用于制備放射性藥物的試劑盒的制作方法
本發明涉及用于制備放射性藥物的穩定的試劑盒。尤其,本發明涉及用于穩定試劑盒的配體組分的非水性溶劑的用途。
必須在有限的時間內制備和施用放射性藥物,因為應用中使用的大部分放射性核素的短的半衰期。其通常由在GMP條件下產生的試劑盒配制。試劑盒通常包含待與放射性核素,比如99mTc絡合的適用的配體;足夠量的還原劑;調整pH以適合最佳標記條件的緩沖劑;穩定試劑;和賦形劑。以凍干或冷凍干燥的形式制備試劑盒,其增加了穩定性和貨架期。試劑盒可容易運輸并且儲存,然后使用指定的放射性核素重構。冷凍干燥的試劑盒簡化了標記并確保了用于標記的更穩定的條件。
冷凍干燥的試劑盒制劑的可用性有利于負責的醫院人員容易制備用于施用的放射性藥物,因為其僅僅涉及添加放射性核素和必要時的加熱。所以,這些制備步驟在醫院責任人員的能力范圍內。
放射性藥物的例子是99m锝-乙烯雙半胱氨酸脫氧葡糖胺(99mTc-ECDG)1。99mTc-ECDG是單光子發射計算機斷層成像術(SPECT)/計算機斷層成像術(CT)(SPECT/CT)成像劑,其對于檢測肺癌基本損害的能力,目前在美國處在三期臨床試驗1。99mTc-ECDG的成像能力與正電子成象術(PET)/CT成像劑,18F-氟脫氧葡萄糖(18F-FDG)相當2,其廣泛用于(大于95%的掃描)檢測冬眠心肌和代謝活性癌組織2。相對于18F-FDG潛在實施99mTc-ECDG的背后主要驅動力是相比PET放射性指示劑與采用SPECT放射性指示劑相關的顯著更低的成本和在肺癌成像中實現相同水平的質量和效率相關的顯著更低的成本3。
提出99mTc-ECDG的作用機制是經氨基己糖途徑發生,原因是包含兩個葡糖胺取代基。葡糖胺通過氨基己糖生物合成途徑進入細胞并且其葡糖胺-6-磷酸酯的調節產物介導胰島素活化下游和標志糖基化以及癌癥生長2。在氨基己糖途徑中,上調葡萄糖轉運蛋白(glucose transporter)促進了谷氨酰胺:6-磷酸-果糖酰基轉移酶(fructose-6-phosphate amidotransferase)(GFAT)的過表達。磷酸化的葡糖胺結合尿苷二磷酸(UDP)以形成UDPN-乙酰葡糖胺(UDP-GLcNAc)。通過O連接的蛋白質N-乙酰葡糖胺(O-GlcNAc)轉移酶,細胞核和細胞溶質蛋白質上絲氨酸和蘇氨酸殘基的糖基化在所有多細胞真核生物中是常見的。糖基化是翻譯后修飾的一部分并且好像修飾許多核質蛋白質。O-GlcNAc轉移酶活性與細胞內UDP-GLcNAc和UDP濃度高度相關,其又與葡萄糖濃度和其他刺激高度相關。在細胞核內,普遍存在的轉錄因子Sp1被O-GlcNAc高度修飾。響應高血糖或升高的葡糖胺,Sp1經歷高糖基化(hyperglycosylation)。因為O-GlcNAc參與氨基己糖途徑和核活性,對于腫瘤的辨別診斷,其成為吸引人的成像劑。
對于公開的合成的文獻調查(參考文獻[5]、[6]、[7]和[8])總結了如何產生ECDG 3的數個實驗方法。不幸的是,這些公開的程序都未成功可重現的(reproducible),因為這些合成涉及將ECDG暴露于水性介質,這證明了是無效的,因為ECDG已經顯示對于空氣、光水和溫度敏感9。已經描述了ECDG的合成是一項具有挑戰性的任務,原因是該配體非常易變的性質9。因為ECDG被期望用作成像劑,材料必須是藥學級別的,這意味著需要進行純化步驟,而基本上不實質損失產率。這證明是非常困難的,因為ECDG的低穩定性。
