茂金屬透明薄膜樹脂的制備方法與流程
本發明涉及一種茂金屬樹脂,尤其涉及一種耐沖擊型茂金屬透明薄膜樹脂。
背景技術:
茂金屬薄膜的透明性好,薄膜強度高,縱橫強度均衡性好,起封溫度低、熱封強度高、熱封溫度范圍寬,耐沖擊性和綜合性能高,表面平整性好,在包裝業占據相當重要的地位,主要用于生產重包裝膜、保鮮膜、纏繞膜及農用棚膜等產品,也可應用于共混領域。
Exxon-Mobil公司茂金屬催化劑的開發與茂金屬聚乙烯生產一直處于世界領先的地位。1991年Exxon公司首次實現了茂金屬催化劑用于聚烯烴工業化生產,生產出第一批茂金屬聚乙烯(mPE),其商品名是“Exact”。1995年正式推出Exceed系列以己烯-1為共聚單體的茂金屬線型低密度聚乙烯(mLLDPE)產品,主要用于薄膜領域。最近Exxon-Mobil公司又推出了一種新的mLLDPE產品-Enable mLLDPE,稱該產品可以在幫助生產商在保持優異的薄膜性能的同時,強化薄膜的擠出加工性能。這種單一而獨特的樹脂將薄膜加工性能與高α-烯烴的優良物理性能結合在一起,適用于一系列軟包裝薄膜應用。2004年初,Exxon-Mobile又致力于Nexxstar產品的市場推廣,以提高公司mPE產品在市場中的占有率。Nexxstar是一種三層共擠壓薄膜,外面兩層是mPE材料,核心層是Escorene特高強度乙酸乙烯酯共聚物。該公司1996年與UCC聯合成立一家合資公司-Univation,開發并轉讓Unipol工藝采用Exxpol的技術。
Univation公司利用茂金屬催化劑與改進的Unipol工藝相結合,已開發生產出高性能產品HPR mLLDPE和易加工產品EZP mLLDPE。前者的加工性比常規的Z-N催化劑生產的LLDPE難度大,在加工時需添加易加工助劑來改善其加工性能;而后者可以不添加易加工助劑或共混就可以很容易加工成制品。 Univation公司于1996年初在Texas 45kt/a LLDPE-HDPE裝置上進行了mLLDPE生產。該公司的Unipol技術在世界的轉讓專利許可證最多,目前聲稱要發放更多使用茂金屬催化劑的許可證。新建的300kt/a Unipol新裝置均可用混合催化劑生產SSC-1和SSC-2兩個系列mLLDPE產品。
Dow公司開發成功的Insite技術,自1993年在57kt/a溶液法聚合裝置上生產mLLDPE后,生產能力已翻番,1996年西班牙又建57kt/a裝置。該技術利用限制幾何構型的催化劑生產兩種系列產品,一種是Affinity聚烯烴塑性體,另一種是Elite聚烯烴彈性體。目前Dow公司已采用茂金屬催化劑開發領先及技術轉讓領先的戰略,1996年宣布與英國BP化學公司聯合發展氣相mPE技術,采用共同技術轉讓的方式,將BP公司“Innovene”氣相聚合工藝與Dow公司Insite催化劑結合生產新型聚乙烯。
2001年,Basell公司推出了其最先進的催化劑體系Avant M。在產品應用開發上,Avant M催化劑技術比傳統的Z-N催化劑更為有效地擴展聚合物的特性,Avant M與氫良好的反應性,可無需過氧化物降解,便得到熔體質量流動速率(MFR)為12g/10min和20g/10min的粒狀茂金屬聚合物,這些聚合物可加工生產工業和衛生用無紡布。新產品的加工性得到改善、揮發性降低、阻隔性較好,長絲纖度降低。Avant M固有的單中心特性,也可以生產具有高純度、高強度、翹曲性極低的聚合物。而且,這些產品均可消毒,具有極好的透明性和光澤度。
