一種核酸擴增反應引物的設計方法及其應用與流程
本發明屬于生物技術領域,涉及一種核酸擴增反應引物的設計方法及其應用,所述方法是一種既可以在特定基因或區域,也可以在全基因組范圍內設計出具有通用性和特異性的核酸擴增反應引物的方法。
背景技術:
核酸擴增技術已成為當前分子生物學研究和應用中使用最多、最廣泛的手段之一,核酸擴增的目的在于針對目標序列,利用引物組擴增一段不含任何副產物(非特異性DNA)的特異DNA片段,因此引物設計是核酸擴增技術中至關重要的一環。基于核酸擴增技術的目的,擴增反應的引物組首先需要具備特異性,即該引物組不能擴增非目標序列;為了提高效率,有時同時需要引物組具有通用性,即引物組可以應用于多個目標序列(多個目標基因或區域,或多個目標基因組)。
目前常用的引物設計軟件一般只能在預先設定的一個基因區域內設計引物,也就是說,用戶必須首先尋找到具有特異性和通用性的序列(通常的做法是,從文獻中獲取物種特異性基因,并根據其保守的范圍確定其通用性),然后將其輸入引物設計軟件如Primer、PrimerExplorer V4等。然而,從文獻中獲得的所謂“物種特異性基因”往往基于滯后的知識,并未基于不斷增長的基因組數據進行必要的更新,導致基于該基因序列獲得的引物在實際應用中不一定能確保其特異性和通用性,導致傳統方法引物設計的成功率偏低。表1舉例展示了現有技術阪崎克羅諾桿菌檢測方法中存在的引物特異性不夠的問題。也就是說,現有技術方法中所使用的阪崎克羅諾桿菌特異序列實際上并非阪崎克羅諾桿菌所特有,即,有可能將非阪崎克羅諾桿菌錯誤地認定為阪崎克羅諾桿菌。出現這類問題的原因之一是未利用現有基因組數據庫中對設計的引物進行包括諸如特異性檢測和判定等的有效性檢驗。類似的問題同樣也存在于通用性的確認。
表1阪崎克羅諾桿菌的現有檢測方法中引物的特異性分析
注:a)將專利中引物F3和B3間的序列與阪崎克羅諾桿菌的三個基因組(GI號分別為156932229、449306535和389839000)進行Bowtie比對,確定檢測區域在GI號156932229基因組中的位置(星號*表示引物F3和B3間的序列可比對到GI號156932229基因組中的7個重復區域,本表中僅列出1個區域)。專利CN201310287426.5中四套引物的擴增區域無法定位,文本中也沒有給出擴增靶基因為何種基因。b)將檢測區域序列在公共數據庫資源中進行Blast比對,引物區域匹配程度越高,特異性越差。
因此,亟需一種新的方法,不但可以在特定基因或區域,而且可以在全基因組范圍內高效率地設計出具有通用性和特異性的引物,滿足檢測、擴增等對確保具有特異性和通用性的引物的需求。
技術實現要素:
本發明創新提出了一種核酸擴增反應引物的設計方法。該方法既可以針對特定基因或區域的序列,也可以針對全基因組序列進行引物設計。該方法設計得到的引物能夠同時通過物理化學性質檢測和通用性、特異性檢測。本發明方法得到的引物可以擴增多個目標序列(包括含多個目標基因、多個目標區域、或多個目標基因組);同時,得到的引物無法擴增非目標序列。本方法充分利用了公共資源中的基因組學數據,并采納恰當的數據分析方法和工具,能夠在目標序列(包括目標基因、目標區域、或目標基因組)上高效率地進行引物設計,獲得具有通用性和特異性的引物,其可靠性超過傳統的僅針對特定基因序列的引物設計方法, 本發明確保了所獲得的針對該目標序列的引物的特異性和通用性。
本發明提出一種新的核酸擴增反應引物設計方法,該方法可以在特定的目標基因或區域、或者目標全基因組范圍內設計出滿足通用性和特異性的引物,能夠廣泛地應用于PCR、實時定量PCR、等溫擴增(如LAMP等)、以及其它核酸擴增反應類型所需要引物的設計。
