用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與流程
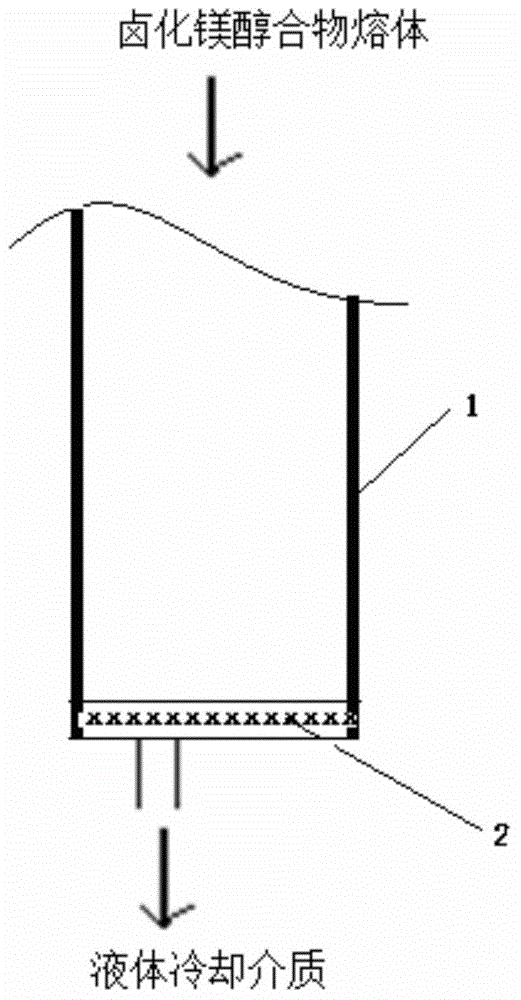
本發明涉及一種用于烯烴聚合的催化劑組分的制備方法,以及由該方法得到的用于烯烴聚合的催化劑組分及其在烯烴聚合反應中的應用;和用于烯烴聚合的催化劑及其在烯烴聚合反應中的應用。技術背景聚丙烯球形催化劑廣泛地應用于環管工藝聚丙烯生產裝置,具有聚合物表觀密度高、粒形規整,催化劑聚合活性高、立構定向性高等優點。載體技術是球形催化劑的技術核心之一。氯化鎂醇合物球形載體在聚烯烴催化劑合成中的應用在聚烯烴催化劑領域中是公知的。目前,氯化鎂醇合物球形載體的制備方法包括噴霧干燥法、噴霧冷卻法、高壓擠出法、高速攪拌法和超重力旋轉床法。US4421674和CN1765940A公開了一種噴霧干燥制備法制備烯烴聚合催化劑載體(載體醇含量小于14%,表觀密度0.32g/ml,載體的粒徑小于20微米),將氯化鎂的乙醇溶液(大約100-300g/L)預熱至90-100℃后噴霧在熱氮氣流的氣化室中,脫除大部分醇后形成氯化鎂醇合物的球形載體。該方法先制備氯化鎂的乙醇溶液,乙醇用量大,能耗較高。US6020279公開了一種氯化鎂醇合物噴霧冷卻制備法。將氯化鎂醇合物的熔融物經過噴嘴噴霧至冷卻介質中形成氯化鎂醇合物載體。該方法制備的載體的粒徑小,設備負荷低,粒徑大小調節難。CN1330086A和US6686307公開了氯化鎂醇合物的高攪制備工藝,先在惰性介質中制備氯化鎂醇合物的懸浮液,通過高速攪拌將氯化鎂醇合物熔體 液珠分散后放入冷卻介質中形成氯化鎂醇合物顆粒。該方法制備的載體的粒徑分布較寬。CN1463990A公開了一種氯化鎂醇合物的乳化冷卻制備工藝。在惰性介質中制備氯化鎂醇合物的懸浮液,懸浮物通過乳化機后進入冷卻介質形成氯化鎂醇合物顆粒。該方法制備的載體的粒徑小,設備負荷低,粒徑大小調節難。CN1267508C公開了一種鹵化鎂/醇加合物及其制備方法和應用。在惰性介質中制備氯化鎂醇合物的懸浮液,懸浮物通過超重力旋轉床后進入冷卻介質形成氯化鎂醇合物顆粒。該方法制備較小粒徑載體困難,制備的大粒徑載體的規整度較差(易出現異形料)。上述制備的載體均通過與四氯化鈦和內給電子體化合物反應制備聚丙烯球形催化劑。氯化鎂醇合物載體的粒形決定催化劑的粒形,而其制備關鍵是氯化鎂醇合物熔體液珠的分散技術,它控制著載體粒徑大小和分布。技術實現要素:本發明的目的在于克服現有技術的缺陷,提供一種操作簡單,能穩定地控制鹵化鎂醇合物載體的粒徑大小和大小分布,以及得到的載體制備的烯烴聚合催化劑具有氫調敏感性和/或立構定向性高等優勢的鹵化鎂醇合物載體的制備方法。如
背景技術:
:所述,鹵化鎂醇合物球形載體的制備方法包括噴霧干燥法、噴霧冷卻法、高壓擠出法、高速攪拌法和超重力旋轉床法。(1)噴霧干燥法制備用于聚烯烴催化劑的載體的方法:用氮氣將MgCl2-醇混合體系通過特別設計的噴嘴噴入干燥室,并在預熱的惰性氣體中被干燥成球形氯化鎂載體;然而,本發明的發明人在研究中發現,使用噴霧干燥法在載體制備過程 中,耗費大量乙醇、氮氣、能量,且最終得到的載體顆粒形態不佳,且載體中醇含量不足;同時噴霧干燥制備的載體對催化劑有如下影響:由于噴霧干燥儀的出口溫度較高,致使得到的球形載體醇含量較低,尤其是球形載體顆粒表面醇過少,在后續負載過程中,活性組分不能和載體有效作用并結合在表面上,使催化劑中活性成份含量低而影響最終催化聚合活性。(2)噴霧冷卻法用于聚烯烴催化劑的載體的方法:用氮氣將氯化鎂醇混合體系通過特別設計的噴嘴噴入干燥室,并在冷卻介質中冷凝形成球形氯化鎂載體;然而,該過程工藝較為復雜,儀器設備要求較高,不易制得粒徑小的球形載體,且制備的載體中醇含量過高;同時噴霧冷卻制備的載體對催化劑有如下影響:球形載體中,由于平均粒徑較大,n大于3,醇含量較高,在載體與含鈦化合物反應時,由于反應過于激烈,常使載體破碎,引起最終細粉增加。(3)高壓擠出冷卻法用于制備聚烯烴催化劑的載體的方法:以粘度較小的煉油、石蠟、白油等反應介質,反應體系溫度升至120-130℃并維持一段時間后,向反應釜內充入高純氮氣,使反應釜內壓力達到10-15大氣壓;之后,氯化鎂醇合物熔體和反應介質的混合物通過一根卸料管卸入冷卻介質中;卸料管的長度為3-10m,管內徑為1-2mm,混合液在管內的流動速率約為4-7m/s;該工藝具有如下缺點:對設備要求較高,而且所得到的氯化鎂醇合物顆粒的形態欠佳,從而使得最終制備的催化劑顆粒形態欠佳,聚合物的顆粒形態不夠理想,聚合物的堆積密度不高。(4)高速攪拌法用于制備聚烯烴催化劑的載體的方法:將無水氯化鎂與醇按一定比例加入到與其不相溶的惰性液體介質中,在攪拌下加熱,使 EtOH/MgCl2形成復合物熔化分散在介質中,然后在高速攪拌器或乳化機中進行乳化,并轉移到低溫的介質中,將MgCl2-醇復合物在其中迅速冷凝固化成球形MgCl2載體;該工藝具有如下缺點:得到的載體顆粒較大,粒度分布較寬,所制備的催化劑的活性也不令人滿意。