國內茂金屬聚烯烴的研究與開發始于上世紀90年代初,近幾年加入該工作的科研單位和院校逐步增多,其中主要有中國石油石油化工研究院、石油化工科學研究院、中科院化學所、北京化工研究院、浙江大學等。而在工業化生產方面,大慶石化分公司于2007年在90kt/a LLDPE裝置上,通過改造首次生產了mLLDPE薄膜產品。也是國內首家引進國外茂金屬催化劑技術的企業,mLLDPE的產量為20kt/a,引進牌號有5個,均為HPR系列產品,主要用于制作高檔薄膜制品。目前已有兩個引進的mLLDPE牌號,在裝置上進行了工業化試生產,實現了國內第一套在低壓氣相法裝置開發成功的先例。
然而無論是國外茂金屬薄膜產品還是國內茂金屬薄膜產品,均致力于開發綜合性能較高的專用樹脂,在某單一方面的性能并不比目前市場上的現有LLDPE/LDPE專用樹脂優異,在產品使用過程中,需通過摻混或結合PP使用 來達到特定目的,亟需開發出在保證綜合性能基礎上,單一性能更加突出的產品,來擴大茂金屬產品的應用領域。
技術實現要素:
針對現有技術中的不足,本發明提供了一種具有獨特分子鏈結構的耐沖擊型茂金屬透明薄膜樹脂的制備方法,由該樹脂加工制得的薄膜具有良好的透明性和
/耐沖擊性,可用于有特殊需求的包裝膜、纏繞膜等薄膜產品。
本發明提供一種茂金屬透明薄膜樹脂的制備方法,包括如下步驟:
乙烯與α-烯烴在氫氣、惰性氣體及新型茂金屬催化劑的存在下,在單一反應器內進行聚合。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述α-烯烴優選為1-丁烯和/或1-己烯。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述α-烯烴摩爾含量優選為1.0~5.0%。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述α-烯烴與乙烯的摩爾比優選為0.01:1~0.06:1。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述反應器中氫氣的濃度優選為100~1000ppm。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所新型茂金屬催化劑優選由負載在載體上的茂金屬化合物與助催化劑組成。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述茂金屬化合物優選為非橋聯的有機金屬化合物,其結構如下:
其中:R1優選為甲基、叔丁基,R2優選為叔丁基、金剛烷基或三芳甲基,Cp' 優選為環戊二烯基或茚基;所述助催化劑優選為甲基鋁氧烷、乙基鋁氧烷、丁基鋁氧烷、[C6H5NH(CH3)2B(C6F5)4]或B(C6F5)3;所述載體優選為無機物載體或高聚物載體。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述金屬原子優選為Ti、Zr或Hf;所述載體優選為硅膠。
茂金屬化合物具有結構和性能易調變的特點,通過改變過渡金屬中心原子,改變配體的取代基團和橋聯基團,可制得不同對稱性、電子效應和空間位阻的催化劑,達到分子設計與分子剪裁的功能。其中,非橋聯茂金屬催化劑最顯著特點是可以通過修飾配體實現對催化劑分子結構的設計。選擇適當的取代基和取代位置,改變配體的電子效應和空間效應,是設計催化劑分子、開發優異催化性能的關鍵。