本發明提出的核酸擴增反應引物的設計方法,包括:(1)確定引物的目標序列和非目標序列;(2)針對目標序列設計單條引物;(3)引物序列比對;(4)根據核酸擴增反應的類型及其對引物組合方式的要求,將單條引物組合成引物組;(5)對引物組進行物理化學性質、通用性以及特異性檢測和判定;(5)輸出符合條件的引物組。
本發明核酸擴增反應引物的設計方法,包括以下步驟:
(1)初始化:將由引物的所有目標序列組成的集合定義為集合G_1,將由引物的所有非目標序列組成的集合定義為集合G_2;
(2)設計單條引物:根據核酸擴增反應的類型,設定描述引物特性的各項參數,以集合G_1中的某一目標序列為引物設計的候選序列,計算得出符合條件的單條引物,并記錄每條引物的信息;
(3)引物序列比對:將設計出的單條引物分別與集合G_1和集合G_2進行序列比對,記錄比對信息;
(4)單條引物組合:根據核酸擴增反應的類型及其對引物組合方式的要求,將單條引物組合成引物組;
(5)引物的檢測:對引物組進行物理化學性質、通用性以及特異性檢測和判定;
(6)輸出符合條件的引物組,即得到所述核酸擴增反應的引物。
其中,步驟(1)中,所述集合G_1是由引物的所有目標序列組成,所述集合G_2是由引物的所有非目標序列組成。
所述目標序列是指引物設計的目標基因或目標區域、或目標基因組序列。
所述非目標序列是指除目標基因或目標區域之外的序列、或除目標基因組之外的其它基因組序列。
在具體實施方案中,當核酸擴增反應的檢測對象為特定某物種時,目標序列 可以是待測物種全基因組,非目標序列則是其它物種或其它相關物種的基因組。
在具體實施方案中,當核酸擴增反應目標為特定基因或區域時,目標序列為待測基因或區域的DNA序列。根據實施方案對特異性的不同要求,非目標序列可以是該目標序列之外的其它任意所有序列,也可以是所在基因組中除目標序列之外的其它序列。
例如,當實驗目的為檢測混合的微生物基因組中是否存在某種特定微生物X,目標序列是X微生物的各菌株基因組序列,而非目標序列是除X微生物之外的其它微生物基因組序列。例如,當實驗目的為設計恰當的引物以擴增出小鼠、大鼠和人的某一基因序列X,則目標序列是小鼠X基因、大鼠X基因和人X基因,非目標序列是小鼠基因組中除X基因之外的其它序列,大鼠基因組中除X基因之外的其它序列,和人基因組中除X基因之外的其它序列。
其中,步驟(2)中,所述根據核酸擴增反應的類型,是指引物的應用范圍,可以為聚合酶鏈式反應(PCR),實時定量PCR,也可以為等溫擴增反應,包括環介導等溫擴增(Loop-Mediated Isothermal Amplification,LAMP)等,以及其它核酸擴增反應類型。所述“描述引物特性的各項參數”,包含但不限于引物的長度、GC含量、Tm值、3’端穩定性、5’端穩定性和/或二級結構穩定性等物理化學性質,在具體實施方案中根據實際情況確定適當的一項或多項參數的組合。所述“記錄每條引物的信息”,是指記錄包括引物在所述特定目標序列中的位置、正負鏈信息(即來源于正鏈還是負鏈)和/或引物的長度等信息,在具體實施方案中根據實際情況確定適當的一項或多項參數的組合。
其中,步驟(3)中,所述序列比對,可以使用Bowtie、Bowtie2、BWA、SOAP等短序列快速比對軟件,也可以為Blast、Blat等長序列比對軟件,優選地,為Bowtie、Bowtie2、BWA、SOAP等快速比對軟件。所述“記錄比對信息”,是指每條引物在集合G_1和集合G_2中每個序列上面的位置、正負鏈信息(即來源于正鏈還是負鏈)等信息。