(5)超重力旋轉床技術制備用于制備聚烯烴催化劑的載體的方法:將無水氯化鎂與醇按一定比例加入到與其不相溶的惰性液體介質中,在攪拌下加熱,使EtOH/MgCl2形成復合物熔化分散在介質中,進入超重力旋轉床,經過設置在轉子中心處的靜態分布器均勻地噴灑在調整旋轉著的填料的內緣上,物料被調整旋轉的填料剪切、分散后,氯化鎂/醇加合物熔體以細小液滴的形式分散于惰性介質中;該工藝的缺點:超重力旋轉床在生產較小(<30微米)粒徑和較大粒徑(>65微米)載體時,載體粒徑分布較寬;穩定生產過程中,必須使用混合介質(一般為白油和硅油混合)才能正常生產,但隨著生產過程的不斷消耗,需要不斷補充新鮮的惰性介質,保持二者的比例,這樣對于維持載體生產的重復穩定性,增加了困難。隨著載體技術的逐步發展,北京化工研究院的技術已經發展到超重力機技術聚烯烴載體,但這種技術存在前述的弊端,需要前瞻性研究及開發:而本發明的發明人首次提出了一種操作簡單、重復穩定性強,能穩定地控制鹵化鎂醇合物載體的粒徑大小和大小分布,以及得到的載體制備的烯烴聚合催化劑具有氫調敏感性和/或立構定向性高等優勢的鹵化鎂醇合物載體的制備方法,該方法僅需通過加壓,將鹵化鎂醇合物熔體穿過具有孔的結構單元與液體冷卻介質接觸即可實現前述目的。由此,根據本發明的第一方面,本發明提供了一種用于烯烴聚合的催化劑組分的制備方法,該方法包括:將鹵化鎂醇合物載體、鈦化合物以及內給 電子體進行接觸反應,所述內給電子體含有醇酯化合物;所述鹵化鎂醇合物載體的制備步驟包括:在加壓下,將鹵化鎂醇合物熔體穿過具有孔的結構單元與液體冷卻介質接觸。根據本發明的第二方面,本發明提供了本發明所述的制備方法制備得到的用于烯烴聚合的催化劑組分。根據本發明的第三方面,本發明提供了一種用于烯烴聚合的催化劑,該催化劑含有:(i)催化劑組分,該催化劑組分為本發明所述的用于烯烴聚合的催化劑組分;(ii)至少一種烷基鋁化合物;以及(iii)任選地,至少一種外給電子體。根據本發明的第四方面,本發明提供了本發明所述的用于烯烴聚合的催化劑組分或所述的用于烯烴聚合的催化劑在烯烴聚合反應中的應用。本發明的方法具有如下優勢:第一,相比于目前先進的技術超重力機技術,超重力機技術穩定生產過程中,必須使用混合介質(通常為白油和硅油的混合介質)才能正常生產,但隨著生產過程的不斷消耗,需要不斷補充新鮮的惰性介質,保持二者的比例,這樣對于維持載體生產的重復穩定性,增加了困難;而本發明的技術除了可以使用混合的惰性介質可以正常生產,還可以使用單一的介質例如單獨的白油或硅油生產載體,效果更佳,顯而易見,使用單一介質對于載體生產無疑改善了其生產的重復穩定性。第二,相比前面各種載體的制備方法,本發明的技術的設備簡單,能耗和物耗低。第三,通過調整孔的目數、結構單元的層數及壓力等要素,比較容易實現對粒徑、粒形的控制,也就是說,可以根據市場需要,進行量體裁衣式的生產,轉產方便易行。第四,本發明的技術制備的載體合成的催化劑,相比現有生產的催化劑,具備良好的氫調敏感性和/或立構定向性,這對于開發高附加值的聚丙烯樹脂奠定了基礎。本發明的其他特征和優點將在隨后的具體實施方式部分予以詳細說明。附圖說明附圖是用來提供對本發明的進一步理解,并且構成說明書的一部分,與下面的具體實施方式一起用于解釋本發明,但并不構成對本發明的限制。在附圖中:圖1為根據本發明的一種實施方式的具有孔結構單元的安裝方式的示意圖;圖2為根據本發明的一種實施方式的具有孔結構單元的安裝方式的示意圖。附圖標記說明1:鹵化鎂醇合物熔體物料輸送管線;2:具有孔結構的單元。具體實施方式以下對本發明的具體實施方式進行詳細說明。應當理解的是,此處所描述的具體實施方式僅用于說明和解釋本發明,并不用于限制本發明。如前所述,本發明提供了一種用于烯烴聚合的催化劑組分的制備方法,該方法包括:將鹵化鎂醇合物載體、鈦化合物以及內給電子體進行接觸反應,所述內給電子體含有醇酯化合物;所述鹵化鎂醇合物載體的制備步驟包括:在加壓下,將鹵化鎂醇合物熔體穿過具有孔的結構單元與液體冷卻介質接 觸。根據本發明的方法,優選所述加壓的條件包括:壓力為0.05-1MPa,更優選為0.1-0.5MPa,進一步優選為0.2-0.5MPa。采用該優選的壓力,能夠進一步優化粒徑大小和大小分布,同時得到的載體制備的烯烴聚合催化劑具有氫調敏感性和/或立構定向性高等優勢。根據本發明的方法,所述壓力為以大氣壓為0MPa計的相對壓力,即,以現有大氣壓為基準的相對壓力。本發明的方法,只需通過加壓,無需在超重力條件下以及無需使用噴霧噴嘴等設備即可實現本發明的目的,由此可見,本發明的方法,相比前面各種載體的制備方法,設備簡單,操作簡單易行、能耗和物耗低。根據本發明的方法,優選所述孔的孔徑為10-1800μm,更優選為20-800μm,進一步優選為30-150μm。根據本發明的方法,所述具有孔的結構單元可以為各種形式,例如為具有孔的板、具有孔的網等等。針對本發明,優選為具有孔的網,根據實際生產的需要及材料的易得性考慮,最優選直徑為0.025-4mm的網絲。根據本發明的方法,所述具有孔的結構單元的材質無特殊要求,例如為金屬材質、布料材質、塑料材質和陶瓷材質中的一種或多種。根據實際生產的需要及材料的易得性,優選為金屬材質。根據本發明的一種優選實施方式,所述具有孔的結構單元為網層。根據本發明的方法,采用多層大孔徑結構的金屬網與采用相對少的層數的小孔徑結構金屬網,可以獲得粒子形貌相當的加合物固體顆粒。