配體的電子效應可通過茂環上的取代基團進行調節,茂環上的給電子取代基能增加茂環和中心金屬離子周圍的電子云密度,從而降低烯烴雙鍵與金屬離子之間的配位鍵和金屬離子與碳原子所形成的C-M鍵的穩定性,有利于烯烴單體的插入及聚合鏈的增長,吸電子基團可降低鏈增長速率,使之對乙烯聚合的催化活性下降,聚合物的相對分子量也下降;而取代基的位阻效應可阻止雙中心的失活,提高催化劑活性和所得聚合物的分子量,但體積過大空間擁擠會對催化劑活性起反作用。例如,Giannetti等用Cp2ZrMe2/MAO,Ind2ZrMe2/MAO,Flu2ZrMe2/MAO三種催化體系對乙烯聚合的催化活性進行了比較,催化活性順序:Ind2ZrMe2/MAO>Cp2ZrMe2/MAO>Flu2ZrMe2/MAO,原因是茚環的給電子能力更強,而芴環雖然也有較強的給電子能力,但由于體積太大,單體與中心金屬離子配位、插入反應的空間位阻太大,因而其活性相對較低。
本發明所采用的非橋聯茂金屬催化劑,優選了具有特殊配體結構的非橋聯有機金屬化合物,從電子效應和空間位阻效應角度出發,設計了適合的取代基及取代位置,令它起到提高聚合物產率、穩定催化劑活性中心、抑制失活的作用,并且可對共聚單體在聚合物分子鏈上的分布進行調控,使聚合物分子鏈具有特殊結構。
本發明所述的茂金屬透明薄膜樹脂的制備方法,其中,所述聚合為氣相單一反應器聚合,其中聚合溫度優選為80~90℃,聚合壓力優選為1.8~2.5MPa,循環氣速優選為0.60~0.82m/s,停留時間優選為1~8h。
本發明提供方法制得的茂金屬薄膜樹脂,其分子鏈段中的共聚單體支化分布的非均一性,即存在一部分分子鏈段的支化點間距相對較近,一部分分子鏈段的支化點間距相對較遠,使得該樹脂中共聚單體含量多的分子鏈段,兩共聚單體插入點間的鏈端長度小,不易結晶,形成的片晶厚度小;共聚單體含量少的分子鏈段,兩共聚單體插入點間的鏈端長度大,鏈規整,易結晶,形成的片晶厚度大,而結晶形態決定產品的物理性能,這種獨特的晶粒共存方式及晶體形態有利于提高產品的光學性能和沖擊性能,使得該樹脂加工制得的薄膜霧度<8%,落錘沖擊強度>900g。
本發明所述的茂金屬透明薄膜樹脂的制備方法中,對各個工藝參數的控制范圍進行了細化,并提出了循環氣速這個至關重要的工藝參數的控制范圍。因為茂金屬生產控制要求循環氣速很大,以達到最佳的流化狀態,消除反應器中局部熱點,這也是茂金屬生產控制與傳統生產控制的一個主要不同之處。茂金屬生產工藝控制的核心要素是聚合反應的流化狀態控制,良好的流化狀態有利于加速傳質、傳熱,減少局部過熱現象的發生,同時良好的流化狀態能確保對反應器各部位的實時沖刷,防止粉料沉積造成結片。高氣速使超過10%的粉末在系統中循環,循環的粉末對反應器擴大段、循環氣管線、換熱器和分布板形成沖刷,防止粉料吸壁沉積現象,使整個循環系統的阻力減少,以達到最佳的流化狀態和傳質傳熱效果。因此,對聚合工藝參數控制范圍的細化與流化狀態的集中控制至關重要,再輔以本發明所選用的具有特殊配體結構的非橋聯茂金屬催化劑,能夠將本發明的提供的茂金屬透明薄膜樹脂及其制備效果達到最佳。
附圖說明
圖1-4為本發明提供的制備方法生產所得樹脂的分子鏈結構表征圖,以其中5組表征圖為例進行說明;
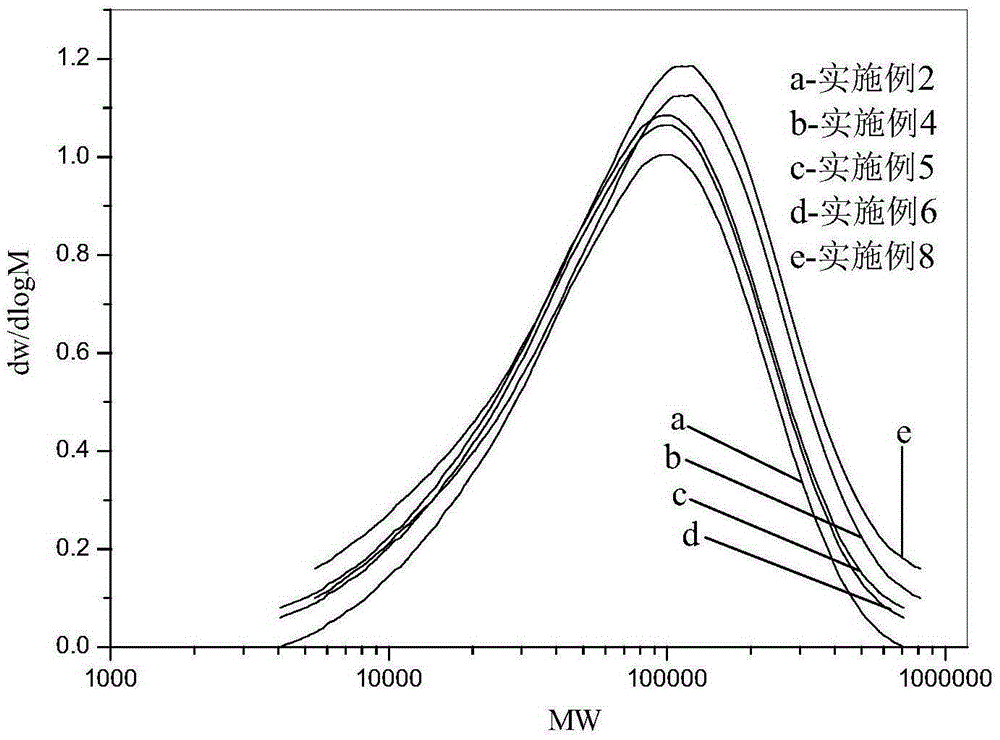
圖1茂金屬薄膜樹脂的GPC表征譜圖;
圖2茂金屬薄膜樹脂的NMR表征譜圖;
圖3茂金屬薄膜樹脂的SSA表征譜圖;
圖4茂金屬薄膜樹脂的TREF表征譜圖。
具體實施方式