其中,步驟(4)中,所述根據核酸擴增反應的類型,是指引物的應用范圍,可以為聚合酶鏈式反應(PCR),實時定量PCR、也可以為等溫擴增反應,包括環介導等溫擴增(Loop-Mediated Isothermal Amplification,LAMP)等,以及其它核酸擴增反應類型。所述對引物組合方式的要求,是指單條引物在所述特定目 標序列上的位置、正負鏈信息(即來源于正鏈還是負鏈),以及單條引物間的距離等。
其中,步驟(5)中,所述對引物組進行物理化學性質檢測,是指檢測各條引物在每個序列上的位置關系與正負鏈信息(即來源于正鏈還是負鏈)、引物的GC含量與擴增區域的GC含量的關系、引物的Tm值與擴增區域的Tm值的關系、引物之間是否發生相互作用等。所述通用性檢測和判定,是指檢測該引物組能否根據檢測需求應用在G_1集合中的多個目標序列,若符合檢測需求,則通用性檢測結果為“通過”;若不符合檢測需求,則通用性檢測結果為“未通過”。所述特異性檢測和判定,是指檢測該引物組能否用于集合G_2中的任意一個非目標序列,若不能用于集合G_2中的任意一個非目標序列,則特異性檢測結果為“通過”,若能用于集合G_2中的一個或多個非目標序列,則特異性檢測結果為“未通過”。若未通過上述物理化學性質、通用性及特異性三項檢測的,則放棄該引物組,繼續篩選合適的引物組。
其中,步驟(6)中,所述輸出符合條件的引物組,是指輸出前步驟中獲得的能夠同時通過物理化學性質檢測、通用性和特異性檢測的引物組中每一條引物的位置、長度、正負鏈信息(即來源于正鏈還是負鏈)、以及該引物組的通用性情況(即可以用于哪些目標序列上)等信息。系統將符合條件的引物組按照通用性程度排序,通用性越強,排名越靠前,系統按此順序輸出符合條件的引物組。其中,引物組的數量可預先設定。
本發明還提出了按上述方法設計得到的引物,所述引物包括引物組,所述引物組為:
F3:5’-GGATTAGGAAATATACGACGC-3’(SEQ ID NO:1)
FIP:5’-AGCAAAACCCATTGGTACCTTATACTGTCTATGGGCACAA(SEQ ID NO:2)
BIP:5’-TGAATATGATCCCCCGGCTTCTGGGAGGAAATATCGCTAA-3’(SEQ ID NO:3)
B3:5’-AGCATATATGATACGGGGAA-3’(SEQ ID NO:4)。
本發明還提出了按上述方法設計得到的引物,所述引物包括引物組,所述引物組為:
P1:5’-GGAGCCTGCTCAGTCTGTT-3’(SEQ ID NO:5)
P2:5’-TAACTGGGCCAAGGAAGGT-3’(SEQ ID NO:6)。
本發明還提出了按上述方法設計得到的引物,所述引物包括引物組,所述引物組為:
P3:5’-GACTCACGAAAGCCAGCTG-3’(SEQ ID NO:7)
P4:5’-AGTCAGCACGCCATACCA-3’(SEQ ID NO:8)
本發明還提出了將上述引物設計方法或引物分別在阪崎克羅諾桿菌和志賀氏菌相關基因(或區域)的擴增或檢測中的應用。
本發明方法充分運用了基因組學數據以及相關方法、工具,能夠在目標序列,(可以是特定基因或區域、也可以是全基因組)上高效率地進行引物設計,獲得具有高通用性和高特異性的引物,其可靠性超過傳統的僅針對特定基因序列的引物設計方法。本發明可以廣泛應用于PCR、實時定量PCR、等溫擴增,包括LAMP,以及其它核酸擴增反應類型中的引物設計。
附圖說明
圖1本發明引物設計方法的流程圖。
圖2實施例1的阪崎克羅諾桿菌LAMP引物的位置及正負鏈關系示意圖。
圖3利用實施例1設計的阪崎克羅諾桿菌LAMP引物進行核酸擴增反應后產物的電泳圖。
圖4實施例2的志賀氏菌PCR引物的位置、正負鏈關系示意圖。