制備小粒徑(如小于30μm)加合物載體時優選采用1-20層小孔徑結構(如孔徑為30-50μm)的金屬網結構,在制備大粒徑(如大于65μm)加合物載體時優選采用1-20層相對大孔徑(如孔徑為65-200μm)結構的金屬網結構。根據本發明的一種優選實施方式,所述網層包括1-20層,優選包括2-8 層;每層厚度各自為0.01-0.35mm,優選為0.05-0.25mm,更優選為0.1-0.15mm。根據本發明的一種優選實施方式,所述網層為金屬網層。本發明的方法,通過調整孔的目數、結構單元的層數及壓力等要素,容易實現對粒徑、粒形的控制,也就是說,本發明的方法可以根據市場需要,進行量體裁衣式的生產,轉產方便易行。根據本發明,本發明對所述載體的組成無特殊要求,針對本發明,優選所述載體的通式如下式所示:MgXY–mROH–nE–pH2O;其中,m為1-5,優選為2.4-3.5;其中,n為0-0.3,優選為0.005-0.2;其中,p為0-0.08;其中,X為鹵素,優選為氯或溴,更優選為氯;其中,Y為鹵素、C1-C14的烷基、C1-C14的烷氧基、C6-C14的芳基或C6-C14的芳氧基;其中,R為C1-C12的烷基、C3-C10的環烷基或C6-C10芳基,優選為C1-C4的烷基;其中,E為除醇以外的含氧原子的給電子體化合物。根據本發明的一種優選的實施方式,所述MgXY為二氯化鎂、二溴化鎂、氯化苯氧基鎂、氯化異丙氧基鎂和氯化丁氧基鎂中的一種或多種,優選為二氯化鎂。根據本發明一種優選的實施方式,所述ROH為甲醇、乙醇、丙醇、異丙醇、正丁醇、異丁醇、戊醇、異戊醇、正己醇正辛醇、2-乙基己醇、乙二醇和丙二醇中的一種或多種。根據本發明,優選所述E為醚、酯和酮化合物。根據本發明,上式中的H2O為反應原料和反應介質中所帶的水。根據本發明,本發明對所述鹵化鎂醇合物熔體的制備方法無特殊要求,可以按照常規技術進行制備,針對本發明,優選所述鹵化鎂醇合物熔體的制備步驟包括:在密閉條件下,將鹵化鎂MgXY、醇ROH、任選地給電子體化合物E和惰性液體介質混合,在攪拌下將混合物加熱升溫到100-160℃;其中,X為鹵素,優選為氯或溴,更優選為氯;其中,Y為鹵素、C1-C14的烷基、C1-C14的烷氧基、C6-C14的芳基或C6-C14的芳氧基;其中,R為C1-C12的烷基、C3-C10的環烷基或C6-C10芳基,優選為C1-C4的烷基;其中,E為除醇以外的含氧原子的給電子體化合物。根據本發明的一種優選的實施方式,所述MgXY為二氯化鎂、二溴化鎂、氯化苯氧基鎂、氯化異丙氧基鎂和氯化丁氧基鎂中的一種或多種,優選為二氯化鎂。根據本發明的一種優選的實施方式,所述ROH為甲醇、乙醇、丙醇、異丙醇、正丁醇、異丁醇、戊醇、異戊醇、正己醇正辛醇、2-乙基己醇、乙二醇和丙二醇中的一種或多種。根據本發明,優選所述E為醚、酯和酮化合物。根據本發明,所述的惰性液體介質為不與鹵化鎂、醇、給電子體化合物E反應的化合物,優選所述惰性液體介質為硅油和/或惰性液體烴溶劑;優選地,所述惰性液體介質為C4-C10的烷烴、煤油、石蠟油、凡士林油、白油、甲基硅油、乙基硅油、甲基乙基硅油、苯基硅油和甲基苯基硅油中的一種或多種;優選地,所述惰性液體介質為硅油。本發明的方法,相比于目前先進的技術超重力機技術,超重力機技術穩 定生產過程中,必須使用混合介質(通常為白油和硅油的混合介質)才能正常生產,但隨著生產過程的不斷消耗,需要不斷補充新鮮的惰性介質,保持二者的比例,這樣對于維持載體生產的重復穩定性,增加了困難;而本發明的技術除了使用混合的惰性介質可以正常生產,還可以使用單一的介質例如單獨的白油或硅油生產載體,效果更佳。顯而易見,使用單一介質對于載體生產無疑增加了其生產的重復穩定性。根據本發明,在鹵化鎂醇合物熔體的制備中,加料順序不分先后。根據本發明的一種優選實施方式,以1mol以鎂計的MgXY為基準,醇ROH的用量為1-5.5mol,優選為2-3.7mol,更優選為2.4-3.7mol;E的用量為0.001-0.32mol,優選為0.005-0.17mol,更優選為0.015-0.12mol。本發明中,所述惰性液體介質的用量可以根據MgXY的具體用量來選擇。一般地,相對于1摩爾以鎂計的MgXY,惰性液體介質的用量為0.2-13L;優選地,相對于1摩爾以鎂計的MgXY,惰性液體介質的用量為0.6-6.5L。根據本發明,所述液體冷卻介質為惰性烴溶劑,優選為戊烷、己烷、庚烷、汽油和石油醚中的一種或多種;優選所述液體冷卻介質的溫度為(-10℃)-(-40℃)。本發明的制備載體的方法簡單易行,例如可以將所述具有孔結構的單元安裝在鹵化鎂醇合物熔體物料輸送管線內或者安裝在鹵化鎂醇合物熔體物料輸送管線與液體冷卻介質儲罐之間,具體的例如如圖1所示,將所述具有孔結構的單元2安裝在鹵化鎂醇合物熔體物料輸送管線1內,鹵化鎂醇合物熔體與冷卻介質接觸前穿過所述具有孔結構的單元。又如如圖2所示,將所述具有孔結構的單元2安裝在鹵化鎂醇合物熔體物料輸送管線1與液體冷卻介質儲罐(圖中未示出)之間,鹵化鎂醇合物熔體與冷卻介質接觸前穿過所述具有孔結構的單元。根據本發明的方法,優選本發明的所述鹵化鎂醇合物載體的制備步驟還 包括:將得到的球形鹵化鎂醇合物顆粒經惰性烴類溶劑洗滌、干燥的步驟。