以下對本發明的實施例作詳細說明:本實施例在以本發明技術方案為前提下進行實施,給出了詳細的實施方式和過程,但本發明的保護范圍不限于下述的實施例,下列實施例中未注明具體條件的實驗方法,通常按照常規條件。
本發明提供的耐沖擊型茂金屬透明薄膜樹脂制備方法,其內容包括:乙烯與α-烯烴在氫氣、惰性氣體及負載型茂金屬催化劑作用下,在單一反應器內進行聚合。
所述聚合包括淤漿聚合、氣相聚合和溶液聚合。所述聚合為氣相聚合時,聚合溫度為80~90℃,優選82~86℃;聚合壓力為1.8~2.5MPa,優選2.0~2.2MPa;循環氣速為0.60~0.82m/s,優選0.66~0.74m/s;停留時間為1~8h,優選4~6h。
所述聚合過程中,α-烯烴與乙烯的摩爾比為0.01:1~0.06:1,優選0.02:1~0.04:1;反應器中氫氣的濃度為100~1000ppm,優選200~600ppm。
本發明所述的α-烯烴為1-丁烯、1-己烯及其混合物等,用于調節聚乙烯的密度;所述氫氣用于調節聚乙烯的分子量;所述惰性氣體為氮氣。
所述負載型茂金屬催化劑由負載在載體上的茂金屬化合物與助催化劑組成。
所述茂金屬化合物為非橋聯的有機金屬化合物,也可以為橋聯的有機金屬化合物,有機金屬化合物需含有一個以上茂環配體,茂環配體為含有環戊二烯類的配體;所述金屬為Ti、Zr或Hf,也可以選自第4、5和6副族金屬。
所述助催化劑為甲基鋁氧烷,還可以為烷基鋁氧烷及含硼陽離子型助催化劑,如乙基鋁氧烷(EAO)、丁基鋁氧烷(BAO)、[C6H5NH(CH3)2B(C6F5)4]、B(C6F5)3等,目前最有效的、活性最高的助催化劑仍然是甲基鋁氧烷。
所述載體主要分為無機物載體和高聚物載體兩類。無機物載體中,絕大多數是靠表面羥基與過渡金屬配合物或有機鋁化合物進行鍵合,如SiO2、MAO、分子篩、黏土等,僅有少部分載體是直接與過渡金屬配合物鍵合,如MgC12。其中優選無機氧化物、無機氯化物。所述無機氧化物如氧化鋁;所述無機氯化物如氯化鎂。所述載體也可以為高聚物,高聚物載體可分為三類:一類是含有羥基的高聚物,如環糊精等;第二類是在惰性高聚物表面通過輻射接枝不同單體,使其表面含有利-OH、-NHR、-CH2COOCH3等基團;第三類是在惰性高聚物(如聚苯乙烯)的合成過程中加入少量含有一定官能團的單體,如丙烯酰胺 等,從而形成能夠與過渡金屬配合物鍵合或配位的高聚物載體。
由該方法制得的茂金屬薄膜樹脂的熔融指數范圍在1.0±0.2g/10min,密度范圍為0.918±0.002g/cm3。該樹脂的相對分子質量范圍為5000~200000,優選50000~150000;其中數均分子質量范圍為5000~60000,優選20000~50000;重均分子質量范圍為80000~200000,優選100000~150000;相對分子質量分布為2.0~5.0,優選2.0~3.5。該茂金屬薄膜樹脂的分子鏈具有獨特結構:
其分子鏈結構具有明顯的非均一性。
分子鏈結構的非均一性又包括分子量大小和分布不同引起的非均一性和支鏈長短、含量與分布不同引起的非均一性。支鏈含量與分布引起的非均一性又包括分子間支鏈含量不同的分子間非均一性和分子內支鏈分布不同而引起的分子內非均一性,其中分子內支化非的均一性,對聚乙烯產品質量具有決定性作用。
由圖1的對比曲線可以看出,本發明提供的茂金屬薄膜樹脂的相對分子質量分布均較窄,樹脂間的相對分子質量略有差異,分子鏈長短均一性較好,體現除了茂金屬產品的特點。