使99mTc EsCDG用作核醫學環境中的放射性藥物的問題復雜化的第二個因素是其在試劑盒制劑中的存在。
產生ECDG——一種水不穩定的配體——的試劑盒是存在問題的,因為一般的試劑盒程序包括在水相中的凍干步驟,其中純的配體活性藥學成分(API)溶于包含還原劑、添加劑和緩沖劑的每個的至少一種的水/生理鹽水,分發在小瓶中并且被冷凍干燥。在醫院,生理鹽水中的99mTc被添加至試劑盒并且重構。然后,99mTc與ECDG配體螯合并且準備99mTc-ECDG放射性藥物用于注射。發明人已經發現ECDG在水中幾乎立即分解。僅僅當金屬離子與ECDG,比如在99mTc-ECDG的情況下螯合時,其在水中是穩定的。
所以,仍需要包括穩定的組分的試劑盒系統,其允許適于診斷、治療或其他指示應用的簡單的、可重復和穩定的標記技術。此外,仍需要在管理認可的可接受的放射化學純度水平并且同時保持高的穩定性、純度和產率的有效的配體的放射性標記。
技術實現要素:
根據本發明的第一方面,提供用于制備放射性藥物的試劑盒,試劑盒包括:
a)溶解在非水性溶劑中的配體,配體能夠結合放射性核素并且其中溶劑選自己烷至丙三醇的相對極性范圍;
b)還原劑;
c)緩沖溶液;
d)和任選地添加劑比如弱螯合劑、抗氧化劑、增溶劑或膨松劑并且其中組分a)、b),c)和d)各自為凍干形式。
在本發明優選的實施方式中,還原劑是SnCl2或SnF2或酒石酸亞錫、鹽酸和水的混合物,并且緩沖溶液是磷酸鹽或檸檬酸或乙酸鹽緩沖溶液。可選地,緩沖劑是磷酸鹽、檸檬酸或乙酸鹽緩沖溶液的任何一種的組合。
優選地,弱螯合劑選自DTPA、葡庚糖酸鹽、酒石酸鹽和亞甲基二膦酸鹽(medronate)或任何組合。抗氧化劑選自龍膽酸、抗壞血酸和對氨基苯甲酸或其組合。增溶劑選自明膠或環糊精或其組合,并且膨松劑選自甘露醇、肌醇、葡萄糖和乳糖或其組合。
組分a)、b)、c)和d)可包含在一個小瓶中。可選地,組分b)、c)和d)包含在第一小瓶中并且組分a)包含在第二小瓶中。
配體可選自ECD、HMPAO、MAG3和MIBI或其堿金屬鹽或其堿土金屬。優選地,配體是ECDG或其堿金屬鹽。溶劑選自:甲醇、乙醇、乙酸乙酯、己烷、氯仿、二氯甲烷、甲苯、醚、四氫呋喃和乙腈或其組合。優選地,溶劑選自甲醇或乙醇。更優選地,溶劑是甲醇。
金屬放射性核素可選自99mTc、188Re、186Re、153Sm、166Ho、90Sr、90Y、89Sr、67Ga、68Ga、111In、153Gd、59Fe,52Fe、225Ac、212Bi、45Ti、60Cu、61Cu、62Cu、64Cu、67Cu、195mPt、191mPt、193mPt、117mSn、103Pd、103mRh、89Zr、177Lu、169Er、44Sc、155Tb、140Nd、140Pr、198Au、103Ru、131Cs、223Ra、224Ra和62Zn。
優選地,放射性核素是99mTc、103Pd、103mRh、195mPt、193mPt、191Pt。更優選地,放射性核素是99mTc。
試劑盒進一步包括使用說明書。
附圖說明
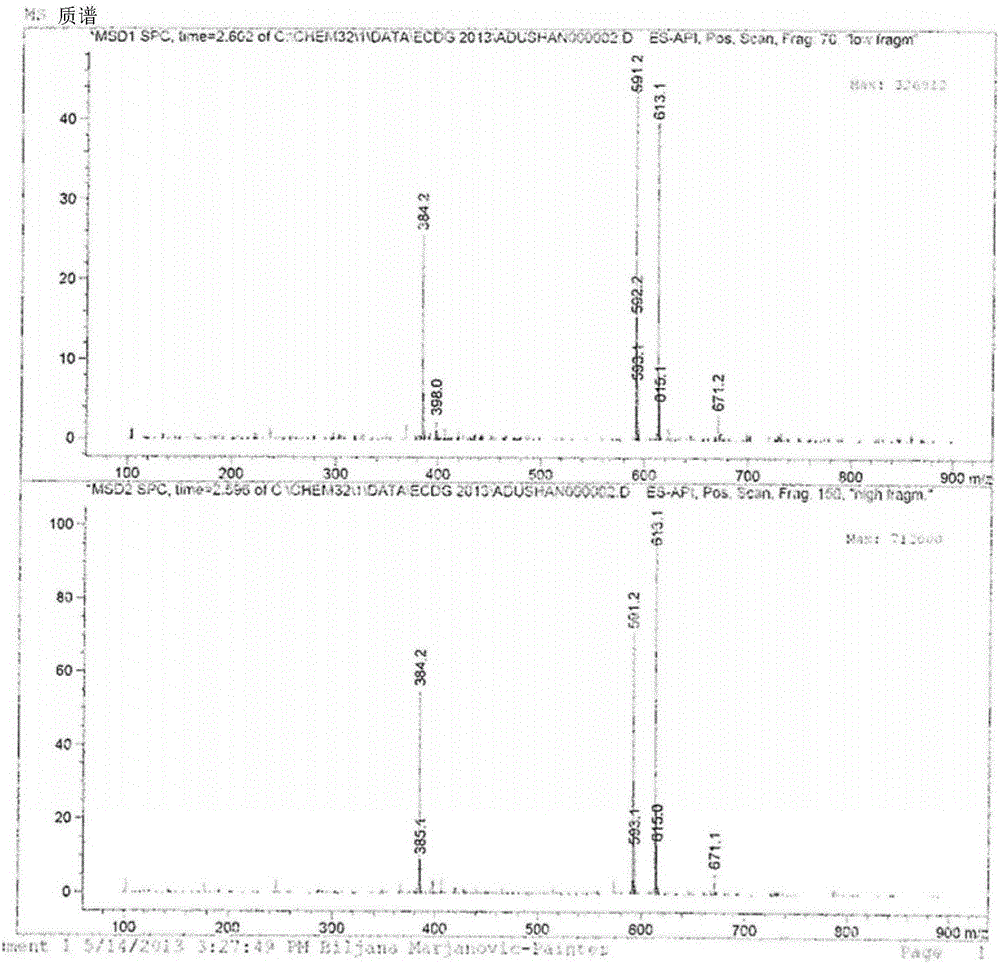
圖1是ECDG產生的質譜。
具體實施方式
根據下述制備試劑盒。
在Ar(g)條件下制備下述溶液以確保沒有CO2或O2:
a)足夠量的ECDG或其鹽溶于己烷至甘油的相對極性范圍內的非水性溶劑。
b)在適當的pH下用于最佳標記條件的磷酸鹽/檸檬酸緩沖溶液。
c)在中性或酸性介質中的亞錫鹽溶液,所述介質用作VII氧化態的高锝酸鹽離子(99mTcO4-)至IV的還原劑,以確保99mTc是化學活性的,以與配體,ECDG結合。
對于兩個小瓶試劑盒配制,使用上述溶液的冷凍干燥程序涉及下述:
a)小瓶1:將足夠體積的ECDG溶液添加至小瓶1,冷凍并且然后在Ar(g)條件下冷凍干燥。
b)小瓶2:將預定體積的制備的磷酸鹽/檸檬酸緩沖溶液添加至充滿Ar(g)的小瓶2,冷凍并冷凍干燥過夜,隨后添加Sn溶液(60-100μg Sn(II)),隨后在Ar(g)條件下凍干。
將所有的小瓶儲存在制冷器的黑暗條件下。標記方案需要小瓶1的重構或溶解,添加小瓶1至小瓶2,隨后立即添加足夠的99mTc活性。將反應混合物加熱(60-80℃)有限的時間,以確保標記。用TLC和HPLC的質量控制應記錄>90%的標記并且放射化學純度大于95%。
對于一個小瓶試劑盒配制,使用上述溶液的冷凍干燥程序涉及下述:
a)首先,將預定體積的制備的磷酸鹽/檸檬酸緩沖劑冷凍并冷凍干燥。接著,將Sn溶液(60-100μg Sn(II))添加至充滿Ar(g)的小瓶并且冷凍,隨后在Ar(g)條件下冷凍干燥。
b)最后,將純的ECDG溶于a)的冷凍干燥材料上方的非水性溶劑、冷凍并冷凍干燥。該標記方案需要僅僅通過添加足夠的99mTc活性重構。將反應混合物加熱(60-80℃)有限的時間,以確保標記。用TLC和HPLC的質量控制應記錄>90%標記并且放射化學純度大于95%。添加至試劑盒并且構建準備注射。
在制備試劑盒時,申請人合成制備了ECDG。從商業上可得的L-噻唑烷-4-羧酸開始,在五個合成步驟中成功進行了產生ECDG的合成路徑。合成路徑可簡單總結如下。
從結構角度,99mTc-ECDG可視為由三個組分組成,即:(i)在其核心處的L,L-乙烯二半胱氨酸(EC)配體,(ii)兩個靶向癌癥的D-葡糖胺基團和(iii)99mTc放射性核素。EC可獲得自商業上可得的L-4-噻唑烷羧酸的自由基促進的二聚化反應[10]。EC的硫醇和仲胺官能團是活性位點并已經顯示分別被芐基(Bn)[11]和氯甲酸芐酯(Cbz)保護基團有效和高效保護(mask)。兩個葡糖胺基團理論上可通過采用試劑氯甲酸乙酯經混合酸酐偶聯反應與EC的酸部分結合。接著,可通過鈉/氨溶液中偶聯反應產物的全部脫保護(global deprotection)產生ECDG[8]。該反應可用苯基乙酸銨淬滅,其產生2-丙醇可溶性苯基乙酸鈉鹽,這使得從反應副產物適當純化ECDG。該合成的ECDG可接著用99mTc標記并且根據需要使用。
如文獻規定[10],由L-噻唑烷-4-羧酸精確進行EC 4的合成并且產生期望的產物,產率是38%。
一旦反應結束,使氨快速蒸發(沸點是-33℃)并且將所得殘渣溶于水,以產生高堿性的(pH=12.0)溶液。因此,添加5M HCl,以使堿化的EC配體質子化并且使分子作為其二鹽酸化合物鹽沉淀,這在pH 3.0–2.0內實現。將開始材料L-噻唑烷-4-羧酸溶于酸性介質中并且保留在溶液中并且所以該步驟用作EC 4純化的第一階段。接著,將沉淀EC 4過濾并且發現該粗EC 4立即從沸騰乙醇中重結晶,隨后在高真空度下干燥材料,產生純的EC 4,為粉末狀白色固體。在D2O中進行EC 4的NMR,必要時添加6.0當量的K2CO3,以(i)中和二鹽酸化合物鹽和(ii)使硫醇和酸官能團(functionality)去質子化,這使得EC4溶解并被分析。EC 4的質子和碳NMR數據與文獻數據精確一致,以及確定的熔點。該數據也說明EC 4的純度大于99%。