此為本領域的公知技術,在此不詳細描述。本發明的技術制備的載體粒徑大小和粒徑分布容易控制,且采用載體合成的催化劑,相比現有生產的催化劑,具備良好的氫調敏感性和/或立構定向性,這對于開發高附加值的聚丙烯樹脂奠定了基礎。本發明的方法制備的載體合成的催化劑,相比現有技術生產的催化劑,具備良好的氫調敏感性和/或立構定向性,這對于開發高附加值的聚丙烯樹脂奠定了基礎。根據本發明,當所述內給電子體含有醇酯化合物即可實現本發明的目的,優選所述醇酯化合物的總用量可以為80-100重量%。根據本發明,所述醇酯化合物可以為一元或多元醇酯,優選為式Ⅰ所示的化合物,式Ⅰ中,R1和R2相同或不同,各自獨立地為C1-C10的直鏈或支鏈烷基、C3-C20的取代或未取代的環烷基、C6-C20的取代或未取代的芳基、C7-C20的取代或未取代的芳烷基和C7-C20的取代或未取代的烷芳基中的一種,所述芳基、芳烷基或烷芳基中的芳環任選地被選自鹵素、C1-C6的直鏈或支鏈烷基和C1-C6的烷氧基中的一種或多種取代;式Ⅰ中,中括號“[]”的內容表示有n個碳原子依次鍵連,且每個碳原子還與2個取代基鍵連,即中括號內共有n個碳原子和R1、R2、R3…R2n等2n個取代基。R3、R4、R5、R6和R1-R2n相同或不同,各自獨立地為氫、鹵素、C1-C20的直鏈或支鏈烷基、C3-C20的取代或未取代的環烷基、C6-C20的取代或未取 代的芳基、C7-C20的取代或未取代的烷芳基、C7-C20的取代或未取代的芳烷基、C2-C10的直鏈或支鏈烯烴基和C10-C20的稠環芳基中的一種,R3、R4、R5、R6和R1-R2n任選地含有雜原子,所述雜原子為氮、氧、硫、硅、鹵素和磷中的一種或多種;或者,R3、R4、R5、R6和R1-R2n中的兩個或兩個以上相互鍵合,以形成飽和的或不飽和的環;n為0-10的整數,當n=0時,式Ⅰ所示的二醇酯化合物中,取代基為R3、R4的碳原子直接與取代基為R5、R6的碳原子鍵連;本發明中,C1-C20的直鏈或支鏈烷基的實例可以包括但不限于:甲基、乙基、正丙基、異丙基、正丁基、仲丁基、異丁基、叔丁基、正戊基、1-乙基丙基、2-甲基丁基、3-甲基丁基、2,2-二甲基丙基、正己基、2-甲基戊基、3-甲基戊基、4-甲基戊基、正庚基、2-甲基己基、3-甲基己基、4-甲基己基、5-甲基己基、正庚基、正辛基、正壬基、正癸基、四氫香葉基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十八烷基、正十九烷基和正二十烷基。本發明中,C3-C20的取代或未取代的環烷基的實例可以包括但不限于:環丙基、環丁基、環戊基、環己基、4-甲基環己基、4-乙基環己基、4-正丙基環己基、4-正丁基環己基、環十一烷基和環十二烷基。本發明中,C6-C20的取代或未取代的芳基的實例可以包括但不限于:苯基、甲基苯基、乙基苯基、4-叔丁基苯基等。在本發明中,C7-C20的取代或未取代的芳烷基是指碳原子數為7-20的具有芳基取代基的烷基基團。C7-C20的取代或未取代的芳烷基的實例可以包括但不限于:3-苯基丙基、芐基等。在本發明中,C7-C20的取代或未取代的烷芳基是指碳原子數為7-20的具有烷基取代基的芳基基團。C7-C20的取代或未取代的烷芳基的實例可以包括 但不限于:甲基苯基、乙基苯基等。在本發明中,C1-C6的烷氧基的實例可以包括但不限于:甲氧基、乙氧基、正丙氧基、異丙氧基、正丁氧基、仲丁氧基、異丁氧基、叔丁氧基、正戊氧基、異戊氧基、叔戊氧基和己氧基。在本發明中,C2-C10的直鏈或支鏈烯烴基的實例可以包括但不限于:乙烯基、丙烯基、丁烯基、戊烯基、辛烯基等。在本發明中,C10-C20的稠環芳基的實例可以包括但不限于:萘基、蒽基、菲基、芘基等。根據本發明,所述醇酯化合物的實例可以包括但不限于:1,3-丙二醇二苯甲酸酯、2-甲基-1,3-丙二醇二苯甲酸酯、2-乙基-1,3-丙二醇二苯甲酸酯、2-丙基-1,3-丙二醇二苯甲酸酯、2-丁基-1,3-丙二醇二苯甲酸酯、2,2-二甲基-1,3-丙二醇二苯甲酸酯、2-乙基-2-丁基-1,3-丙二醇二苯甲酸酯、2,2-二乙基-1,3-丙二醇二苯甲酸酯、2-甲基-2-丙基-1,3-丙二醇二苯甲酸酯、2-異丙基-2-異戊基-1,3-丙二醇二苯甲酸酯、2,4-戊二醇二苯甲酸酯、3-甲基-2,4-戊二醇二苯甲酸酯、3-乙基-2,4-戊二醇二苯甲酸酯、3-丙基-2,4-戊二醇二苯甲酸酯、3-丁基-2,4-戊二醇二苯甲酸酯、3,3-二甲基-2,4-戊二醇二苯甲酸酯、2-甲基-1,3-戊二醇二苯甲酸酯、2,2-二甲基-1,3-戊二醇二苯甲酸酯、2-乙基-1,3-戊二醇二苯甲酸酯、2-丁基-1,3-戊二醇二苯甲酸酯、2-甲基-1,3-戊二醇二苯甲酸酯、2-乙基-1,3-戊二醇二苯甲酸酯、2-丙基-1,3-戊二醇二苯甲酸酯、2-丁基-1,3-戊二醇二苯甲酸酯、2,2-二甲基-1,3-戊二醇二苯甲酸酯、2-甲基-1,3-戊二醇二苯甲酸酯、2,2-二甲基-1,3-戊二醇二苯甲酸酯、2-乙基-1,3-戊二醇二苯甲酸酯、2-丁基-1,3-戊二醇二苯甲酸酯、2,2,4-三甲基-1,3-戊二醇二苯甲酸酯、3-甲基-3-丁基-2,4-戊二醇二苯甲酸酯、2,2-二甲基-1,5-戊二醇二苯甲酸酯、1,6-己二醇二苯甲酸酯、6-庚烯-2,4-庚二醇二苯甲酸酯、2-甲基-6-庚烯-2,4-庚二醇二苯甲酸酯、3-甲基-6-庚烯-2,4-庚二醇二苯甲酸酯、4-甲基-6-庚烯-2,4-庚二醇 