圖2的對比曲線顯示,共聚單體在譜圖上的出峰位置相同,屬于相同種類,經譜圖各峰歸屬及區域劃分,計算其三單元序列分布數據見表1。
表1 三單元序列分布/%
三單元序列分布數據對比可以看出,各組樹脂的共聚單體在分子鏈中 的含量相當,但共聚單體在分子鏈中的分布存在非均一性。
目前國外對茂金屬聚乙烯分子鏈結構的非均一性研究主要集中在用溶液抽起分級和升溫溶解分級及兩者并用的交叉分級方法來表征茂金屬聚乙烯的非均勻性,這類工作主要在大型石化公司進行(如杜邦、菲利蒲公司等)。而在高等院校,如美國阿克隆大學,則主要用DSC多步結晶分級方法表征支化非均勻性及其對結晶結構形態的影響。本工作是通過連續自成核退火熱分級(SSA)和升溫淋洗分級(TREF)兩種分析方法相互印證對本發明提供的茂金屬薄膜樹脂的分子鏈結構進行表征,表征譜圖見圖3-圖4。
SSA表征曲線通常會有多重較窄的熔融峰,不同的熔融峰代表了不同厚度片晶的熔融結果,即對應著不同分子尺寸的鏈結構單元形成的片晶。這是因為經過第1次熔融降溫后再升到退火溫度時,只有一部分的晶片能夠被熔融,不熔的部分為結晶較完善的部分,它們為比較厚的片晶。在第2個退火溫度時,又有另外一部分片晶沒有被熔融。這樣,不同厚度的晶片便可被分級,而所形成的不同厚度的晶片與分子鏈的結構有關。在這些熔融峰中,較高溫度的峰對應的是結構規整性較好的分子,其片晶較厚;而較低溫度的峰對應的是結構規整性較差的分子,其片晶較薄,共聚單體含量相對較高。這樣,經SSA分級后再升溫的曲線上每個熔融峰基本上就代表了支鏈含量非常接近的一類分子所形成的晶體。
在以往文獻檢索及工作中所表征的國內外同類樹脂的SSA表征曲線上各熔融峰基本呈正態分布,中間峰高,兩端峰低,說明絕大部分級份都比較集中,但本發明的樹脂其最高熔融溫度處的級分含量較大,且遠大于其他各級份,級份分布異于其它樹脂,這說明該樹脂中存在一部分級份其在結晶過程中形成了較厚片晶,為驗證該樹脂確實存在這樣一部分級份,對其進行TREF表征,進一步對該樹脂的級份分布及結晶狀態進行分析。
TREF表征結果顯示,本發明提供的茂金屬薄膜樹脂明顯存在兩種結晶形式,部分級份在結晶過程中形成了較厚尺寸的片晶,結論與SSA分析一致。結晶形成的片晶厚度與分子鏈中的支鏈分布狀態有關,兩共聚單體插入點間的鏈端長度小,不易結晶,形成的片晶厚度小;共聚單體含量少的分子鏈段,兩共聚單體插入點間的鏈端長度大,鏈規整,易結晶,形成的片晶厚度大。因此該茂金屬樹脂的分子鏈段中的共聚單體分布并不是一致 的,存在一部分分子鏈段的支化點間距相對較近,一部分分子鏈段的支化點間距相對較遠。
實施例1
催化劑制備
在氮氣氛圍下加入500g在干燥空氣下600℃活化4小時的硅膠載體,然后加入1250ml 10%的甲基鋁氧烷的甲苯溶液,然后加入10g茂金屬化合物(R1可為甲基,R2為叔丁基,Cp'為環戊二烯基;金屬原子M為Ti),反應5小時后,過濾、洗滌干燥,得到流動性很好的粉末,即負載型茂金屬催化劑。
聚合物制備
將乙烯、1-己烯、氫氣、氮氣以及由上述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-己烯/乙烯摩爾比0.015:1,氫氣濃度100ppm,在聚合溫度為80℃,聚合壓力為1.8MPa,循環氣速為0.60m/s,停留時間為1h的工藝條件下進行聚合反應。
實施例2
催化劑制備
