滌階段。為了在HCl洗滌階段中使有機氯化合物反應,同樣也可以使用單酚代替碳酸二芳基酯作為洗滌介質,因為要洗出的副產物在單酚中的物理溶解度也足夠高。這種單酚可以是例如單酚反應物料流的子流。如果需要含氯酯(Chlorester)或光氣反應以形成碳酸二芳基酯,用于該洗滌的單酚可以以任意方式含有催化劑。使用碳酸二芳基酯或使用單酚的HCl洗滌優選在高于碳酸二芳基酯的熔點的溫度下進行;在碳酸二苯酯的制備中,80-95℃的熔融溫度特別優選。該HCl洗滌可以在常壓下或8至25巴(絕對)的升高的壓力下進行;在碳酸二苯酯的制備中,12巴(絕對)特別優選。
在這種洗滌中,可以獲得具有大于99.8重量%的純度的氯化氫氣體。優選地,光氣的含量低于500體積ppm,氯甲酸酯的含量低于檢出限,并將酚的含量降至低于10體積ppm。
這一HCl洗滌階段不是絕對必要的,并在其它方法步驟的任意互相組合的情況下也可避開。
氯化氫蒸餾特別好地適用于實現氯化氫氣體的高純度。為了能以能量有效的方式進行這樣的蒸餾,在上游的第二冷凝階段中將待提純的氯化氫預先冷卻至較低溫是有用但不是絕對必要的。如果省略這一階段,在后續的氯化氫蒸餾中需要在低溫下的相應較高能量。在也可任選在多種不同溫度和/或壓力水平下運行的這種第二冷凝階段中,分離出仍包含于氯化氫氣體中的痕量的較高沸點副產物,特別是在使用8至25巴(絕對)的較高壓力時,在碳酸二苯酯的情況下優選12巴(絕對)時。該溫度可根據技術情況在加25℃至減50℃的極寬范圍內變化。當已使用單酚進行HCl洗滌階段中的洗滌時,這種第二冷凝階段特別值得推薦,因為由此可顯著降低該HCl氣體流中存在的單酚濃度并由此降低HCl蒸餾中的負擔。如果省略這種第二冷凝階段,對HCl蒸餾中的能量需求的要求相應地遠遠更高。冷凝物同樣可以如第一冷凝階段中那樣供入該反應。
作為方法區段II)中的氯化氫后處理的第四和最后階段,氯化氫的蒸餾在一個特別優選的實施方案中特別好地適用于制備高純度的氯化氫。其優選應在升高的壓力下進行,因為否則用于設定替代性所需的足夠低溫的能量消耗會不合比例地高。如果之前的提純階段已在常壓下進行,最晚在這一提純階段中將氯化氫料流壓縮到8至25巴(絕對)的較高壓力是非常值得推薦的;對于碳酸二苯酯的制備,12巴(絕對)特別優選。在這些條件下可獲得具有99.95重量%的純度的氯化氫氣體。
方法區段II)中的氯化氫提純的所有四個上述階段在所述順序下特別好地適合根據本發明用于制備高純度的氯化氫氣體。不是絕對必須遵守特定的順序或實施所有方法階段,而是取決于從反應中分離出的氯化氫的污染程度和作為最終產物的氯化氫氣體的所需純度。因此,如下文以HCl蒸餾為例所示,使用各個提純階段或單個提純階段就任選完全可能實現所需結果。
如果來自方法區段I)(反應)的進料流沒有預先提純就直接供入氯化氫蒸餾,在相同溫度和壓力條件下同樣可獲得具有99.95重量%的純度的氯化氫氣體。
提純階段的組合完全有可能以不依賴于上述列舉內容的特定順序進行以實現特定純度。
