聯苯類液晶材料的制備方法與流程
本發明涉及液晶材料領域,具體而言,涉及一種聯苯類液晶材料的制備方法。
背景技術:
立體顯示是顯示領域的發展方向,尤其是裸眼3D顯示無需佩戴眼鏡,將會是3D顯示的主流趨勢。
裸眼3D顯示裝置中,需要一種分光的器件將顯示器中有區別的像素信息準確的傳送到人的左、右眼,常用的分光器件有透鏡、狹縫等。透鏡可以大體分為兩種,一種是具有透鏡常規的物理形狀,比如曲面結構,常使用可聚合各向異性的光學材料通過UV聚合或熱聚合等方法制備,但是不適合大規模量產,且良率較低;另一種為液晶透鏡,將液晶材料夾在兩層電極基板中間,利用液晶材料的電響應特性,施加電壓改變液晶層的光學性質,通過電場梯度作用可實現透鏡的效果,且液晶透鏡可以使用現有液晶顯示屏的生產線來進行生產,利于大規模量產和推廣。
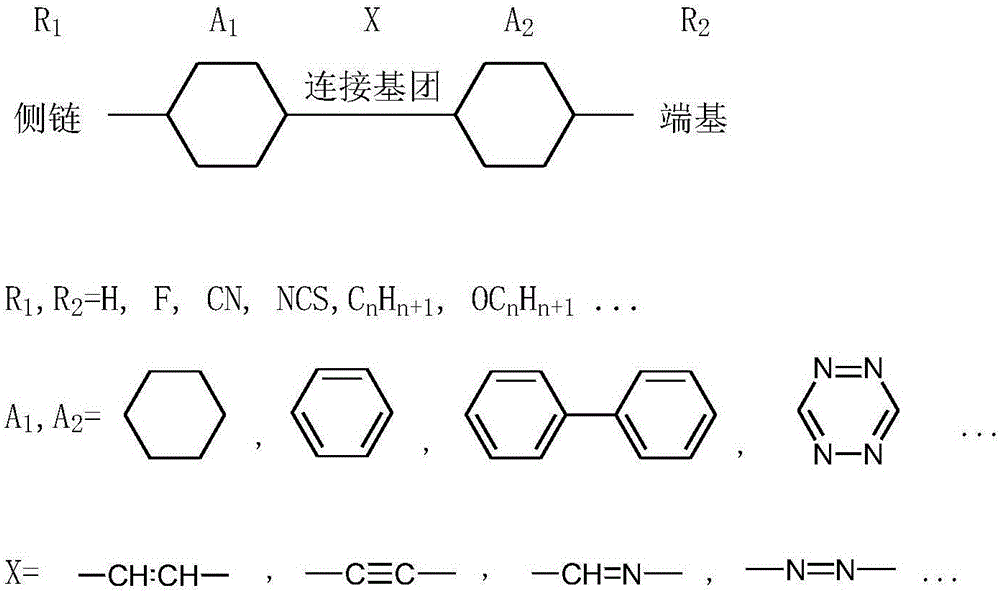
液晶透鏡中常用到的高雙折射率液晶材料一般為有特定結構的小分子混合物,圖1為高雙折射率液晶材料的常見分子結構,兩個帶側鏈(R1)或端基(R2)的環結構(A1,A2)由連接基團(X)連接起來。
圖2為高雙折射率液晶材料中一類常用的聯苯類分子結構,其化學結構對溫度和紫外光等相對穩定,且其對提升液晶材料的雙折射率和清亮點具有顯著的效果。因此被廣泛應用于液晶材料中,也是研究最多的一類液晶材料。
對于圖2的聯苯類液晶分子結構的合成,現有技術中一般采用suzuki反應(鈴木反應)制備此類單體液晶,如圖3所示。
suzuki反應在此類結構制備中具有一些不可替代的優勢,如反應產率 較高,條件相對溫和,反應步驟簡單等。但是,suzuki反應也有一些不可避免的缺點,如催化劑金屬鈀不易去除,殘留的金屬鈀將會較大地降低液晶材料的電阻率,并影響其在顯示器件中的響應電壓;又如反應底物硼酸在suzuki反應條件下會產生自身偶合的副產物,使后處理純化變得復雜。特別是在制備氰基(CN)與異硫腈基(NCS)類液晶單體時,由于自身偶合副產物與最終產物CN,NCS類液晶單體的極性非常接近,自身偶合產物與最終產物混合在一起時,用柱層析色譜法很難將他們分離,導致最終產物純化變得復雜,因此在制備最終反應產物CN,NCS類液晶單體步驟前,必須把自身偶合副產物除去,保證最終產物的純度。
目前,用于suzuki后處理的常用純化方法是柱層析色譜法,即利用產物與副產物的不同極性來對產物和副產物進行分離,從而達到純化目的,但由于柱層析色譜法在制備量變大時,所需花費的準備時間,分離時間和洗脫溶劑大大增加,并且相對應的柱層析柱子也要加大,非常不利于工業化的大量生產。
因此需提供一個簡便有效的方法來制備聯苯類液晶材料,簡化產物提純方法,并且保證產率和純度較高,便于大量生產。
技術實現要素:
為了簡化聯苯類液晶材料的提純方法,本發明提出一種聯苯類液晶材料的制備方法,所述聯苯類液晶材料如式I所示:
所述聯苯類液晶材料的制備包括如下步驟:
1)合成:在第一有機溶劑里,將式II的化合物 式III的化合物和堿,在催化劑的作用下加熱發生鈴木反應:
其中,R1為-F或-NH2,R2為具有3-7個碳原子的烷基、具有3-7個碳原子的不飽和烴基或具有3-7個碳原子的烷氧基,L1,L2,L3和L4分別為H或F,m為0,1或2,X為-Cl,-Br或-I,為
2)提純:上述反應完成后,用索式提取器對步驟1)反應產物進行提純。
進一步的,所述步驟2)的所述提純包括以下步驟:
a.將所述反應液倒入冰水,攪拌并滴加鹽酸至中性,使所述反應液里的所述堿全部反應完;
b.用第二有機溶劑萃取所述步驟a所得的液體,收集有機相,先用鹽水洗所述有機相,接著用無水硫酸鈉干燥所述有機相,最后,旋轉蒸發干所述有機相得褐色固體;
c.在索氏提取器的提取瓶中加入石油醚,提取管加入所述步驟b所得的褐色固體,加熱回流1~2小時后,將提取瓶的石油醚冷卻至室溫,產物固體析出,利用抽濾裝置過濾得到提純產物。
進一步的,所述第二有機溶劑選自二氯甲烷、乙酸乙酯和乙醚中的一種。
進一步的,所述步驟1)的反應在惰性氣氛中進行。