二苯甲酸酯、5-甲基-6-庚烯-2,4-庚二醇二苯甲酸酯、6-甲基-6-庚烯-2,4-庚二醇二苯甲酸酯、3-乙基-6-庚烯-2,4-庚二醇二苯甲酸酯、4-乙基-6-庚烯-2,4-庚二醇二苯甲酸酯、5-乙基-6-庚烯-2,4-庚二醇二苯甲酸酯、6-乙基-6-庚烯-2,4-庚二醇二苯甲酸酯、3-丙基-6-庚烯-2,4-庚二醇二苯甲酸酯、4-丙基-6-庚烯-2,4-庚二醇二苯甲酸酯、5-丙基-6-庚烯-2,4-庚二醇二苯甲酸酯、6-丙基-6-庚烯-2,4-庚二醇二苯甲酸酯、3-丁基-6-庚烯-2,4-庚二醇二苯甲酸酯、4-丁基-6-庚烯-2,4-庚二醇二苯甲酸酯、5-丁基-6-庚烯-2,4-庚二醇二苯甲酸酯、6-丁基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,5-二甲基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,5-二乙基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,5-二丙基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,5-二丁基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,3-二甲基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,3-二乙基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,3-二丙基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,3-二丁基-6-庚烯-2,4-庚二醇二苯甲酸酯、3,5-庚二醇二苯甲酸酯、2-甲基-3,5-庚二醇二苯甲酸酯、3-甲基-3,5-庚二醇二苯甲酸酯、4-甲基-3,5-庚二醇二苯甲酸酯、5-甲基-3,5-庚二醇二苯甲酸酯、6-甲基-3,5-庚二醇二苯甲酸酯、3-乙基-3,5-庚二醇二苯甲酸酯、4-乙基-3,5-庚二醇二苯甲酸酯、5-乙基-3,5-庚二醇二苯甲酸酯、3-丙基-3,5-庚二醇二苯甲酸酯、4-丙基-3,5-庚二醇二苯甲酸酯、3-丁基-3,5-庚二醇二苯甲酸酯、2,3-二甲基-3,5-庚二醇二苯甲酸酯、2,4-二甲基-3,5-庚二醇二苯甲酸酯、2,5-二甲基-3,5-庚二醇二苯甲酸酯、2,6-二甲基-3,5-庚二醇二苯甲酸酯、3,3-二甲基-3,5-庚二醇二苯甲酸酯、4,4-二甲基-3,5-庚二醇二苯甲酸酯、6,6-二甲基-3,5-庚二醇二苯甲酸酯、2,6-二甲基-3,5-庚二醇二苯甲酸酯、3,4-二甲基-3,5-庚二醇二苯甲酸酯、3,5-二甲基-3,5-庚二醇二苯甲酸酯、3,6-二甲基-3,5-庚二醇二苯甲酸酯、4,5-二甲基-3,5-庚二醇二苯甲酸酯、4,6-二甲基-3,5-庚二醇二苯甲酸酯、4,4-二甲基-3,5-庚二醇二苯甲酸酯、6,6-二甲基-3,5-庚二醇二苯甲酸酯、2-甲基-3-乙基-3,5-庚二醇二苯甲酸酯、2-甲基-4-乙基-3,5-庚二醇二苯甲酸 酯、2-甲基-5-乙基-3,5-庚二醇二苯甲酸酯、3-甲基-3-乙基-3,5-庚二醇二苯甲酸酯、3-甲基-4-乙基-3,5-庚二醇二苯甲酸酯、3-甲基-5-乙基-3,5-庚二醇二苯甲酸酯、4-甲基-3-乙基-3,5-庚二醇二苯甲酸酯、4-甲基-4-乙基-3,5-庚二醇二苯甲酸酯、4-甲基-5-乙基-3,5-庚二醇二苯甲酸酯、2-甲基-3-丙基-3,5-庚二醇二苯甲酸酯、2-甲基-4-丙基-3,5-庚二醇二苯甲酸酯、2-甲基-5-丙基-3,5-庚二醇二苯甲酸酯、3-甲基-3-丙基-3,5-庚二醇二苯甲酸酯、3-甲基-4-丙基-3,5-庚二醇二苯甲酸酯、3-甲基-5-丙基-3,5-庚二醇二苯甲酸酯、4-甲基-3-丙基-3,5-庚二醇二苯甲酸酯、4-甲基-4-丙基-3,5-庚二醇二苯甲酸酯、4-甲基-5-丙基-3,5-庚二醇二苯甲酸酯等。優選情況下,所述醇酯化合物為式Ⅱ所示的二醇酯化合物,式Ⅱ中,R7、R8、R9、R10、R11和R12相同或不同,各自獨立地為氫或C1-C20的直鏈或支鏈烷基。最優選情況下,所述醇酯化合物為2,4-戊二醇二苯甲酸酯和/或3,5-庚二醇二苯甲酸酯。根據本發明,在用于烯烴聚合的催化劑組分的制備過程中,鈦化合物可以為本領域的常規選擇,例如,所述鈦化合物可以為通式為Ti(OR′)3-aZa和/或Ti(OR′)4-bZb的物質,其中,R′為C1-C20的烷基,Z為F、Cl、Br或I,a為1-3的整數,b為1-4的整數。優選情況下,所述鈦化合物為四氯化鈦、四溴化鈦、四碘化鈦、三丁氧基氯化鈦、二丁氧基二氯化鈦、丁氧基三氯化 鈦、三乙氧基氯化鈦、二乙氧基二氯化鈦、乙氧基三氯化鈦和三氯化鈦中的一種或多種,優選為四氯化鈦。