在氮氣氛圍下加入500g在干燥空氣下600℃活化10小時的硅膠載體,然后加入1250ml 10%的[C6H5NH(CH3)2B(C6F5)4]的甲苯溶液,然后加入10g茂金屬化合物(R1為叔丁基,R2為金剛烷基,Cp'為茚基;金屬原子M為Hf),反應5小時后,過濾、洗滌干燥,得到流動性很好的粉末,即負載型茂金屬催化劑。
聚合物制備
將乙烯、1-己烯、氫氣、氮氣以及由上述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-己烯/乙烯摩爾比0.02:1,氫氣濃度200ppm,在聚合溫度為82℃,聚合壓力為1.9MPa,循環氣速為0.62m/s,停留時間為2h的工藝條件下進行聚合反應。
實施例3
催化劑制備
在氮氣氛圍下加入500g在干燥空氣下600℃活化8小時的硅膠載體,然后加入1250ml 10%的B(C6F5)3的甲苯溶液,然后加入10g茂金屬化合物(R1為甲基,R2為三芳甲基,Cp'為環戊二烯基;金屬原子M可為Zr),反應5小時后,過濾、洗滌干燥,得到流動性很好的粉末,即負載型茂金屬催化劑。
聚合物制備
將乙烯、1-己烯、氫氣、氮氣以及由上述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-己烯/乙烯摩爾比0.022:1,氫氣濃度300ppm,在聚合溫度為83℃,聚合壓力為2.0MPa,循環氣速為0.64m/s,停留時間為3h的工藝條件下進行聚合反應。
實施例4
將乙烯、1-己烯、氫氣、氮氣以及由實施例1所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-己烯/乙烯摩爾比0.025:1,氫氣濃度400ppm,在聚合溫度為84℃,聚合壓力為2.1MPa,循環氣速為0.66m/s,停留時間為3.5h的工藝條件下進行聚合反應。
實施例5
將乙烯、1-丁烯及1-己烯、氫氣、氮氣以及由實施例1所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-丁烯及1-己烯/乙烯摩爾比0.03:1,1-丁烯/1-己烯摩爾比為2:1,氫氣濃度500ppm,在聚合溫度為85℃,聚合壓力為2.2MPa,循環氣速為0.68m/s,停留時間為4h的工藝條件下進行聚合反應。
實施例6
將乙烯、1-丁烯及1-己烯、氫氣、氮氣以及由實施例2所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-丁烯及1-己烯/乙烯摩爾比0.035:1,1-丁烯/1-己烯摩爾比為4:1,氫氣濃度600ppm,在聚合溫度為86℃,聚合壓力為2.3MPa,循環氣速為0.72m/s,停留時間為 4.5h的工藝條件下進行聚合反應。
實施例7
將乙烯、1-丁烯及1-己烯、氫氣、氮氣以及由實施例2所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-丁烯及1-己烯/乙烯摩爾比0.04:1,1-丁烯/1-己烯摩爾比為6:1,氫氣濃度700ppm,在聚合溫度為87℃,聚合壓力為2.4MPa,循環氣速為0.74m/s,停留時間為5h的工藝條件下進行聚合反應。
實施例8
將乙烯、1-丁烯、氫氣、氮氣以及由實施例3所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-丁烯/乙烯摩爾比0.045:1,氫氣濃度800ppm,在聚合溫度為88℃,聚合壓力為2.5MPa,循環氣速為0.76m/s,停留時間為6h的工藝條件下進行聚合反應。
實施例9
