化合物及其制備方法和用途與流程
本發明涉及生物醫藥領域,具體而言,本發明涉及一種化合物及其制備方法和用途,更具體的,本發明涉及艾氟康唑藥物的新雜質,該雜質的合成方法和在艾氟康唑藥物檢測中的用途。
背景技術:
艾氟康唑(efinaconazole,商品名為jublia)是由dowpharmaceutical研發。2014年6月6日獲得fda批準,對人和動物的真菌疾病有效治療,臨床上用10%的溶液來治療灰指甲(onychomycosis)。
然而,目前的艾氟康唑藥物仍有待改進。
技術實現要素:
本發明旨在至少在一定程度上解決相關技術中的技術問題之一。為此,本發明的一個目的在于提出一種能夠有效地用于艾氟康唑藥物檢測的手段。在本發明的第一個方面,本發明提供了一種化合物。根據本發明的實施例,該化合物為式1所示化合物。
發明人驚奇地發現,采用該化合物作為對照品,能夠有效地用于對艾氟康唑藥物進行質量控制。
在本發明的第二方面,本發明提出了一種制備式1所示化合物的方法,所述方法包括:(1)將式2所示化合物與三甲基碘化亞砜接觸,以便獲得式3所示化合物;(2)將式3所示化合物的環氧進行開環反應,以便獲得式4所示化合物;(3)將式4所示化合物的氨基進行環合反應,以便獲得式5所示化合物;(4)將式5所示化合物進行脫四氫吡喃保護基反應,以便獲得式6所示化合物;(5)將式6所示化合物進行分子內環氧化反應,以便獲得式7所示化合物;(6)將式7所示化合物與和式8所示化合物接觸,以便獲得式1所示化合物。
發明人發現,通過采用該方法可以有效地制備式1所示化合物,且合成路線短、環境友好、目標產物的收率和純度較高,操作簡單,原料易得,適合工業化大生產。如前所述,根據本發明的方法所制備的化合物作為對照品,能夠有效地用于對艾氟康唑藥物進行質量控制。
同時,發明人發現,通過采用該方法能得到1,3,4構型的三氮唑官能團,且該方法簡單易行,收率高,性質穩定,從而為三氮唑異構體的合成提供了新方法。
在本文中所使用的術語“接觸”應做廣義理解,其可以是任何能使得至少兩種反應物發生化學反應的方式,例如可以是將兩種反應物在適當的條件下進行混合。在本文中,“化合物n”在本文中有時也稱為“式n所示化合物”,在本文中n為1-8的任意整數,例如“化合物2”在本文中也可以稱為“式2所示化合物”。
在本發明的描述中,需要理解的是,術語“第一”、“第二”僅用于描述目的,而不能理解為指示或暗示相對重要性或者隱含指明所指示的技術特征的數量。由此,限定有“第一”、“第二”的特征可以明示或者隱含地包括一個或者更多個該特征。在本發明的描述中,“多個”的含義是兩個或兩個以上,除非另有明確具體的限定。
根據本發明的實施例,上述制備式1所示化合物的方法還可以進一步包括如下附加技術特征至少之一:
根據本發明的實施例,在步驟(2)中,所述開環反應是通過將式3所示化合物與氨水在第一有機溶劑中進行接觸完成的。由此,可提高制備式4所示化合物的效率,進而進一步提高制備式1所示化合物的效率,另外,可以進一步提高利用所得到的式1所示化合物作為對照品檢測艾氟康唑藥物的效率。
根據本發明的實施例,所述第一有機溶劑為包括選自甲醇、乙醇和四氫呋喃中的至少之 一,優選為甲醇。由此可以縮短反應時間,提高制備式4所示化合物的效率,進而進一步提高制備式1所示化合物的效率,另外,可以進一步提高利用所得到的式1所示化合物作為對照品檢測艾氟康唑藥物的效率。