圖5利用實施例2的志賀氏菌PCR引物進行核酸擴增反應后產物的電泳圖。
具體實施方式
結合以下具體實施例和附圖,對本發明作進一步的詳細說明,本發明的保護內容不局限于以下實施例。在不背離發明構思的精神和范圍下,本領域技術人員能夠想到的變化和優點都被包括在本發明中,并且以所附的權利要求書為保護范圍。實施本發明的過程、條件、試劑、實驗方法等,除以下專門提及的內容之外,均為本領域的普遍知識和公知常識,本發明沒有特別限制內容。
實施例1阪崎克羅諾桿菌(Cronobacter sakazakii)檢測用LAMP引物的設計
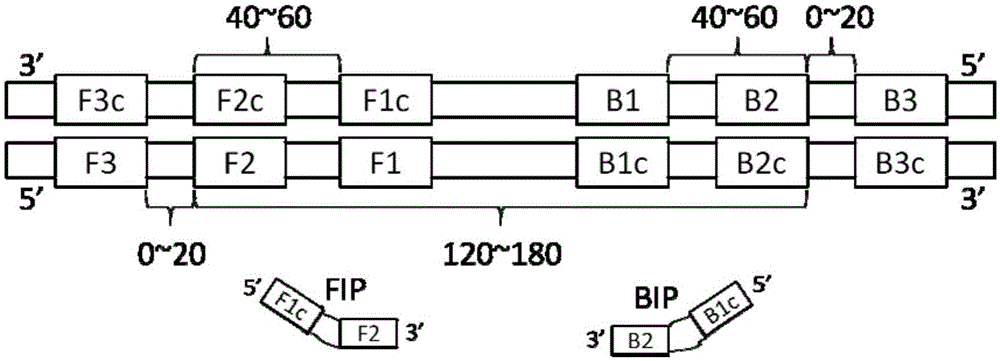
LAMP是榮研化學株式會社研發的核酸擴增方法,LAMP引物一般由來自基因組6個不同位置(分別為F3、F2、F1c、B1c、B2和B3)序列的4條單獨的 引物(F3,B3,F2和F1c組成的FIP,B2和B1c組成的BIP)組成。
本發明針對阪崎克羅諾桿菌設計具有通用性和特異性的LAMP引物的方法,包括如下步驟:
(1)初始化:
使用2013年8月5號從NCBI的FTP上下載的有完整基因組序列的所有細菌和古菌的數據,共2,599個全基因組序列。設定集合G_1,其包含目標基因組序列(即,阪崎克羅諾桿菌的3個全基因組序列),其GI號分別為156932229、449306535和389839000,所設計引物應當能夠檢測出對應的3株阪崎克羅諾桿菌,即具有通用性;設定集合G_2,其包含非目標基因組序列(即,除阪崎克羅諾桿菌的前述3個目標基因組序列之外的2596個全基因組序列),所設計的引物應當不能夠檢測到除阪崎克羅諾桿菌之外的細菌和古細菌,即,具有特異性。
(2)設計單條引物:
根據LAMP引物的特性及本發明的需求,設定引物的GC含量、Tm值、3’端穩定性、5’端穩定性(見表2)及引物長度(18~25bp)等特性參數,同時設定單條引物不能產生發卡結構、自身不能發生相互作用等條件,以GI號為156932229的阪崎克羅諾桿菌ATCC BAA-894的全基因組序列作為單條引物設計的候選序列,計算得出符合上述設定條件的單條引物。并記錄每條引物在156932229基因組序列上的位置、正負鏈信息(即來源于正鏈還是負鏈)以及引物的長度等信息。
表2 LAMP反應中單條引物的特性要求
(3)引物序列比對:
使用比對軟件Bowtie,將上一步設計出的單條引物分別與集合G_1中的所 有目標基因組和集合G_2中的所有非目標基因組進行序列比對。為了確保獲得的引物的通用性,當該單條引物與集合G_1中的目標序列進行比對時,使用參數設置“-a -n 0”,即要求該單條引物與所有目標序列完全匹配。為了確保獲得的引物的特異性,當該單條引物與集合G_2中的非目標序列進行比對時,使用參數“-a -n 3”,即,該單條引物與所有非目標序列不超過3個錯配。比對結束后,獲得符合上述比對條件的單條引物,并記錄每條引物在集合G_1和集合G_2中的每個基因組上的位置、正負鏈信息(即來源于正鏈還是負鏈)等信息。
(4)單條引物組合:
從上述步驟中產生的候選的單條引物F3、F2、F1c、B1c、B2和B3,按照圖2中的條件設定這六條單條引物之間的位置以及在正負鏈上的關系,將單條引物組合成引物組。單條引物組合成引物組可按通用方法操作。
(5)引物組的檢測:
首先對引物組進行物理化學性質檢測,包括:①引物組在目標序列或者非目標序列上能否滿足引物之間的位置關系、正負鏈關系;②引物組的GC含量、Tm值等與其擴增區域的GC含量是否滿足相互關系;③每一條引物與其它單條引物之間是否存在相互作用。