將乙烯、1-丁烯、氫氣、氮氣以及由實施例3所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-丁烯/乙烯摩爾比0.05:1,氫氣濃度900ppm,在聚合溫度為89℃,聚合壓力為2.4MPa,循環氣速為0.78m/s,停留時間為7h的工藝條件下進行聚合反應。
實施例10
將乙烯、1-丁烯、氫氣、氮氣以及由實施例1所述方法制備的負載型茂金屬催化劑加入到單一氣相流化床反應器中,按照1-丁烯/乙烯摩爾比0.06:1,氫氣濃度1000ppm,在聚合溫度為90℃,聚合壓力為2.4MPa,循環氣速為0.82m/s,停留時間為8h的工藝條件下進行聚合反應。
根據實施例1-10的制備方法進行聚合試驗,收集聚合所得的茂金屬線型低密度聚乙烯樹脂,對其進行物性測試,結果列于表2。
表2 實施例聚合產品物性測試
從表2中可以得知,利用本發明提供的方法,制備得到的茂金屬薄膜樹脂,其拉伸性能、透明性和耐沖擊性均得到良好體現,薄膜制品的霧度均小于8%,落錘沖擊強度均在900g以上,并且多僅需少量的共聚單體和氫氣在單一反應器中就能夠直接聚合得到,聚合操作條件溫和、工藝流程短、現有裝置經簡單改造即可進行生產,具有較好的應用前景。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 樹脂組合物、薄膜、偏振片保護膜、偏振片和液晶顯示裝置、雙型脂環Cardo酚化合物的制作方法
- 樹脂薄膜、金屬端子部件、以及二次電池的制作方法
- 一種樹脂組合物和樹脂組合物粒料以及薄膜的制作方法
- 二次電池用端子被覆樹脂薄膜、二次電池用接頭片部件、以及二次電池的制作方法
- 顯示器基板用樹脂組合物、顯示器基板用樹脂薄膜及顯示器基板用樹脂薄膜的制造方法
- 一種利用芳綸樹脂制備芳綸絕緣薄膜的方法
- 顯示器基板用樹脂組合物、顯示器基板用樹脂薄膜及顯示器基板用樹脂薄膜的制造方法
- 一種封裝薄膜、顯示器件及其封裝方法
- 一種五氧化二釩光致變色柔性環氧樹脂薄膜的制備方法
- 顯示器基板用樹脂薄膜的制造方法及顯示器基板用樹脂薄膜形成用組合物的制作方法
- 靜電吸附片材和使用了其的顯示物的制造方法與工藝
- 樹脂前體及含有其的樹脂組合物、聚酰亞胺樹脂膜、樹脂薄膜及其制造方法與制造工藝
- 氫氟酸蝕刻用樹脂薄膜形成用組合物和氫氟酸蝕刻用樹脂薄膜的制造方法與工藝
- 一種BPADA型13BDAPB支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝
- 一種HQDA型14BDAPB支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝
- 一種BPADA型14BDAPB支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝
- 一種PMDA型雙酚A四胺支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝
- 一種ODPA型13BDAPB支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝
- 一種6FDA型14BDAPB支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝
- 一種BPDA型13BDAPB支化聚酰亞胺樹脂薄膜及其制備方法與制造工藝