根據本發明的實施例,所述氨水與式3所示化合物的體積質量比為10-20ml:1g,優選為15ml:1g;由此可以提高反應速度,降低副產物的形成。發明人發現,所述氨水與式3所示化合物的體積質量比小于10ml:1g,則應速度下降,所述氨水與式3所示化合物的體積質量比高于20ml:1g,,則反應副產物增多。
根據本發明的實施例,所述第一有機溶劑與式3所示化合物的體積質量比為5ml~25ml):1g,優選為15ml:1g。由此可以提高化合物4的含量,進而進一步提高化合物1的含量。發明驚奇的發現,第一有機溶劑與式3所示化合物的體積質量比小于5ml:1g,則反應副產物增多,第一有機溶劑與式3所示化合物的體積質量比大于25ml:1g,則反應速度下降。
根據本發明的實施例,所述開環反應是在50~70攝氏度的溫度下進行的,優選為60攝氏度。由此可以調高化合物4的含量,進而進一步提高化合物1的產量。發明人發現。開環反應溫度低于50攝氏度,則反應速度過低,溫度高于70攝氏度,則副產物顯著增多。
根據本發明的實施例,在步驟(3)中,所述環合反應是通過將式4所示化合物與n-甲酰肼和原甲酸三乙酯在第二有機溶劑中進行接觸完成的。由此可以提高制備式5所示化合物的效率,進而進一步提高制備式1所示化合物的效率,另外,可以進一步提高利用所得到的式1所示化合物作為對照品檢測艾氟康唑藥物的效率。
根據本發明的實施例,所述第二有機溶劑為選自甲醇和乙醇中的至少一種,優選為甲醇。由此可以提高反應的效率。
根據本發明的實施例,所述第二有機溶劑與式4所示化合物的體積質量比為(5ml~15ml):1g,優選為10ml:1g。由此可以進一步提高式5所示化合物的產量,進而進一步提高式i所述化合物的效率。發明人經過大量的篩選實驗發現,所述第二有機溶劑與式4所示化合物的體積質量比小于5ml:1g,則反應副產物增多,所述第二有機溶劑與式4所示化合物的體積質量比大于15ml:1g,反應原料剩余多。
根據本發明的實施例,所述n-甲酰肼與式4所示化合物的摩爾比為6:1到2:1,優選為4:1;由此可以滿足原料完全消耗,產物含量較高。發明人發現,所述n-甲酰肼與式4所示化合物的摩爾比低于2:1,則原料剩余,所述n-甲酰肼與式4所示化合物的摩爾比高于6:1,則化合物5的產量顯著降低。
根據本發明的實施例,所述原甲酸三乙酯與式4所示化合物的摩爾比為6:1到2:1優選為 4:1。由此可以滿足原料完全消耗,產物含量較高。同樣,發明人發現,所述原甲酸三乙酯與式4所示化合物的摩爾比低于2:1,則原料剩余,所述原甲酸三乙酯與式4所示化合物的摩爾比高于6:1,則化合物5的產量顯著降低。
根據本發明的實施例,所述環合反應是在氮氣保護下進行。氮氣化學性質不活潑,只有在高溫高壓或者是放電的情況下才進行反應,在本發明實施例的環化反應條件下,氮氣可作為保護氣避免反應物與周圍物質的反應,從而有效避免了副產物的生成,提高了化合物5的產量,進而進一步提高了化合物1的產量。
根據本發明的實施例,步驟(3)進一步包括:(3-1)先預先將所述n-甲酰肼和原甲酸三乙酯接觸混合,以便得到第一混合體系;(3-2)將所述式4所示化合物與所述第一混合體系接觸,以便獲得式5所示化合物。由此,可以有效保證反應按照預期進行環合反應,發明人發現,改變步驟(3)中改變式4所示化合物與n-甲酰肼和原甲酸三乙酯的接觸順序,導致反應無法得到預期產物,且反應的產物含量偏低。