作為用于進行第一和第二冷凝階段的裝置,具有對工藝條件而言足夠高的熱交換器表面積和用于將冷凝物進給到該反應中的裝置的經典冷阱是合適的。這樣的冷阱也可制造成多級的并任選不同調溫的。適用于HCl洗滌階段的裝置特別是連續運行的裝置,如泡罩塔、泡罩板式塔、填料塔、填充塔(Packungskolonnen)、具有固定內裝件的塔,其中洗滌液體可以從上方與上行氯化氫氣體逆向傳送。連續運行的攪拌裝置,例如混合器-沉降器,或不連續運行的攪拌裝置原則上也合適。
氯化氫蒸餾可以在具有合適的塔內裝件的常規蒸餾塔或精餾塔中進行。
用于上述裝置的材料必須滿足對氯化氫的耐受性的高要求并優選選自黑鋼、不銹鋼、鋼合金、鎳基合金(例如Hastelloy C)、陶瓷、石墨、搪瓷涂覆的材料、PTFE包覆的材料。
在方法區段III)(產物提純和催化劑分離)中,收集在反應I)中形成的較高沸點組分,分離并將催化劑以游離堿形式或以鹽酸鹽形式再循環至該反應。由此將主要產物提純至獲得具有大于99.0重量%,優選大于99.8重量%、特別優選大于99.95重量%的純度的碳酸二芳基酯的程度。
已經令人驚訝地發現,可以借助蒸餾從反應混合物中分離和再循環催化劑,而不發生該催化劑的凝華。
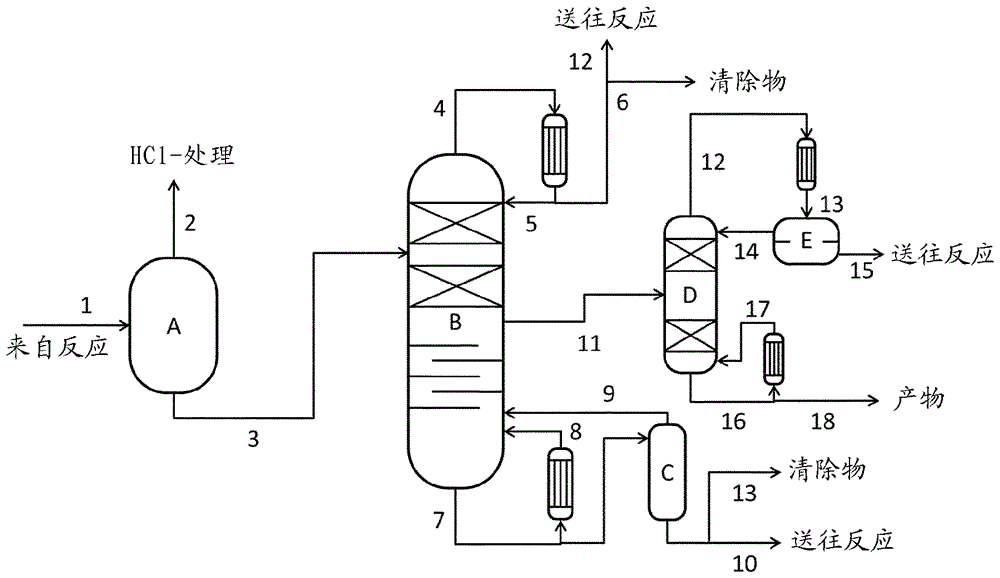
這一方法區段的圖示綜述顯示在圖1中。
在液體反應混合物的第一分離步驟中,在脫氣階段中基本分離出溶解的氯化氫。這可以借助閃蒸器(圖1中的A)、蒸餾塔、這些裝置的組合或另一常規的脫氣技術(例如汽提)實現。
優選使用閃蒸階段(A),其中通過降低壓力將溶解的氯化氫脫氣。在此選擇20毫巴至1巴(絕對)的壓力和140℃-205℃的溫度,優選0.1巴至1巴(絕對)的壓力和165-205℃的溫度,特別優選0.3-0.7巴(絕對)的壓力和180-200℃的溫度。
或者,可以將在200毫巴至2巴(絕對),優選0.5至1巴(絕對),特別優選0.8-1巴(絕對)的壓力下運行的蒸餾塔用于氯化氫的分離。
在閃蒸器的氣相中或在替代性的蒸餾塔的頂部獲得氯化氫、單酚和游離的任選取代的吡啶的混合物。優選將這一混合物在方法區段B)中添加到用于氣體處理的主要氣體料流中。
閃蒸器或替代性的塔的底部產物基本不含氯化氫,并在優選變體中單酚含量低。該底部產物因此由碳酸二芳基酯、單酚、游離形式或鹽酸鹽形式的任選取代的吡啶和副產物形成。
在另外的變體中,閃蒸階段和蒸餾塔可以組合用于分離氯化氫或可以使用另一脫氣技術(例如汽提)。或者,也可以省略第一分離步驟。但是這不優選,因為由此對于第二分離步驟產生較大的工藝料流,并且運送到進一步分離階段中的氯化氫可能造成腐蝕問題。
在第二分離步驟中,通過分離出單酚、吡啶鹽酸鹽和次要組分從第一分離步驟的預提純的料流(圖1中的料流3)中分離出碳酸二芳基酯。在實驗中已經發現,由碳酸二芳基酯和吡啶鹽酸鹽構成的二元混合物的蒸氣-液體平衡具有非均相共沸行為(見圖2)。在85重量%的吡啶-HCl的質量含量下測得最低共沸物。該液體分離成含吡啶鹽酸鹽的相和含碳酸二芳基酯的相。因此看起來難以借助其原理基于蒸氣-液體平衡的分離技術,例如蒸餾將吡啶鹽酸鹽與碳酸二芳基酯分離。此外,140-146℃的吡啶鹽酸鹽的高熔點在蒸餾中可能構成阻礙,因為在冷凝器中的再升華會造成操作問題。
令人驚訝地,在實驗中已證實通過蒸餾將碳酸二芳基酯與酚、吡啶鹽酸鹽和次要組分分離的可行性。
