苯并惡唑并惡嗪酮類化合物WA1-089的晶型C及其制備方法與流程
本發明涉及一種新型Xa因子抑制劑———苯并惡唑并惡嗪酮類化合物WA1-089的新晶型及制備方法,具體涉及5-氯-N-(((3S,3aS)-1-酮-7-(3-酮嗎啡啉)-1,3,3a,4-四氫苯并[b]惡唑并[3,4-d][1,4]惡嗪-3-基)甲基)噻吩-2-甲酰胺的新晶型及其制備方法。
背景技術:
一種全新的Xa因子抑制劑———苯并惡唑并惡嗪酮類化合物(簡稱WA1-089)由結構式(I)表示,化學名稱為5-氯-N-(((3S,3aS)-1-酮-7-(3-酮嗎啡啉)-1,3,3a,4-四氫苯并[b]惡唑并[3,4-d][1,4]惡嗪-3-基)甲基)噻吩-2-甲酰胺,可用于預防術后深靜脈血栓形成(DVT)和肺栓塞(PE),預防在心房顫動時的中風,治療急性冠脈綜合征(ACS)等。
晶型是化合物的一個重要理化性質,同一藥物不同的晶型由于晶型類型和純度的差異,其一些理化性質可能會存在顯著差異,從而影響藥物的穩定性、生物利用度及療效的發揮。化合物的晶型可以通過成熟的技術如:X-射線衍射光譜法、紅外光譜法、差示掃描量熱法、熱重分析以及熔點分析來加以區別和表征。
WO2015/043364A1“作為凝血因子Xa因子抑制劑的苯并惡唑并惡嗪酮類化合物”公開了結構式(I)所示化合物,但未涉及其晶型研究。
技術實現要素:
本發明的目的之一在于提供一種穩定性好的式(I)化合物的晶型C。
本發明的目的之二在于提供前述式(I)化合物的晶型C的制備方法。
本發明的目的之一是這樣實現的:
本發明公開的式(I)化合物的晶型C具有如下特征:
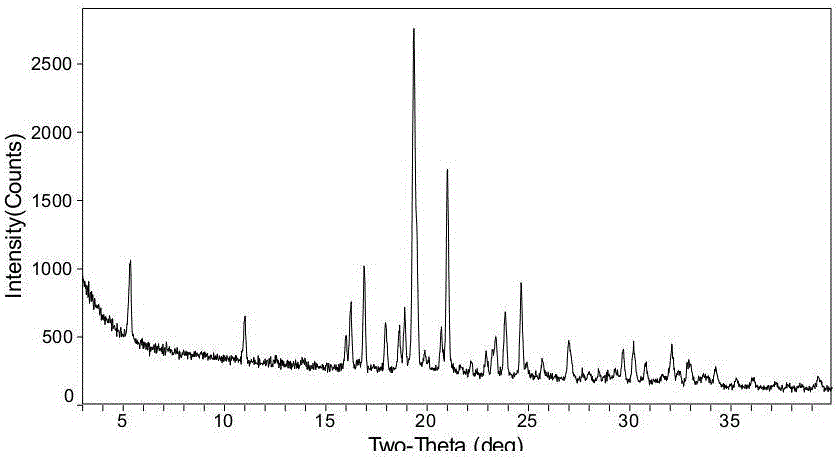
晶型C可通過包含2θ角約為5.35、11.00、16.00、16.24、16.89、17.95、18.64、18.89、19.35、20.71、21.01、22.91、23.39、23.85、24.63、25.68、27.00、29.69、30.19、30.81、32.07、32.92、34.24的X-射線粉末衍射(XRPD)圖譜進行描述,2θ值的一般精度約在±0.2°的范圍內。
優選地,所述XRPD圖基本與圖1一致。
晶型C還可進一步通過包括在約190℃~205℃,優選在約201.6℃(均±2℃)處的吸熱峰的差示掃描量熱(DSC)跡線來表征。
優選地,所述DSC的圖基本與圖2一致。
或者,晶型C還可進一步通過具有如圖3所示的熱重分析(TGA)跡線來表征,顯示該晶型不含結晶水。
或者,晶型C還可進一步通過衰減的總反射紅外光譜進行表征,包括在約為以下波數的吸收譜帶:636、715、807、881、994、1024、1039、1113、1151、1185、1235、1342、1383、1424、1506、1558、1623、1669和1760
cm-1。波數值的一般精度為約±2cm-1的范圍內。
優選地,所述的衰減的總反射紅外光譜圖即傅里葉紅外(FT-IR)圖譜,基本與圖4一致。
本發明的目的之二是這樣實現的:
制備所述式(I)化合物的晶型C的方法,包括下列步驟:
1)將式(I)化合物升溫至熔融狀態;
2)維持熔融狀態一段時間;
3)再降溫至室溫可得到結晶固體。
所述式(I)化合物晶型C的制備方法,其中步驟(1)所述的熔融狀態溫度為190℃~205℃之間。
所述式(I)化合物晶型C的制備方法,其中步驟(2)所述的維持在熔融狀態的時間為3~10min。
所述式(I)化合物晶型C的制備方法,其中步驟(3)所述的室溫為10℃~30℃。
本發明的有益效果:
本發明公開的晶型C有良好的性能,如水溶性好、穩定性好、不易吸水潮解等特點;另外,本晶型通過熔融結晶的方法制得,該方法具有產品純度高、能耗低,不需要加入其它溶劑,對環境污染小等特點。
附圖說明
圖1是WA1-089晶型C的粉末衍射(XRPD)圖譜
圖2.是WA1-089晶型C的差示掃描量熱(DSC)圖譜