根據參考文獻信息[10],EC 4被芐化(benzylate)并且沒有觀察到來自此的偏離(deviation)。該保護步驟是必要的,因為硫醇基也在計劃的葡糖胺偶聯反應中反應并且因此需要被保護。但是關于EC-Bn5的質子
并且因此,必須確定分析的溶劑系統和方法。實驗發現EC-Bn 5完全溶于連同添加4.0當量的K2CO3的以6:4v/v比例的D2O和氘化的DMF的混合物中,K2CO3用于中和EC-Bn 5的二鹽酸化合物鹽并且使兩個酸部分去質子化。這使得產生EC-Bn5的NMR數據并且用作首次報道的有關該化合物的質子和碳NMR圖譜。質子圖譜與親本EC 4化合物的非常相似,但是包含芐基CH2質子作為4.69ppm處的單峰并且在7.16ppm處出現的十個芳族質子作為多峰。碳NMR圖譜與質子NMR圖譜的發現相關,因為在35.9ppm處觀察到CH2碳原子并且在127.1ppm、128.6ppm、128.8ppm和138.6的信號源自芳族環。該數據,以及匹配在預期文獻范圍內的確定的熔點,確認了成功實現了芐基保護。
用氯甲酸芐酯保護基團保護EC-Bn 5的仲胺部分。與硫醇基團類似,這些仲氨基也在計劃的葡糖胺偶聯反應中反應并且所以也需要封端(cap)。EC-Bn Cbz保護起初在0℃下進行2h并且然后在室溫(RT)下持續16h。需要二乙醚洗滌步驟,以去除任何未反應的CbzCl,隨后使水性介質酸化至pH 3.0,以使EC-Bn-Cbz 6的羧酸基團質子化,
其產生產物的沉淀,為白色固體。發現,產物溶于大量的有機溶劑并且用乙酸乙酯提取酸化溶液使得EC-Bn-Cbz6被分離。隨后有機相的分離和溶劑去除產生期望產物,為無定形固體。EC-Bn-Cbz 6必須在存在高真空度的情況下充分干燥,以確保材料完全沒有痕量的溶劑或水。EC-Bn-Cbz 6產物在二氧化硅凝膠上快速分解并且因此可不用另外純化,發現這與公開的數據[12]相反。用于EC-Bn-Cbz 6的NMR分析的溶劑系統可能不是確定的并且這也與在CDCl3中產生NMR數據的文獻信息不一致。該產物的LC-MS分析也證明是不成功的,原因是用于MS測定的非常有問題的芐基保護基團。因此粗EC-Bn-Cbz 6直接用于接下來的步驟。
采用氯甲酸乙酯作為偶聯劑進行EC-Bn-Cbz 6和四-乙酰葡糖胺的偶聯反應。反應條件和流程以與現有技術中發現的那些相同的方式進行,但是確定了新的柱純化溶劑系統。發現以(1-5):(10-90):(10-80)范圍內的比例的甲醇(MeOH)、乙酸乙酯(EtOAc)和己烷的三組分溶劑組合使得完全保護的ECDG 7以較高的純度被分離。
最后的步驟是充分保護的ECDG7的鈉/氨促進的完全脫保護,以產生ECDG 3。充分保護的ECDG 7與20.0當量的鈉金屬反應,以完全去除酸酯、Cbz和Bn保護基團。接著,添加12.0當量的苯基乙酸銨——其使得形成苯基乙酸鈉作為副產物——淬滅反應。一旦在氬氣氣氛蒸發了氨液體,通過2-丙醇洗滌步驟,從反應混合物去除苯基乙酸鈉。苯基乙酸鈉高度溶于2-丙醇,但是ECDG 3不是,并且因此在惰性氣氛下過濾有機介質,以產生ECDG 3,為乳白色、強烈氣味的固體。接著,用二乙醚洗滌該ECDG 3,并且然后在高真空度下避光干燥1h。通過MS確定該ECDG的鑒定和純度(圖1)并且在591.1單位觀察到ECDG 3必要的MS峰。在氬下,避光-20℃儲存ECDG 3。