根據本發明的實施例,所述n-甲酰肼和原甲酸三乙酯混合是在75~85攝氏度的溫度下進行3小時,優選為80攝氏度。由此可以反應產物含量較高。且發明人發現,混合溫度低于75攝氏度,則反應速度過低,混合溫度高于85設施度,則反應副產物過多。
根據本發明的實施例,式4所示化合物與所述第一混合體系接觸是在75~85攝氏度的溫度下進行2~6小時,優選為4小時。由此可以有效保證產物含量較高。發明人發現,式4所示化合物與所述第一混合體系接觸的時間短于2小時,則反應速度顯著下降,式4所示化合物與所述第一混合體系接觸的時間長于6小時,則副產物顯著增加。
根據本發明的實施例,在步驟(6)中,式7所示化合物與和所述式8所示化合物在接觸是在第三有機溶劑中進行接觸,且所述第三有機溶劑且中存在堿和碘化鉀,由此,可以可進一步提高制備式1所示化合物的效率。
根據本發明的實施例,所述第三有機溶劑為乙腈或乙醇,優選為乙腈。由此產物可進一步提高式1所示化合物的產量。
根據本發明的實施例,所述堿為一水氫氧化鋰。由此可進一步提高式1所示化合物的產量。
根據本發明的實施例,所述接觸是在溫度為80~85攝氏度的溫度下進行12~24小時。由此可進一步提高式1所示化合物的產量。
根據本發明的實施例,所述堿與式7所示化合物的摩爾比為2:1~2.5:1,優選為2.2:1。由此可進一步提高式1所示化合物的產量。
根據本發明的實施例,所述碘化鉀與式7所示化合物的摩爾比為1.2:1~2:1,優選為1.3:1。由此可進一步提高式1所示化合物的產量。
根據本發明的實施例,式8所示化合物與式7所示化合物的摩爾比1.2:1~1.8:1,優選為1.5:1。由此可進一步提高式1所示化合物的產量。
根據本發明的實施例,所述第三有機溶劑與式7所示化合物的體積質量比為(1ml~10ml):1g,優選為5ml:1g。由此可進一步提高式1所示化合物的產量。
根據本發明的第三方面,本發明提出式1所示化合物在控制艾氟康唑藥物質量中的用途。根據本發明的實施例,式1所示化合物為制備艾氟康唑過程中的工藝雜質。本發明實施例的制備式1所示化合物的方法可高效制備式1所示化合物,從而實現定向制備艾氟康唑雜質,即化合物1,從而為工業化生產艾氟康唑產品中的質量控制及雜質定量提供了可靠的雜質對照品。另外,本發明實施例的制備式1所示化合物的方法起始原料廉價易得,工藝操作易控制,生產效率高,能滿足工業化生產艾氟康唑產品中的質量控制質量的需求。另外,本發明實施例的制備式1所示化合物的方法,反應操作過程簡單,條件溫和,無需特殊反應設備、在制備過程中沒有難以分離的化合物,工藝操作簡便,生產效率高,適于工業化大生產。進而,采用化合物1作為對照品,能夠有效地用于在工業生產艾氟康唑藥物中對艾氟康唑藥物進行質量控制。
本發明附加方面和優點將在下面的描述中部分給出,部分將從下面的描述中變得明顯,或通過本發明的實踐了解到。
附圖說明
圖1顯示了根據本發明的實施例的所得化合物1的高效液相色譜圖;
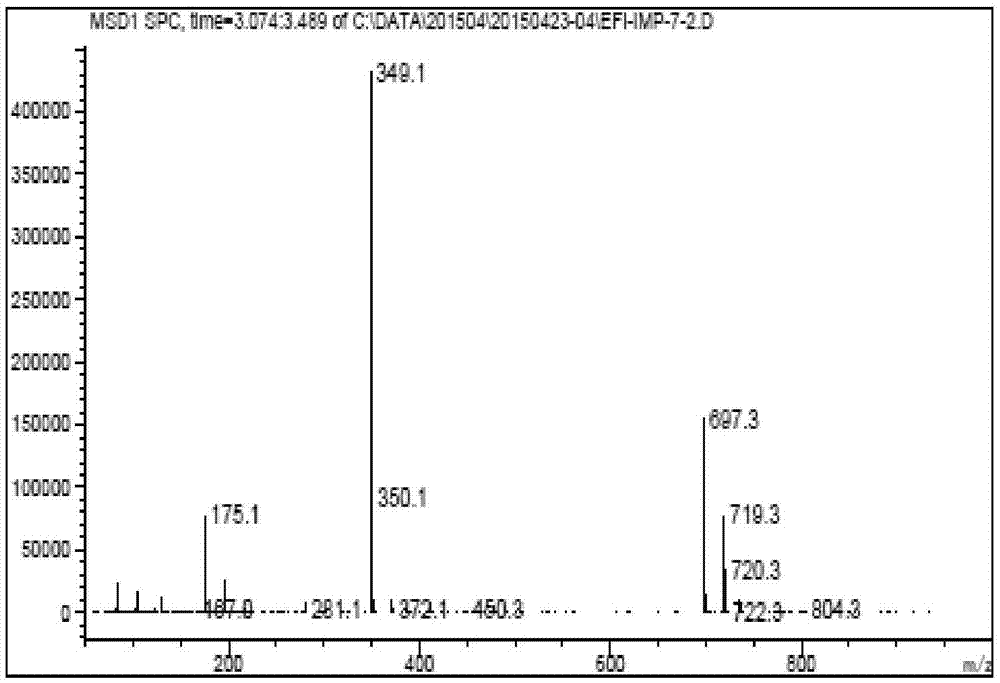
圖2顯示了根據本發明的實施例的所得化合物1的質譜圖;
圖3顯示了根據本發明的實施例的所得化合物1的核磁共振氫譜圖;以及
圖4顯示了根據本發明的實施例的所得化合物1的核磁共振碳譜圖。
具體實施方式