根據本發明,在所述用于烯烴聚合的催化劑組分中,鈦元素、鎂元素與內給電子體的重量比可以為1:(3-15):(2-10),優選為1:(3-12):(2-7),更優選為1:(3-5):(2-4)。根據本發明的一種優選的實施方式,該方法包括:將鹵化鎂醇合物載體與鈦化合物反應,并在所述鹵化鎂醇合物載體與鈦化合物反應之前、期間和之后的一個或多個時間段內加入所述內給電子體進行所述接觸反應。具體地,所述鹵化鎂醇合物載體與鈦化合物的反應可以按照與現有技術相同的方式進行,例如,可以將鈦化合物冷卻至0℃以下(優選為-30℃至10℃,優選為-30℃至-10℃),然后加入鹵化鎂醇合物載體,并在該溫度下攪拌混合0.2-3小時,優選0.5-2小時;之后升溫至反應溫度(即約60-130℃),并在該反應溫度下維持0.5-10小時。在所述用于烯烴聚合的催化劑組分的制備方法中,所述內給電子體化合物在所述鹵化鎂醇合物載體與鈦化合物的反應之前、期間和之后中的一個或多個時間段內加入。所述鹵化鎂醇合物載體與鈦化合物的反應之前的時間段是指在所述鹵化鎂醇合物載體加入反應器中之后且在升溫至反應溫度之前的時間段。根據本發明,在所述用于烯烴聚合的催化劑組分的制備過程中,以鎂元素計的鹵化鎂醇合物載體、以鈦元素計的鈦化合物與內給電子體的用量的摩爾比可以為1:(2-200):(0.03-1),優選為1:(5-180):(0.08-0.5),優選為1:(10-150):(0.12-0.4)。如前所述,本發明提供了一種按照本發明所述的方法制備得到的用于烯烴聚合的催化劑組分。如前所述,本發明提供了一種用于烯烴聚合的催化劑,該催化劑含有:(i)催化劑組分,該催化劑組分為本發明所述的用于烯烴聚合的催化 劑組分;(ii)至少一種烷基鋁化合物;以及(iii)任選地,至少一種外給電子體。根據本發明,所述烷基鋁可以為本領域的常規選擇,例如,所述烷基鋁的通式可以為AlR16R16′R16″,其中,R16、R16′、R16″各自獨立地為C1-8的烷基,且其中一個或兩個基團可以為鹵素;所述C1-8的烷基的具體實例可以包括但不限于:甲基、乙基、丙基、正丁基、異丁基、戊基、己基、正庚基、正辛基,所述鹵素可以為氟、氯、溴、碘。具體地,所述烷基鋁例如可以選自三乙基鋁、三丙基鋁、三異丁基鋁、三正丁基鋁、三正己基鋁、三正辛基鋁、一氫二乙基鋁、一氫二異丁基鋁、一氯二乙基鋁、一氯二異丁基鋁、一氯二正丁基鋁、一氯二正己基鋁、二氯一乙基鋁、二氯一異丁基鋁、二氯一正丁基鋁、二氯一正己基鋁、Al(n-C6H13)3、Al(n-C8H17)3、AlEt2Cl中的一種或多種,優選為三乙基鋁和/或三異丁基鋁。根據本發明,所述外給電子體可以為本領域常用的各種外給電子體,例如,所述外給電子體可以為羧酸、酸酐、酯、酮、醚、醇、有機磷化合物和有機硅化合物中的一種或多種;優選地,所述外給電子體為含有至少一個Si-OR鍵、且通式為(R17)a(R18)bSi(OR19)c的硅化合物,其中,R17、R18和R19為C1-C18的烴基、任選地含有雜原子,a和b各自獨立為0-2的整數,c為1-3的整數,且a、b和c的和為4。優選地,R17、R18為C3-C10的烷基、環烷基,任選地含有雜原子;R19為C1-C10的烷基,任選地含有雜原子。具體地,所述外給電子體例如可以選自環己基甲基二甲氧基硅烷、二異丙基二甲氧基硅烷、二正丁基二甲氧基硅烷、二異丁基二甲氧基硅烷、二苯基二甲氧基硅烷、甲基叔丁基二甲氧基硅烷、二環戊基二甲氧基硅烷、環己基三甲氧基硅烷、叔丁基三甲氧基硅烷、叔己基三甲氧基硅烷、2-乙基哌啶基-2-叔丁基二甲氧基硅烷、(1,1,1-三氟-2-丙基)-2-乙基哌啶基二甲氧基硅烷和(1,1,1-, 三氟-2-丙基)-甲基二甲氧基硅烷中的一種或多種。此外,一般地,在所述用于烯烴聚合的催化劑中,以鈦元素計的所述用于烯烴聚合的催化劑組分和以鋁元素計的所述烷基鋁用量的摩爾比可以為1:(1-1000),優選為1:(10-300),更優選為1:(20-200);所述外給電子體和以鋁元素計的所述烷基鋁用量的摩爾比可以為1:(2-500),優選為1:(5-200)。根據本發明,在用于烯烴聚合的催化劑的制備過程中,烷基鋁和任選的外給電子體化合物可以分別與用于烯烴聚合的催化劑組分混合后反應,或者也可以將烷基鋁和任選的外給電子體事先混合后再與用于烯烴聚合的催化劑組分混合并反應。根據本發明,在將用于烯烴聚合的催化劑用于烯烴聚合時,所述用于烯烴聚合的催化劑組分、烷基鋁、以及任選的外給電子體可分別加入聚合反應器中,也可混合后加入聚合反應器中,也可采用本行業公知的預聚合方法將烯烴預聚后加入到聚合反應器中。如前所述,本發明提供了本發明所述的用于烯烴聚合的催化劑組分或本發明所述的用于烯烴聚合的催化劑在烯烴聚合反應中的應用。本發明的改進之處在于采用了一種新的用于烯烴聚合的催化劑、用于烯烴聚合的催化劑組分,而烯烴的具體種類、烯烴的聚合反應方法和條件均可以與現有技術相同。根據本發明,上述催化劑特別適用于通式為CH2=CHR(其中,R是氫、C1-C6的烷基或C6-C12芳基)的烯烴的均聚和共聚反應,具體地例如為乙烯、丙烯、1-正丁烯、1-正戊烯、1-正己烯、1-正辛烯和4-甲基-1-戊烯。