實施例
實施例1–雙小瓶試劑盒
凍干方案
a)向磷酸氫二鈉(sodium phosphate dibasic)(0.284g,0.002mol)的水溶液(脫氧的(de-oxynated))添加檸檬酸(0.201g,0.001mol),以產生pH 5.5de磷酸鹽/檸檬酸緩沖溶液。將855μl制備的磷酸鹽/檸檬酸緩沖溶液添加至第一填充氬的小瓶,封閉并且冷凍,然后冷凍干燥過夜。
b)將鹽酸(0.10ml,0.1M)添加至氯化錫(II)二水合物溶液(0.01g,0.04mmol),并且用水(脫氧的)稀釋至10ml。接著,將100μl Sn溶液(=60μg Sn(II))添加至小瓶1,冷凍,隨后冷凍干燥。
c)將甲醇(1.5ml)添加至第二個填充氬的具有ECDG(10mg,0.017mmol)的小瓶。將小瓶浸入液氮,以冷凍溶劑并冷凍干燥。小瓶應保持在黑暗和制冷器中。
注意,所以的小瓶應填充Ar,以確保沒有CO2或O2。
標記方案
a)將355μl H2O添加至冷凍干燥的ECDG小瓶。
b)轉移至冷凍干燥的緩沖劑/Sn小瓶并且加入小的磁力攪拌棒。形成漩渦(Vortex),以溶解緩沖鹽。
c)隨后立即添加500μl TcO4-(或等體積,用于約40mCi活性)。
d)放置在加熱板上并且在70℃下攪拌15分鐘。
e)進行TLC和HPLC-QC。
實施例2–一個小瓶試劑盒
凍干方案
a)向磷酸氫二鈉(0.284g,0.002mol)的水溶液(脫氧的)添加檸檬酸(0.201g,0.001mol),以產生pH 5.5的磷酸鹽/檸檬酸緩沖溶液。將855μl制備的磷酸鹽/檸檬酸緩沖溶液添加至第一個填充氬的小瓶,封閉并且冷凍,然后冷凍干燥過夜。
b)將鹽酸(0.10ml,0.1M)添加至氯化錫(II)二水合物溶液(0.01g,0.04mmol)并且用水(脫氧的)稀釋至10ml。接著,將100μl Sn溶液(=60μg Sn(II))添加至小瓶1,冷凍隨后冷凍干燥。
c)將甲醇(1.5ml)添加至第二個填充氬的具有ECDG(10mg,0.017mmol)的小瓶。這被定量轉移至包含Sn/緩沖劑的小瓶1。將小瓶浸入液氮,以冷凍溶劑并冷凍干燥。小瓶應保持在黑暗和制冷器中。
注意,所有的小瓶應填充Ar,以確保沒有CO2或O2。
標記方案
a)將500μl TcO4-(或等體積,用于約40mCi的活性)添加至包含ECDG、Sn和緩沖劑的小瓶。
b)放置在加熱板上并且在70℃下攪拌15分鐘。
c)進行TLC和HPLC-QC。
實施例3–合成ECDG
合成L,L-乙烯雙半胱氨酸.2HCl