進一步的,所述步驟1)中,所述第一溶劑選自乙醇、異丙醇、1,4-二氧六環、N,N-二甲基甲酰胺和甲苯中的一種。
6進一步的,所述步驟1)中,所述堿選自Cs2CO3、K2CO3、Na2CO3、Li2CO3、K3PO4和Ba(OH)2中的一種。
進一步的,所述步驟1)中,所述催化劑為鈀催化劑。
更進一步的,所述催化劑包括Pd(Pph3)4,Pb(dppf)Cl2和Pd(Pph3)2Cl2中的一種。
進一步的,所述步驟1)的反應時間為2~5小時。
進一步的,所述制備方法進一步包括步驟3):在第三有機溶劑里,加入步驟2)所得的提純產物與CSCl2,所述步驟2)的提純產物與CSCl2發生如下反應:
其中,R1為-NH2,R2為具有3-7個碳原子的烷基、具有3-7個碳原子的不飽和烴基或具有3-7個碳原子的烷氧基,L1,L2,L3和L4分別為H或F,m為0,1或2,為
更進一步的,所述第三有機溶劑選自氯仿和二氯甲烷中的一種。
進一步的,所述制備方法進一步包括步驟3):加入步驟2)所得的提純產物與亞硝酸鈉水溶液,并向反應液滴加溶于氨與氯化銨緩沖溶液的鎳氰化鉀,所述步驟2)的提純產物與鎳氰化鉀發生如下反應:
其中,R1為-NH2,R2為具有3-7個碳原子的烷基、具有3-7個碳原子的不飽和烴基或具有3-7個碳原子的烷氧基,L1,L2,L3和L4分別為H或F,m為0,1或2,為
更進一步的,所述制備方法具體包括:
a:在反應瓶中加入步驟2)所得的提純產物和稀鹽酸,并將反應置于冰浴中,緩慢滴加亞硝酸鈉水溶液,在冰浴條件下反應1~4小時;
b:上述反應完成后,用碳酸鈉溶解將上述反應液調至中性,并向反應液滴加溶于氨與氯化銨緩沖溶液的鎳氰化鉀,然后將反應升至60℃。
附圖說明
圖1為高雙折射率液晶材料常見分子結構圖。
圖2為高雙折射率液晶材料聯苯類分子結構圖。
圖3聯苯類分子的suzuki合成方法。
圖4為實施例1利用索氏提取法提純產物過程圖。
具體實施方式
為了能夠更清楚地理解本發明的上述目的、特征和優點,下面結合附圖和具體實施方式對本發明進行進一步的詳細描述。需要說明的是,在不沖突的情況下,本申請的實施例及實施例中的特征可以相互組合。
在下面的描述中闡述了很多具體細節以便于充分理解本發明,但是,本發明還可以采用其他不同于在此描述的其他方式來實施,因此,本發明并不限于下面公開的具體實施例的限制。
實施例1
本實施例為的制備:
步驟1),合成目標產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-丙基聯苯硼酸、2,6-二氟-4-溴苯胺、催化劑Pd(Pph3)4,、碳酸銫;密封反應系統,將密封系統里的空氣置換成氮氣;注入有N,N-二甲基甲酰胺和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。4小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)的產物進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包 含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流1-2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度98%以上產物。
索氏提取法與柱層析色譜法對比:
本發明利用索式提取器對步驟1)的產物粗產品進行純化處理,產物粗產品包括目標產物、自身耦合副產物以及其他副產物。由于產物,自身偶合副產物及其他副產物在石油醚里的不同溶解性,溶解性為:自身偶合副產物(易溶)>產物(部分溶解)>其他副產物(難溶),且金屬鈀在石油醚里難溶。
如圖4所示,隨著石油醚的加熱回流,由于自身偶合副產物易溶于石油醚中,首先隨石油醚一起流入提取瓶;部分溶解的目標產物也隨著抽提的次數增加,全部被抽提到提取瓶,而難溶于石油醚的其他副產物(金屬鈀催化劑等)則留在提取管中。
待抽提完畢,冷卻提取瓶,在冷卻過程中,目標產物慢慢從石油醚中析出,此時只需抽濾,收集濾固,即可獲得到較純的產物,若需要更純產物,再次重結晶便可。