其次,對引物組進行通用性檢測和判定,即檢測該引物組能否應用在集合G_1中的多個目標序列。該引物組能應用于集合G_1中的越多的目標序列,通用性越好。
接著,對引物組進行特異性檢測和判定,即檢測該引物組能否用于集合G_2中的任意一個非目標序列。若不能用于集合G_2中的任意一個非目標序列,則特異性檢測結果為“通過”,若能用于集合G_2中的一個或多個非目標序列,則特異性檢測結果為“未通過”。
通過上述物理化學性質、通用性及特異性三項檢測的,則該引物組即為符合條件的引物組。若未通過上述物理化學性質、通用性及特異性三項檢測,則放棄該引物組。繼續或重新篩選,直至獲得符合條件的引物組。
(6)輸出符合條件的引物組:
輸出前步驟中獲得的能夠同時通過物理化學性質檢測、通用性和特異性檢測的引物組中每一條引物的位置、長度、正負鏈信息(即來源于正鏈還是負鏈)、 以及該引物組的通用性情況(即可以用于哪些目標序列上)等信息。系統將符合條件的引物組按照通用性程度排序,通用性越強,排名越靠前,系統按此順序輸出符合條件的引物組。其中,引物組的數量可預先設定,在本實施例中引物組的數量預先設定為5。
例如,經過運行程序在AT富集區設計出LAMP反應引物組5個,隨機選擇一個引物組進行有效性驗證。所述引物組的序列為:
F3:5’-GGATTAGGAAATATACGACGC-3’(SEQ ID NO:1)
FIP:5’-AGCAAAACCCATTGGTACCTTATACTGTCTATGGGCACAA-3’(SEQ ID NO:2)
BIP:5’-TGAATATGATCCCCCGGCTTCTGGGAGGAAATATCGCTAA-3’(SEQ ID NO:3)
B3:5’-AGCATATATGATACGGGGAA-3’(SEQ ID NO:4)
所用的LAMP反應體系如下(反應總體積為25μL):1×Thermopol反應緩沖液,6mmol/L Mg2+,1.6mmol/L dNTP,0.8μmol/L的FIP和BIP引物,0.3μmol/L的F3和B3引物,8U Bst DNA聚合酶,0.8mol/L甜菜堿,和不同濃度的DNA模板(1ng~1pg)。
當使用阪崎克羅諾桿菌(編號為CICC 21560)DNA為模板時,采用設計的引物組成功地進行了擴增,擴增產物的電泳圖如圖3所示,泳道M為DNA分子量標準DL2000,泳道1~4分別為DNA模板量為1ng、100pg、10pg和1pg的阪崎克羅諾桿菌的LAMP擴增產物,呈現出LAMP反應特征性的梯形條帶,泳道5為以滅菌水作陰性對照模板擴增后的產物,不呈現LAMP反應特征性的梯形條帶。
根據該引物組在GI號為156932229的阪崎克羅諾桿菌(Cronobacter sakazakii)ATCC BAA-894上的位置,將F3到B3之間的擴增區域序列(包含F3和B3)與數據庫中阪崎克羅諾桿菌的另外兩個全基因組序列(GI分別為389839000和449306535)進行Blast比對,結果顯示,引物區序列均完全匹配,表明本實施例中的上述引物組可以擴增目前已有的3株阪崎克羅諾桿菌,通用性很好。
對上述引物組進行特異性檢測。首先,從理論上進行分析,將該引物組與其它所有非阪崎克羅諾桿菌的基因組進行Bowtie比對(將參數設置為“-a -n 3”, 即允許不超過3個堿基不匹配),發現上述引物組的序列在非阪崎克羅諾桿菌的基因組中不存在。此外,從對28株非阪崎克羅諾桿菌和1株阪崎克羅諾桿菌菌株進行的擴增實驗結果(見表3)可以看出,使用該引物組僅有阪崎克羅諾桿菌菌株擴增結果為陽性,而其它非阪崎克羅諾桿菌菌株均為陰性。因此,上述引物組具有很好的特異性。