將L-噻唑烷-4-羧酸(30.0g,225mmol)緩慢添加至配備有冷卻冷凝器(填充有液氮)、氬氣體入口和填充油的出口捕集器(outlet trap)的雙頸圓底燒瓶的液氨(150ml)中。強力攪拌混合物,直到所有L-噻唑烷-4-羧酸完全溶解,隨后在15分鐘內逐份添加清潔的鈉金屬(8.00g,349mmol,1.50當量)。一旦完成鈉金屬的添加,觀察到深藍色,并且在室溫下攪拌該溶液20分鐘。接著,以抹刀尖(spatula-tip)部分小心添加氯化銨,直到混合物變成白色,并且已淬滅所有未反應的鈉金屬。接著,使得氨溶劑蒸發并且將所得反應殘留物溶于水(200ml)并且用濃縮的HCl將pH調整至3.0,其產生乙烯雙半胱氨酸的二鹽酸化合物鹽的沉淀,為白色固體。通過真空過濾收集產物,從沸騰乙醇重結晶并且在高真空度下干燥,以產生14.7g(38%)乙烯雙半胱氨酸.2HCl 4
M.p.:252-254℃(參考251-253℃[10]);
1H NMR(400MHz,D2O和6.0當量的K2CO3):
δH=3.27(2H,t,2x CH-COOH),2.70-3.00(8H,m,2x CH2-N和2x CH2-SH重疊),2.62(2H,m,2x NH)2。
13C NMR(400MHz,D2O和6.0當量K2CO3):
δC=177.9(COOH),65.6(CH-N),44.8(CH2-N),26.8(CH2-SH)。
合成S,S’-二芐基乙烯雙半胱氨酸.2HCl 5
在室溫下將乙烯雙半胱氨酸.2HCl 4(2.0g,6.0mmol)溶于2M NaOH(30ml)并且添加乙醇(40ml),并且強力攪拌所得溶液20分鐘。將二噁烷(20ml)中的芐基氯化物(1.48g,11.7mmol,2.0當量)逐滴添加至乙烯雙半胱氨酸溶液并且然后在添加結束之后攪拌另外的30min。接著,真空去除乙醇和二噁烷,并且然后用5M HCl酸化所得水性混合物的pH至pH 3.0。這產生S,S’-二芐基乙烯雙半胱氨酸5的鹽酸鹽沉淀,其在真空下過濾并且在高真空度下干燥,產率為85%(2.7g)。
M.p.:227-228℃(參考251-253℃);
1H NMR(400MHz,D2O/DMF(6:4v/v比)和4.0當量的K2CO3):
δH=7.16(10H,m,2x CH2-C6H5),3.68(4H,s,2x CH2-C6H5)3.14(2H,t,CH-COOH),2.44-2.85(10H,m,2x CH2-N,2x CH2-SH和2x NH重疊);
13C NMR(400MHz,D2O/DMF(6:4v/v比)和4.0當量的K2CO3):
δC=179.5(COOH),138.6(Ar-C),128.0(Ar-C),128.6(Ar-C),127.1(Ar-C),62.7(CH-N),46.6(CH2-N),35.9(CH2-C6H5),34.4(CH2-SH)。
合成充分保護的乙烯雙半胱氨酸脫氧葡糖胺7
將S,S’-二芐基乙烯雙半胱氨酸5(6.0g,11.5mmol)溶于10%K2CO3溶液(150ml)并且在冰浴中冷卻至0℃。接著,將二噁烷(150ml)中的氯甲酸芐酯的混合物快速添加至溶液,其接著在0℃下攪拌2小時。接著,去除冷卻浴并且將混合物在室溫下攪拌16h,然后用二乙醚(2×50ml)萃取。接著,用1M HCl小心酸化水性層至pH 3.0,其產生白色化合物沉淀。添加乙酸乙酯(200ml)并且強烈攪拌將沉淀的固體溶于該有機層。分開有機層,用無水硫酸鎂干燥,過濾并且在旋轉式汽化器上去除溶劑。接著,在高真空度下干燥所得透明殘留物,以產生5.75g(70%產率)粗N,N’-二芐氧基羰基-S,S’-二芐基乙烯雙半胱氨酸6,為無定形的固體。該化合物是對于純化不穩定并且不溶于測試的NMR溶劑,并且因而直接用于接下來的反應。