下面詳細描述本發明的實施例。下面描述的實施例是示例性的,僅用于解釋本發明,而不能理解為對本發明的限制。實施例中未注明具體技術或條件的,按照本領域內的文獻所描述的技術或條件或者按照產品說明書進行。所用試劑或儀器未注明生產廠商者,均為可以通過市購獲得的常規產品。
在以下實施例中對化合物1合成過程進行詳細描述:
實施例1化合物3的制備
將3.4gnah(60%)加入400毫升dmso中,再加入19.6g三甲基碘化亞砜,室溫攪拌1小時,直至無氣泡產生,過程中伴隨放熱現象,加入20.0g化合物2,室溫攪拌一個小時,取樣,薄層色譜監控表明原料已反應完后,加入200毫升水稀釋,再用1000毫升乙酸乙酯萃取,用500毫升水洗滌有機相,用500毫升飽和食鹽水洗滌有機相干燥,濃縮,得到19.6g化合物3,收率93.2%,直接用于下一步。
實施例2化合物4的制備
將19g實施例1所得化合物3加到190毫升甲醇中,再加入285毫升氨水,加熱到60℃并繼續反應,3小時后取樣,薄層色譜監控反應,原料消失,將反應液在40℃下濃縮去除甲醇,加入500毫升乙酸乙酯萃取,水洗,飽和食鹽水洗滌,干燥濃縮得13.1g化合物4,收率65.0%,純度93.2%。
實施例3化合物4的制備
另外,化合物4也可通過如下方法獲得:將5.0g實施例1所得化合物3加到25毫升甲醇中,再加入50毫升氨水,加熱到50℃并繼續反應,3小時后取樣,薄層色譜監控反應,原料消失,將反應液在40℃下濃縮去除甲醇,加入200毫升乙酸乙酯萃取,水洗,飽和食鹽水洗滌,干燥濃縮得2.38g化合物4,收率45.0%,純度92.3%。
實施例4化合物4的制備
另外,化合物4也可通過如下方法獲得:將5.0g實施例1所得化合物3加到75毫升甲醇中,再加入100毫升氨水,加熱到70℃并繼續反應,3小時后取樣,薄層色譜監控反應,原料消失,將反應液在40℃下濃縮去除甲醇,加入200毫升乙酸乙酯萃取,水洗,飽和食鹽水洗滌,干燥濃縮得2.22g化合物4,收率42.0%,純度92.1%。
實施例5化合物5的制備
室溫,在n2保護下將10.4gn-甲酰肼加入到130毫升甲醇中,攪拌下加入25.6g原甲酸三乙酯,攪拌升溫至80攝氏度,回流反應3小時后,加入13.0g實施例2、3或4所得化合物4,4小時后取樣,薄層色譜監控,原料消失,停止反應,降至室溫,轉移并濃縮至無餾分,加入500毫升乙酸乙酯稀釋,用水洗滌,分液,有機相濃縮后得粗品17.0g,粗品經硅膠柱層析(甲醇:二氯甲烷=1:100~1:20,體積比)得到10.8g化合物5,收率87.8%,純度94.1%。
實施例6化合物5的制備
另外,化合物5也可通過如下方法獲得:室溫,在n2保護下將1.2gn-甲酰肼加入到30毫升甲醇中,攪拌下加入2.9g原甲酸三乙酯,攪拌升溫至75攝氏度,回流反應3小時后,加入3.0g實施例2、3或4所得化合物4,2小時后取樣,薄層色譜監控,原料消失,停止反應,降至室溫,轉移并濃縮至無餾分,加入500毫升乙酸乙酯稀釋,用水洗滌,分液,有機相濃縮后得粗品4.1g,粗品經硅膠柱層析(甲醇:二氯甲烷=1:100~1:20,體積比)得到2.04g化合物5,收率57.8%,純度90.1%。
實施例7化合物5的制備