優選情況下,所述由式CH2=CHR表示的烯烴為乙烯、丙烯、1-正丁烯、1-正己烯和4-甲基-1-戊烯中的一種或多種。更優選地,所述由式CH2=CHR表示的烯烴為丙烯。根據本發明,所述烯烴的聚合反應可以按照現有的方法進行,具體地,在惰性氣體的保護下,在液相單體或含聚合單體的惰性溶劑中,或在氣相中,或通過在氣液相中的組合聚合工藝進行聚合反應。所述聚合反應的溫度一般可以為0-150℃、優選為60-100℃。所述聚合反應的壓力可以為常壓或更高;例如可以為0.01-10MPa,優選為0.5-5MPa。聚合的時間為0.1-5小時,優選為0.5-3小時。本發明的壓力均指表壓。在聚合過程中,氫氣可用作聚合物分子量調節劑加入到反應體系中以調節聚合物的分子量和熔融指數。此外,在烯烴的聚合反應過程中,所述惰性氣體、溶劑的種類和用量為本領域技術人員公知,在此將不再贅述。下面的例子用來說明本發明,并不是用來限制本發明的范圍。測試方法:1、聚合物熔融指數:在230℃的溫度和2.16kg的壓力下,根據ASTMD1238-99測定。2、聚合物等規度:采用庚烷抽提法測定(庚烷沸騰抽提6小時):兩克干燥的聚合物樣品,放在抽提器中用沸騰庚烷抽提6小時后,將剩余物干燥至恒重所得的聚合物重量(g)與2的比值即為等規度。3、粒徑分布測試:鹵化鎂醇合物顆粒的平均粒徑和粒度分布用MastersSizer2000(由MalvernInstrumentsLtd生產制造)粒度儀測定。4、聚合物的分子量分布采用島津LC-10AT型凝膠滲透色譜儀(GPC)測定,其中,THF為流動相,窄分布聚苯乙烯為標樣,溫度為25℃。制備實施例1在1L的高壓釜中,加入硅油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.2MPa下,將混合物由下料管線經過2層200目的金屬網(每層厚度0.1mm)后直接放入預先冷卻至-30℃ 的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到50克的球形氯化鎂醇合物固體顆粒S1,醇/鎂摩爾比為2.6,表征結果如表1。制備實施例2在1L的高壓釜中,加入硅油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.5MPa下,將混合物由下料管線經過4層200目的金屬網(每層厚度0.1mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到54克的球形氯化鎂醇合物固體顆粒S2,表征結果如表1。制備實施例3在1L的高壓釜中,加入硅油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.3MPa下,將混合物由下料管線經過3層200目的金屬網(每層厚度0.1mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到60克的球形氯化鎂醇合物固體顆粒S3,表征結果如表1。制備實施例4在1L的高壓釜中,加入硅油500ml、30克氯化鎂、50ml乙醇和3ml鄰甲氧基苯甲酰氯,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.2MPa下,將混合物由下料管線經過4層200目的金屬網(每層厚度0.1mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到56克的球形氯化鎂醇合物固體顆粒S4,表征結果如表1。制備實施例5在1L的高壓釜中,加入硅油300ml和白油200ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.5MPa下,將混合物由下料管線經過6層200目的金屬網(每層厚度0.1mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到51克的球形氯化鎂醇合物固體顆粒S5,表征結果如表1。制備實施例6在1L的高壓釜中,加入白油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.2MPa下,將混合物由下料管線經過4層200目的金屬網(每層厚度0.1mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到49克的球形氯化鎂醇合物固體顆粒S6,表征結果如表1。制備實施例7在1L的高壓釜中,加入硅油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,在0.6MPa下,將混合物由下料管線經過8層150目的金屬網(每層厚度0.15mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到55克的球形氯化鎂醇合物固體顆粒S7,表征結果如表1。制備實施例8在1L的高壓釜中,加入硅油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到120℃,在120℃下攪拌3小時,在0.6MPa下,將混合物由下料管線經過4層300目的金屬網(每層厚度0.09mm)后直接放入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到58克的球 形氯化鎂醇合物固體顆粒S8,表征結果如表1。