在該優選變體中,在第二分離步驟中提純的碳酸二芳基酯因此在蒸餾塔的側流(11)處獲得(見圖1)。在餾出物中取出單酚、游離吡啶、吡啶鹽酸鹽和低沸點次要組分。由于碳酸二芳基酯和吡啶鹽酸鹽之間的共沸物,該餾出物還具有大約5-15重量%的碳酸二芳基酯含量。
在塔底取出高沸點次要組分和熱分解產物。為了在塔底不超過220℃的破壞產物的溫度并使塔底產物保持可泵送,優選在塔底產物中保留10-50重量%,特別優選20-40重量%的碳酸二芳基酯含量。
在蒸餾塔的頂部優選設定5-100毫巴(絕對),特別優選10-40毫巴(絕對)的壓力。根據頂部壓力、單酚的過量和該反應中的催化劑濃度,頂部溫度在60℃和140℃之間變化。在底部,以180-250℃,優選200-220℃的溫度為主導。由于底部的這些高溫,通過熱分解降低次要組分水楊酸苯酯(Salol)的含量。由此可以在側流中獲得具有大于99重量%的純度的碳酸二芳基酯。優選從蒸氣相中取出該側流,但也可從液相中取出。
可以使用側塔進一步提高側流中的碳酸二芳基酯純度。但是,優選運行該蒸餾塔以使側流不經進一步提純就可引入后續的方法步驟,例如引入熔體聚碳酸酯方法中。
將該餾出物再循環至該反應。優選將餾出物的一部分作為清除物輸出以排出低沸點次要組分。棄置該塔底產物或在一個優選變體中在除去高沸點清除物料流后再循環至該反應。在一個特別優選的變體中,在進一步分離操作(圖1中的C),例如薄膜蒸發器中溫和地分離出該塔底產物中包含的碳酸二芳基酯的一部分并重新送回該蒸餾塔,以降低高沸點清除物中的碳酸二芳基酯含量。
上述優選蒸餾的替代性變體安排在蒸餾塔的側流中取出由碳酸二芳基酯和吡啶鹽酸鹽構成的混合物(見圖3)。在附加蒸餾塔中,隨后在底部從這種混合物中獲得純碳酸二芳基酯。
在該替代性變體的第一蒸餾塔(圖3中的B)的頂部,在5-100毫巴(絕對),優選10-40毫巴(絕對)的頂部壓力和60-115℃的頂部溫度下取出酚和低沸點次要組分。塔底產物的組成和溫度類似于蒸餾塔的上述優選變體中的塔底產物的。如上述優選變體中那樣,將餾出物和塔底產物再循環至該反應。
在該替代性變體中用于提純側流的附加蒸餾塔(圖3中的D)優選制造為非均相共沸蒸餾。頂部的潷析器(圖3中的E)將該餾出物分成富含碳酸二芳基酯的相和富含吡啶鹽酸鹽的相。作為塔頂產物取出富含吡啶鹽酸鹽的相并再循環至該反應。將富含碳酸二芳基酯的相作為回流送回該塔。為了增加該回流,也可以將來自潷析器的兩個相的混合物再循環至該塔。在塔底獲得具有大于99重量%的純度的碳酸二芳基酯,其不經進一步提純就可送往后續的方法步驟,例如熔體聚碳酸酯法中。
附圖解釋:
圖1顯示方法區段III)(通過蒸餾的產物提純和催化劑分離)的一個優選實施方案。
圖2顯示在各種溫度(160℃、170℃和180℃)下碳酸二苯酯/吡啶鹽酸鹽混合物的實驗測定的蒸氣-液體-液體平衡 vs 壓力。這在85重量%吡啶鹽酸鹽的質量含量下表現出最低共沸物。
圖3描述方法區段III)(通過蒸餾的產物提純和催化劑分離)的另一優選實施方案。
圖1和3的解釋:
A: 閃蒸階段
B: 蒸餾塔
C: 附加分離操作(例如薄膜蒸發器)
D: 側流蒸餾塔
E: 潷析器
1: 反應混合物
2: 來自閃蒸階段的蒸氣
3: 來自閃蒸階段的底部產物
4: 來自蒸餾塔的蒸氣