另外,化合物5也可通過如下方法獲得:室溫,在n2保護下將3.6gn-甲酰肼加入到30毫升甲醇中,攪拌下加入9.0g原甲酸三乙酯,攪拌升溫至85攝氏度,回流反應3小時后,加入3.0g實施例2、3或4所得化合物4,6小時后取樣,薄層色譜監控,原料消失,停止反應,降至室溫,轉移并濃縮至無餾分,加入500毫升乙酸乙酯稀釋,用水洗滌,分液,有機相濃縮后得粗品7.0g,粗品經硅膠柱層析(甲醇:二氯甲烷=1:100~1:20,體積比)得到2.39g化合物5,收率67.8%,純度91.1%。
實施例8化合物6的制備
將10.4g實施例5、6或7所得化合物5溶于104毫升甲醇中,冰水浴攪拌,在0~5℃下滴加10毫升濃hcl,加完后室溫攪拌,1.5小時后,薄層色譜監控(甲醇:二氯甲烷=1:20,體積比),原料反應完畢。加20毫升水,調節ph至弱堿性,濃縮,萃取,再濃縮有機相,得粗品8.7g,用435毫升乙醇/水(體積比為3:2)混合液,在60℃下溶解樣品并進行重結晶。得到的白色固體為化合物6,45攝氏度下烘干,得到4.3g,收率54.4%,純度95.1%。
實施例9化合物7的制備
氮氣保護下將2.1g實施例8所得化合物6加入到21毫升干燥的thf中,然后加入2.4g的三乙胺。降低體系溫度至-10℃,緩慢滴加1.7g的甲磺酰氯,反應體系穩定在0~5℃下反應30分鐘,薄層色譜監控(乙酸乙酯:石油醚=1:3,體積比)檢測原料反應完,然后加入10.3毫升3nnaoh溶液,1.25g四丁基溴化銨,反應1小時。薄層色譜監控(乙酸乙酯:石油醚=1:3,體積比),反應完全。加入飽和氯化銨溶液淬滅反應,乙酸乙酯萃取兩次,合并的有機相用水洗和飽和食鹽水洗滌,干燥濃縮得到1.7g淡黃色固體,為化合物7,收率86.7%,純度94.6%。
1h-nmr(400mhz,cdcl3)δ8.01(s,2h,1,3,4-triazole-h),7.00-6.94(m,1h,ph-h),6.82-6.71(m,2h,ph-h),4.78-4.74(d,1h,j=15.0hz,-ch2),4.18-4.174(d,j=14.8hz,1h,-ch2),3.19-3.15(q,j=5.6hz,1h,epoxy-h),1.62-1.60(d,j=5.6hz,3h,-ch3)。
實施例10化合物1的制備
室溫下將化合物8(0.8g,6.0mmol)加入到5毫升干燥的乙腈中,攪拌,再依次加入一水氫氧化鋰(0.37g,8.8mmol)和碘化鉀(0.86g,5.2mmol),攪拌5分鐘后加入實施例9所得化合物7(1.0g,4.0mmol),保持良好攪拌,此為非均相反應液。升溫到85℃回流反應24小時,取樣,直到產物含量為77%,停止反應,冷卻到室溫,加入20毫升飽和氯化銨水溶液,乙酸乙酯萃取,飽和食鹽水洗滌,干燥,過濾濃縮得到粗產品1.7g,乙酸乙酯中結晶得到0.7g化合物1,收率50.7%,液相純度98.4%。其中,化合物1的高效液相色譜圖如圖1所示,化合物1的質譜圖如圖2所示,化合物1的核磁共振氫譜圖如圖3所示,化合物1的核磁共振碳譜圖如圖4所示。
化合物1氫譜數據:1h-nmr(400mhz,cdcl3)δ8.12(s,1h,1,3,4-triazole-h),7.55-7.49(m,1h,ph-h),6.83-6.73(m,2h,ph-h),6.19(brs,1h,-oh),4.69-4.65(dd,j=19.4,1.5hz,1h,-ch2),4.64(s,2h,=ch2),4.30-4.26(d,j=14.1,1.2hz,1h,-ch2),2.78-2.73(q,j=7.2hz,1h,-ch),2.34-2.31(m,4h,piperidine-h),2.21-2.16(m,4h,piperidine-h),1.06-1.03(dd,j=7.2,4.6hz,3h,-ch3);