制備對比例1在1L的高壓釜中,加入硅油300ml、白油150ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,將物料放入開啟的超重力機中,通過超重力機進入預先冷卻至-30℃的3L已烷中,濾去液體,用已烷洗滌固體三次,真空干燥,得到58克的球形氯化鎂醇合物固體顆粒DS1,表征結果如表1。制備對比例2在1L的高壓釜中,加入白油500ml、30克氯化鎂、50ml乙醇,在攪拌下升溫到125℃,在125℃下攪拌3小時,將物料放入開啟的超重力機中,通過超重力機進入預先冷卻至-30℃己烷中,濾去液體,用己烷洗滌固體三次,真空干燥,得到56克的球形氯化鎂醇合物固體顆粒DS2,表征結果如表1。表1實施例D10(μm)D50(μm)D90(μm)Span116.945.362.51.0217.144.356.90.9318.342.160.51.0417.544.962.41.0522.543.465.91.0624.150.278.21.1739.870.6108.30.98818.722.339.80.95對比例121.443.263.30.97對比例226.153.4105.91.5注:Span=(D90-D10)/D50由表1的結果可以看出,相比超重力旋轉床技術,本發明可以制備不同粒徑的加合物載體,且粒度分布較窄,特別是制備的大粒徑(D50>60μm)加合物載體,仍具有較窄的粒度分布。實施例1該實施例用于說明用于烯烴聚合的催化劑組分及其制備方法和用于烯烴聚合的催化劑及其應用。(1)使用制備實施例1制備的載體S1用于烯烴聚合的催化劑組分的制備:在0.5L的玻璃反應釜中,加入四氯化鈦100ml,降溫至-20℃,加入上述的氯化鎂醇合物8克,反應0.5小時,升溫至120℃,在升溫過程中加入2,4-戊二醇二苯甲酸酯0.007mol。在110℃下維持0.5小時,濾掉液體,得到固體物。用四氯化鈦90ml在110℃下洗滌二次,每次洗滌0.5小時,再用已烷80ml洗滌5次,最后在真空下干燥0.5小時(50℃)得到催化劑C1。(2)丙烯聚合:在5L高壓反應釜中,加入丙烯2.3L,三乙基鋁2.5mmol,環已基甲基二甲氧基硅烷(CHMMS)0.1mmol,催化劑組分C110mg,氫氣加入量分別為2L,6.5L(標準體積),升溫到70℃,聚合1小時。降溫至室溫,放空未反應的丙烯,得到丙烯聚合物。結果見表2。對比例1該對比例用于說明參比用于烯烴聚合的催化劑組分及其制備方法和用于烯烴聚合的催化劑及其應用。氯化鎂醇合物按制備對比例1公開的方法制備。按照實施例1的方法制備烯烴聚合催化劑組分DC1。按照實施例1的方法制備聚丙烯,結果見表2。表2根據實施例1與對比例1的比較可以看出,采用本發明的載體制備的催化劑,具有更高的氫調敏感性和立構定向性。實施例2-5按照實施例1的方法制備用于烯烴聚合的催化劑組分和進行丙烯聚合反應,所不同的是,使用載體S3-S6制備催化劑組分C2-C5,結果見表3。對比例2按照實施例1的方法制備用于烯烴聚合的催化劑組分和進行丙烯聚合反應,所不同的是,使用載體DS2制備催化劑組分DC2,結果見表3。表3表中,Al/ED指的是聚合時加入的三乙基鋁和環己基甲基二甲氧基硅烷的摩爾比。由表3的結果可以看出采用本發明的載體制備的催化劑,具有更高的氫調敏感性和立構定向性。以上詳細描述了本發明的優選實施方式,但是,本發明并不限于上述實施方式中的具體細節,在本發明的技術構思范圍內,可以對本發明的技術方案進行多種簡單變型,這些簡單變型均屬于本發明的保護范圍。另外需要說明的是,在上述具體實施方式中所描述的各個具體技術特征,在不矛盾的情況下,可以通過任何合適的方式進行組合。為了避免不必要的重復,本發明對各種可能的組合方式不再另行說明。此外,本發明的各種不同的實施方式之間也可以進行任意組合,只要其不違背本發明的思想,其同樣應當視為本發明所公開的內容。當前第1頁1 2 3
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 用于烯烴聚合的催化劑組分的制備方法與制造工藝
- 用于烯烴聚合的催化劑組分的制備方法與制造工藝
- 催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑體系及其應用和烯烴聚合方法與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑體系及其應用和烯烴聚合方法與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑體系及其應用和烯烴聚合方法與制造工藝
- 鹵化鎂醇合物載體及其制備方法和應用以及用于烯烴聚合的催化劑組分及其應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝
- 用于烯烴聚合的催化劑組分及其制備方法和應用和用于烯烴聚合的催化劑及其應用與制造工藝