綜合上述實驗及理論分析可知,根據本發明方法設計出的阪崎克羅諾桿菌的LAMP引物具有很好的通用性和特異性,滿足設計的要求。
表3引物特異性檢測的實驗結果
注:a)CGMCC:中國普通微生物菌種保藏中心,CICC:中國工業微生物菌種保藏管理中心,CMCC:中國醫學細菌菌種保藏管理中心。b)+:陽性結果,-:陰性結果。
實施例2志賀氏菌檢驗用PCR引物的設計
本發明針對志賀氏菌(Shigella spp.)設計具有通用性和特異性的PCR引物,步驟如下:
(1)初始化:
使用2013年8月5號從NCBI的FTP上下載的有完整基因組序列的所有細菌和古菌的數據,共2599個全基因組序列。集合G_1中包含目標基因組序列(即,志賀氏菌的9個全基因組序列),集合G_2中包含非目標基因組序列(即,除前述集合G_1中的9個全基因組序列之外的2590個全基因組序列)。
(2)設計單條引物:
PCR引物來源于基因組中的2個位置(引物P1和引物P2)。根據PCR引物的特性以及本實施例的需求,設定引物的GC含量、Tm值、3’端穩定性、5’端穩定性(見表4)及引物長度(18~25bp)等特性參數,且規定單條引物不能產生發卡結構、自身不能發生相互作用等條件,以GI號為110804074的福氏志賀氏菌(Shigella flexneri 5str.8401)的全基因組序列作為單條引物設計的候選序列,計算得出符合條件的單條引物。并記錄每一個單條引物在110804074基因組序列上的位置、正負鏈信息(即來源于正鏈還是負鏈)以及引物的長度等信息。
表4 PCR反應中單條引物的特性要求
(3)引物序列比對:
使用比對軟件Bowtie,將上一步設計出的單條引物分別與集合G_1中的目標基因組序列和集合G_2中的非目標基因組進行序列比對。為了保證引物的通用性,當該單條引物與集合G_1中的目標序列進行比對時,使用參數設置“-a -n 0”,即要求該單條引物與目標序列完全匹配;為了保證引物的特異性,當該單條引物與集合G_2中的非目標序列進行比對時,使用參數設置“-a -n 3”,即允許該單條引物與非目標序列有不超過3個錯配。比對結束后,記錄每一個單條引物在集合G_1和集合G_2中的每個基因組上的位置、正負鏈信息(即來源于正鏈還是負鏈)。
(4)單條引物組合:
從上述步驟中產生的候選的單條引物中挑選引物P1和引物P2,并按照圖4中的條件設定2條單條引物之間的位置以及在正負鏈上的關系,將單條引物組合成引物組。
(5)引物的檢測:
首先對引物組進行物理化學性質檢測,包括:①引物組在目標序列或者非目標序列上能否滿足引物之間的位置關系、正負鏈關系;②引物組的GC含量、Tm值等與其擴增區域的GC含量是否滿足相互關系;③每一條引物與其它單條引物之間是否存在相互作用。
其次,對引物組進行通用性檢測和判定,即檢測該引物組能否應用在集合G_1中的9個目標序列。該引物組能應用于集合G_1中的越多的目標序列,通用性越好。
接著,對引物組進行特異性檢測和判定,即檢測該引物組能否用于集合G_2中的任意一個非目標序列,若不能用于集合G_2中的任意一個非目標序列,則特異性檢測結果為“通過”,若能用于集合G_2中的一個或多個非目標序列,則特異性檢測結果為“未通過”。
通過上述物理化學性質、通用性及特異性三項檢測的,則該引物組即為符合條件的引物組。若未通過上述物理化學性質、通用性及特異性三種檢測,則放棄該引物組。繼續或重新篩選,直至獲得符合條件的引物組。
(6)輸出符合條件的引物組:
輸出前步驟中獲得的能夠同時通過物理化學性質檢測、通用性和特異性檢測的引物組中每一條引物的位置、長度、正負鏈信息(即來源于正鏈還是負鏈)、以及該引物組的通用性情況(即可以用于哪些目標序列上)等信息。系統將符合條件的引物組按照通用性程度排序,通用性越強,排名越靠前,系統按此順序輸 出符合條件的引物組。其中,引物組的數量可預先設定,在本實施例中引物組的數量預先設定為5。