將EC-Bn-CBz6(1.34g,1.87mmol)溶于具有三乙胺(0.378g,3.74mmol,2.0當量)的干燥氯仿(30ml),并且在氬氣氛下在氯化鈉/冰漿冷卻浴(ice slurry cooling bath)中將溶液冷卻至-15℃。逐滴添加氯甲酸乙酯(0.406g,3.74mmol,2.0當量)并且攪拌所得混合物另外的15分鐘。向該反應混合物,添加四乙酰葡糖胺(1.58g,4.11mmol,2.2當量)和三乙胺(0.416g,4.11mmol,2.0當量)的干燥氯仿(30ml)的溶液,并且在0℃下攪拌組合的反應混合物1h,并且然后在室溫下攪拌12h。接著,將用1M HCl(2×25ml)、5%K2CO3溶液(2×25ml)、H2O(50ml)洗滌溶液,用無水硫酸鎂干燥,過濾并且真空去除溶劑。通過柱色譜(二氧化硅凝膠60;流動相:MeOH/EtOAc/己烷)純化殘留物,以產生1.80(70%產率)g的完全保護的乙烯雙半胱氨酸脫氧葡糖胺7,為白色結晶固體。
1H NMR(400MHz,CDCl3):
δH=8.62(2H,s,2x NH),7.48-7.40(20H,m,2x OCH2-C6H5,2x SCH2-C6H5),6.04(2H,d,四氫吡喃端基質子),5.45-5.20(6H,m,2x OCH2-C6H5,2x四氫吡喃質子重疊),4.48-4.07(6H,m,2x CH-CONH,4x四氫吡喃質子重疊),3.72-3.48(12H,4x四氫吡喃質子,4x CH2-N-,2x CH2-S-重疊),2.20-1.92(24H,8x OCH3)。
合成乙烯雙半胱氨酸脫氧葡糖胺3
在氬氣氛下將充分保護的乙烯雙半胱氨酸脫氧葡糖胺7(1.00g,0.73mmol)溶解在氨液體(100ml)中,并且以小部分添加清潔的鈉金屬(0.334g,14.5mmol,20.0當量)。反應混合物變成深藍色并且在室溫下攪拌15分鐘,然后添加少量的銨苯基乙酸酯,以淬滅未反應的鈉金屬。在氬氣體流下干燥所得乳白色溶液,以提供強烈氣味的乳白色固體的。在惰性氣氛下避光處理粗產物。將2-丙醇(200ml)添加至材料并且強力攪拌10min,然后真空過濾。用二乙醚洗滌所得乳白色沉淀,并且然后在高真空度下干燥2小時,以產生0.230g(53%產率)的乙烯雙半胱氨酸脫氧葡糖胺的鈉鹽(ECDG)3。通過1H NMR、HPLC和MS分析確認產物,其與文獻數據一致。C20H38O12N4S2需要590.665,其中觀察到591.1。
參考文獻
1.http://clinicaltrials.gov/show/NCT01394679,20/08/2013
2.Zhang,Y.H.,Bryant,J.,Kong,F.L.,Yu,D.F.,Mendez,R.,Kim,E.E.&Yang,D.J.,2012,Molecular Imaging of Mesothelioma with 99mTc-ECG and 68Ga-ECG,Journal of Biomedicine and Biotechnology,2012.
3.Zaman,M.,2007,99mTc-EC-deoxyglucose–a poor man’s 18F-FDG:what will be the future of PET in molecular imaging?,European Journal of Nuclear Medicine and Molecular Imaging,34,427-428.