5: 返回蒸餾的回流
6: 再循環至該反應的餾出物
7: 來自蒸餾塔的塔底產物
8: 進入蒸餾塔的蒸發器蒸氣
9: 來自附加分離操作(例如薄膜蒸發器)進入蒸餾塔的蒸氣
10: 再循環至該反應的塔底產物
11: 產物料流(DPC)
12: 用于除去低沸點次要組分的清除物
13: 用于除去高沸點次要組分的清除物。
下列實施例旨在例示用于通過蒸餾分離吡啶鹽酸鹽的可行性的程序,但不構成限制。
實施例:
將碳酸二苯酯(DPC)、苯酚、吡啶鹽酸鹽(氯化吡啶鎓,下文也稱作“吡啶·HCl”或“Py.HCl”)和Salol(水楊酸苯酯,苯酚的直接光氣化的副產物)的各種混合物引入由具有30厘米長維格羅柱(包裹在鋁箔中以隔熱)的蒸餾燒瓶(1L)、李比希冷凝器(使用溫水在80℃下運行)、冷阱(在-80℃下)和真空泵構成的實驗室蒸餾裝置中。測量柱頂部和底部的溫度。熔融混合物在每種情況下在大約20毫巴(絕對)下分餾。單獨收集2或3個餾分,并如同冷阱的底部產物和內容物一樣通過氣相色譜法分析。
實施例1:
在所述裝置中蒸餾下列合成反應混合物:
在蒸餾中形成下列餾分:
令人驚訝地,餾分2在室溫下是液體并也持久保持這樣。
各餾分的分析得出下列組成(組成總量與100%的偏差歸因于測量誤差):
顯而易見的是,各餾分中吡啶與HCl之間的摩爾比在每種情況下為大約1:1。相反,蒸餾柱底產物不含吡啶鹽酸鹽。在冷阱中,重新獲得少量HCl,但沒有以鹽形式的吡啶鹽酸鹽沉積的跡象。這表明吡啶鹽酸鹽可令人驚訝地以鹽形式作為低沸物從粗制碳酸二苯酯(DPC)反應混合物中蒸餾出。
實施例2:
在所述裝置中蒸餾下列合成反應混合物:
在蒸餾柱中形成下列餾分:
由于顯著低的苯酚含量,不同于實施例1,沒有出現主要由苯酚構成的第一餾分。其它觀察結果,特別是在室溫下為液體的餾分2在實施例2的情況中也適用。
各餾分的分析得出下列組成(組成總量與100%的偏差歸因于測量誤差):
又顯而易見的是,特別在餾分2(其中重新獲得所用吡啶鹽酸鹽的主要部分)中,吡啶與HCl之間的摩爾比幾乎理想地為1:1。該蒸餾柱底產物又不含吡啶鹽酸鹽。在冷阱中又重新獲得少量HCl,但在這一實驗中,也沒有以鹽形式的吡啶鹽酸鹽沉積的跡象。由此證實,吡啶鹽酸鹽可令人驚訝地以鹽形式作為低沸物從粗制碳酸二苯酯(DPC)反應混合物中蒸餾出。
對比例1:
作為對比例,蒸餾吡啶鹽酸鹽的下列混合物(含有痕量水):
在蒸餾中形成下列餾分:
各餾分的分析得出下列組成(組成總量與100%的偏差歸因于測量誤差):
在加熱純吡啶鹽酸鹽時,在所選溫度下也幾乎沒有發生該鹽的離解。僅在餾分1中,溫度、在室溫下的物質狀態以及組成都表明消去純吡啶。
在餾分2中,發現少量的大約1:1組成的吡啶·HCl。
吡啶鹽酸鹽的主要部分如預期那樣留在柱底產物中。在冷阱中又重新獲得少量HCl,但在這一實驗中也沒有以鹽形式的吡啶鹽酸鹽沉積的跡象。
對比例2:
制造40重量%吡啶鹽酸鹽和60重量%苯酚的合成混合物(類似于來自實施例1和2的餾分2)。該混合物在室溫下為液體。溶解在苯酚中的吡啶鹽酸鹽的凝固點的降低導致在該蒸餾中出現的混合物也為液體,并因此可預計在工業蒸餾裝置中也沒有在真空管路中等的凝華。
- 還沒有人留言評論。精彩留言會獲得點贊!