例如,經過運行程序在AT富集區設計出PCR反應所需要的引物組5個,隨機選擇一個引物組進行有效性驗證。所述引物組的序列為:
P1:5’-GGAGCCTGCTCAGTCTGTT-3’(SEQ ID NO:5)
P2:5’-TAACTGGGCCAAGGAAGGT-3’(SEQ ID NO:6)
所用的PCR反應體系(反應總體積為25μL)為:1×PCR反應緩沖液(含Mg2+),0.1mmol/L dNTP,0.8μmol/L的P1和P2引物和0.1U/μL Taq DNA聚合酶和50ng/μL DNA模板。所用的反應溫度條件為:95℃預變性3min;95℃變性30s,不同溫度(51℃、53℃、55℃、57℃和59℃)退火30s,72℃延伸30s,循環30次;最后72℃延伸7min;16℃保存。
當使用志賀氏菌(編號為CGMCC1.1868,為福氏志賀氏菌)DNA為模板時,采用設計的引物組成功進行了擴增,擴增產物的電泳圖如圖5所示,泳道M為100bp梯度DNA分子量標準DNAMarker I(100bp~600bp),泳道1~4分別為退火溫度為51℃、53℃、55℃、57℃和59℃的志賀氏菌的PCR擴增產物,其片段大小約為240bp,與預期片段大小(242bp)一致,泳道5為以滅菌水作陰性對照模板擴增后的產物,沒有條帶。
通過將該引物組的P1和P2序列與所有細菌基因組的數據庫進行Bowtie比對(允許不超過3個錯配),在理論上對該引物組的特異性進行分析,結果發現兩條引物僅在志賀氏菌的9個基因組里同時出現,而在所有其它細菌基因組里均不能同時出現,表明采用本發明方法設計的該引物組具有很好的特異性。
綜合上述實驗及理論分析可知,根據本發明方法設計出的志賀氏菌的PCR引物具有很好的通用性和特異性,滿足設計的要求。
實施例3阪崎克羅諾桿菌(Cronobacter sakazakii)OmpA基因擴增用PCR引物的設計
利用本發明設計用以擴增阪崎克羅諾桿菌OmpA基因的PCR引物,步驟如下:
(1)初始化:
使用2013年8月5號從NCBI的FTP上下載的三株阪崎克羅諾桿菌的完整 基因組序列(GI號分別為156932229、449306535和389839000)。設定集合G_1,其包含三株阪崎克羅諾桿菌的OmpA基因序列;設定集合G_2,其包含三株阪崎克羅諾桿菌基因組中除OmpA基因之外的其它序列。
(2)設計單條引物:
根據PCR引物的特性以及本實施例的需求,設定引物(P3和P4)的GC含量、Tm值、3’端穩定性、5’端穩定性(見表4)及引物長度(18~25bp)等特性參數,且規定單條引物不能產生發卡結構、自身不能發生相互作用等條件,以GI號為156932229的阪崎克羅諾桿菌的OmpA基因序列作為單條引物設計的候選序列,計算得出符合條件的單條引物。并記錄每一個單條引物在156932229基因組序列上的位置、正負鏈信息(即來源于正鏈還是負鏈)以及引物的長度等信息。
(3)引物序列比對:
使用比對軟件Bowtie,將上一步設計出的單條引物分別與集合G_1中的目標基因組序列和集合G_2中的非目標基因組進行序列比對。為了保證引物的通用性,當該單條引物與集合G_1中的目標序列進行比對時,使用參數設置“-a -n 0”,即要求該單條引物與目標序列完全匹配;為了保證引物的特異性,當該單條引物與集合G_2中的非目標序列進行比對時,使用參數設置“-a -n 3”,即允許該單條引物與非目標序列有不超過3個錯配。比對結束后,記錄每一個單條引物在集合G_1和集合G_2中的每個基因組上的位置、正負鏈信息(即來源于正鏈還是負鏈)。