4.http://www.health24.com/Medical/Cancer/Facts-and-figures/South-Africa-78-increase-in-cancer-by-2030-20120721,20/08/2013.
5.Yang,D.,Kim,C.,Schechter,N.R.,Azhdarinia,A.,Yu,D.,Oh,C.,Bryant,J.L.,Won,J.,Kim,E.&Podoloff,D.A.,2003,Imaging with 99mTc-ECDG targeted at the multifunctional glucose transport system:feasibility study with rodents,Radiology,226,465-473.
6.Zhang,Y.,Mendez,R.,Kong,F.,Bryant,J.,Yu,D.,Kohanim,S.,Yang,D.&Kim,E.,2011,Efficient synthesis of 99mTc-ECDG for evaluation of mesothelioma,Journal of Nuclear Medicine,52(Suppl.1),1532
7.Ebrahimabadi,H.,Lakouraj,M.M.,Johari,F.,Charkhlooie,G.A.,Sadeghzadeh,M.,2006,Synthesis,characterization and biodistribution of99mTc-(EC-DG),a potential diagnostic agent for imaging of brain tumors,Iranian Journal of Nuclear Medicine,14(Suppl.1),36-37
8.Yang,D.J.,2008,US Patent Application US20080107598
9.Blondeau,P.,Berse,C.,Gravel,D.,1967,Dimerization of an intermediate during the sodium in liquid ammonia reduction of L-thizolidine-4-carboxylic acid,Canadian Journal of Chemistry,45,49-52
10.Assad,T.,2011,Synthesis and Characterization on novel benzovesamicol analogues,Turkish Journal of Chemistry,35,189-200.
11.Mang’era,K.O&Verbruggen,A.,1999,Synthesis and Evaluation ofβ-Homocysteine Derivatives of 99mTc-L,L-EC and 99mTc-L,L-ECD,Journal of Labelled Compounds and Radiopharmaceuticals,42,683-699.
- 還沒有人留言評論。精彩留言會獲得點贊!