圖3.是WA1-089晶型C的熱重分析(TGA)圖譜
圖4.是WA1-089晶型C的傅里葉紅外(FT-IR)圖譜
圖5.是WA1-089晶型C的動態水分吸附圖譜
圖6.是WA1-089晶型C的等溫吸附曲線。
具體實施方式
下述實施例僅用于闡述實現本發明的方法,不應理解為對本發明的限制。
本發明所使用的式(Ⅰ)化合物(WA1-089)來自華北制藥集團新藥研究開發有限責任公司,含量為99%。本發明使用的高效液相色譜(HPLC)為996型檢測器,515泵(Waters公司)。本發明所使用的粉末衍射儀型號為Bruker D8 Advance diffractometer,采用銅靶波長為1.54nm的Kα radiation(40Kv,40mA)、θ-2θ測角儀、Mo單色儀和Lynxeye探測器。儀器在使用前用金剛砂檢測過。采集軟件是Diffrac Plus XRD Commander ,分析軟件示MDI Jade 6。樣品在室溫條件下測試,把需要檢測的樣品放在無反射片上。詳細檢測條件如下,角度范圍:3-40°2θ,步長:0.02°2θ,速度:0.2 s.step-1。除特別說明,樣品在檢測前未經研磨。差熱分析數據采自于TA Instruments Q200 DSC,儀器控制軟件是Thermal Advantage,分析軟件是Universal Analysis。熱重分析數據采自于TA Instruments Q500TGA,儀器控制軟件是Thermal Advantage,分析軟件是Universal Analysis。傅里葉紅外(FT-IR)數據采集于Bruker Tensor 27,儀器控制軟件和數據分析軟件都是OPUS。動態水分吸附(DVS)分析數據采自于TA Instruments Q500TGA,儀器控制軟件是Thermal Advantage,分析軟件是Universal Analysis。除非特別說明,樣品通常檢測條件如下表所示。
表1 DSV實驗參數
實施例1
將10mg WA1-089放置DSC儀器上,以10℃/min升溫至190℃,維持10min,再以10℃/min降溫至10℃可得固體結晶。
XRPD和DSC分析數據證實了,獲得的產物是WA1-089的晶型C,其收率99%,化學純度>99%(通過HPLC測得)。
其X-射線粉末衍射圖(XRPD)如圖1所示,差示掃描量熱圖(DSC)如圖2所示,熱重分析圖如圖3所示,衰減的總反射紅外光譜圖如圖4所示。
實施例2
將10mg WA1-089放置DSC儀器上,以20℃/min升溫至205℃,維持3min,再以10℃/min降溫至30℃可得固體結晶。
XRPD和DSC分析數據證實了,獲得的產物是WA1-089的晶型C,其收率99%,化學純度>99%(通過HPLC測得)。
其XRPD圖譜與圖1基本一致, DSC跡線圖譜與圖2基本一致,TGA跡線圖譜與圖3基本一致, FT-IR圖譜與圖4基本一致。
實施例3
將10mg WA1-089放置DSC儀器上,以20℃/min升溫至200℃,維持7min,再以10℃/min降溫至20℃可得固體結晶。
XRPD和DSC分析數據證實了,獲得的產物是WA1-089的晶型C,其收率99%,化學純度>99%(通過HPLC測得)。
其XRPD圖譜與圖1基本一致, DSC跡線圖譜與圖2基本一致,TGA跡線圖譜與圖3基本一致, FT-IR圖譜與圖4基本一致。
實施例4
取10mg的WA1-089的晶型C放置于TGA的白金坩堝內,TA軟件記錄樣品在濕度變化過程中的重量變化,如圖5所示,圖6為結晶產品在20%~80%相對濕度范圍內重量變化為0.67%,結果顯示晶型C不易吸濕。
實施例5
取實施例1中的產品10.28mg,加入3 mL 37℃水中,恒溫10分鐘,過濾得清液。清液利用 HPLC 進行含量測定,測得溶解度為22.56 μg/ml。
- 還沒有人留言評論。精彩留言會獲得點贊!