(4)單條引物組合:
從上述步驟中產生的候選的單條引物中挑選引物P3和引物P4,并按照圖4中的條件設定2條單條引物之間的位置以及在正負鏈上的關系,將單條引物組合成引物組。
(5)引物的檢測:
首先對引物組進行物理化學性質檢測,包括:①引物組在目標序列或者非目標序列上能否滿足引物之間的位置關系、正負鏈關系;②引物組的GC含量、Tm值等與其擴增區域的GC含量是否滿足相互關系;③每一條引物與其它單條引物之間是否存在相互作用。
其次,對引物組進行通用性檢測和判定,即檢測該引物組能否應用在集合G_1中的9個目標序列。該引物組能應用于集合G_1中的越多的目標序列,通用性越好。
接著,對引物組進行特異性檢測和判定,即檢測該引物組能否用于集合G_2中的任意一個非目標序列,若不能用于集合G_2中的任意一個非目標序列,則特異性檢測結果為“通過”,若能用于集合G_2中的一個或多個非目標序列,則特異性檢測結果為“未通過”。
通過上述物理化學性質、通用性及特異性三項檢測的,則該引物組即為符合條件的引物組。若未通過上述物理化學性質、通用性及特異性三種檢測,則放棄該引物組。繼續或重新篩選,直至獲得符合條件的引物組
(6)輸出符合條件的引物組:
輸出前步驟中獲得的能夠同時通過物理化學性質檢測、通用性和特異性檢測的引物組中每一條引物的位置、長度、正負鏈信息(即來源于正鏈還是負鏈)、以及該引物組的通用性情況(即可以用于哪些目標序列上)等信息。系統將符合條件的引物組按照通用性程度排序,通用性越強,排名越靠前,系統按此順序輸出符合條件的引物組。其中,引物組的數量可預先設定(在本實施例中引物組的數量預先設定為5)。
例如,經過運行程序在GC含量正常區設計出PCR反應所需要的引物組5個,隨機選擇一個引物組進行有效性驗證。所述引物組的序列為:
P3:5’-GACTCACGAAAGCCAGCTG-3’(SEQ ID NO:7)
P4:5’-AGTCAGCACGCCATACCA-3’(SEQ ID NO:8)
所用的PCR反應體系(反應總體積為25μL)為:1×PCR反應緩沖液(含Mg2+),0.1mmol/L dNTP,0.8μmol/L的P3和P4引物,0.1U/μL Taq DNA聚合酶和50ng/μL DNA模板。所用的反應溫度條件為:95℃預變性3min;95℃變性30s,55℃退火30s,72℃延伸30s,循環30次;最后72℃延伸7min;16℃保存。
當使用阪崎克羅諾桿菌(編號為CICC 21560)DNA為模板時,采用設計的引物組進行了擴增,電泳分析結果表明,擴增產物與預期片段大小(230bp)一致。
因此,根據本發明方法設計出的阪崎克羅諾桿菌OmpA基因的PCR引物能夠滿足設計的要求。
- 還沒有人留言評論。精彩留言會獲得點贊!
- 一種引物組及其在擴增siv/shiv基因組中的應用與試劑盒的制作方法
- 檢測f8基因突變的擴增引物、試劑盒及方法
- 擬莖點霉rpb2基因擴增引物及其設計方法和應用
- 一種馬來穿山甲線粒體Cox3基因序列的擴增引物及鑒定方法
- 擬莖點霉gpdh基因擴增引物及其設計方法和應用
- 甲型流感病毒全基因組擴增方法及使用的復合引物和試劑盒的制作方法
- 用于檢測2型糖尿病視網膜病變的miR-2116-5p分子標記物及其擴增引物和應用
- 用于檢測2型糖尿病視網膜病變的miR-3197分子標記物及其擴增引物和應用
- 一種用于魚類線粒體全基因組擴增的引物、試劑盒及擴增方法
- 用Alu重復序列基因擴增引物且無需DNA抽提的檢測血漿游離DNA的定量PCR方法