化合物1碳譜數據:13c-nmr(100mhz,cdcl3)δ164.16-164.03(d,j=12.6hz),161.66-161.54(d,j=12.6hz),159.91-159.79(d,j=11.6hz),157.47-157.35(d,j=11.6hz),144.64,143.66,131.30-131.14(dd,j=6.4,9.3hz),122.72-122.54(dd,j=3.8,13.8hz),112.17-111.94(dd,j=3.0,20.4hz),108.98,104.53-103.99(dd,j=25.2,28.7hz),75.64-75.59(d,j=5.0hz),65.65,52.33-52.253(d,j=7.9hz),34.89,9.15-9.09(d,j=5.9hz)。
實施例11化合物1的制備
另外,化合物1也可以通過如下方法獲得:室溫下將化合物8(0.64g,4.8mmol)加入到1毫升干燥的乙腈中,攪拌,再依次加入一水氫氧化鋰(0.34g,8mmol)和碘化鉀(0.8g,4.8mmol),攪拌5分鐘后加入實施例9所得化合物7(1.0g,4.0mmol),保持良好攪拌,此為非均相反應液。升溫到80℃回流反應12小時,取樣,直到產物含量為77%,停止反應,冷卻到室溫,加入20毫升飽和氯化銨水溶液,乙酸乙酯萃取,飽和食鹽水洗滌,干燥,過濾濃縮得到粗產品1.36g,乙酸乙酯中結晶得到0.56g化合物1,收率50.4%,液相純度98.3%。
實施例12化合物1的制備
另外,化合物1也可以通過如下方法獲得:室溫下將化合物8(0.96g,7.2mmol)加入到10毫升干燥的乙腈中,攪拌,再依次加入一水氫氧化鋰(0.42g,10mmol)和碘化鉀(1.32g,8mmol),攪拌5分鐘后加入實施例9所得化合物7(1.0g,4.0mmol),保持良好攪拌,此為非均相反應液。升溫到83℃回流反應18小時,取樣,直到產物含量為77%,停止反應,冷卻到室溫,加入20毫升飽和氯化銨水溶液,乙酸乙酯萃取,飽和食鹽水洗滌,干燥,過濾濃縮得到粗產品2.04g,乙酸乙酯中結晶得到0.84g化合物1,收率50.5%,液相純度98.2%。
在本說明書的描述中,參考術語“一個實施例”、“一些實施例”、“示例”、“具體示例”、或“一些示例”等的描述意指結合該實施例或示例描述的具體特征、結構、材料或者特點包含于本發明的至少一個實施例或示例中。在本說明書中,對上述術語的示意性表述不必須針對的是相同的實施例或示例。而且,描述的具體特征、結構、材料或者特點可以在任一個或多個實施例或示例中以合適的方式結合。此外,在不相互矛盾的情況下,本領域的技術人員可以將本說明書中描述的不同實施例或示例以及不同實施例或示例的特征進行結合和組合。
盡管上面已經示出和描述了本發明的實施例,可以理解的是,上述實施例是示例性的,不能理解為對本發明的限制,本領域的普通技術人員在本發明的范圍內可以對上述實施例進行變化、修改、替換和變型。
- 還沒有人留言評論。精彩留言會獲得點贊!