步驟3),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入上述步驟2)提純后的產物和溶劑二氯甲烷或氯仿,攪拌溶解,并將反應瓶置于冰浴中,待冷至冰浴溫度,緩慢滴加CSCl2,滴加完畢,移除冰浴,反應液慢慢升至室溫后,開始對反應加熱回流。12個小時后,用TLC檢測反應程度;反應 完畢后,加入碳酸鈉溶液,使過量的CSCl2消耗完,然后分離出有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得紅褐色油狀物,利用柱層析色譜法可分離出純度為99%以上的最終產物。
此外,步驟2)對產物的提純,對于步驟3)的NCS類最終產物(或CN類最終產物)提純比較關鍵。由于步驟1)反應過程產生的自身偶合副產物與步驟3)的最終產物極性非常接近,用柱層析色譜法提純最終產物時,若不對自身偶合副產物提前處理,自身偶合副產物與最終產物很難分離,必須經過多次分離,這必然導致產率降低,甚至部分結構經多次分離也無法分離干凈,導致純度無法提升。因此必須提前除去自身偶合副產物,利用以上索氏提取的方法便可巧妙地解決自身偶合副產物對CN類或NCS類聯苯液晶單體最終產物提純的影響。
本發明提供的聯苯類液晶材料的制備方法,操作方便,儀器裝置簡單,只需索氏提取裝置和使用抽濾裝置即可;節省溶劑,利用索氏提取器,相比柱層析色譜法只需較少的溶劑即可完成抽提;提純損失少,產率在一般80%以上,比柱層析色譜法的產率高;獲取的產物純度高,普遍在97%以上;由于儀器,步驟簡單,所需溶劑少,耗費時間少等優勢,因此,此方法在工業化的大量生產中具有可行性。
實施例2
本實施例為的制備:
步驟1),合成目標產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入2,6-二氟-4-氯苯胺、催化劑Pb(dppf)Cl2和碳酸鉀;密封反應系統,將密封 系統里的空氣置換成氮氣;注入有1,4-二氧六環和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。5小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)的產物進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度98%以上產物。
索氏提取法與柱層析色譜法對比:
步驟3),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入上述步驟2)提純后的產物和溶劑二氯甲烷或氯仿,攪拌溶解,并將反應瓶置于冰浴中,待冷至冰浴溫度,緩慢滴加CSCl2,滴加完畢,移除冰浴,反應液慢慢升至室溫后,開始對反應加熱回流。12個小時后,用TLC檢測反應程度;反應完畢后,加入碳酸鈉溶液,使過量的CSCl2消耗完,然后分離出有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得紅褐色油狀物,利用柱層析色譜法可分離出純度為99%以上的最終產物。
實施例3
本實施例為的制備:
步驟1),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-丙基苯硼酸、4-碘苯胺、催化劑Pd(Pph3)2Cl2、碳酸鈉;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。2小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)的產物進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度99%以上產物。
索氏提取法與柱層析色譜法對比:
本發明提供的聯苯類液晶材料的制備方法,操作方便,儀器裝置簡單,只需索氏提取裝置和使用抽濾裝置即可;節省溶劑,利用索氏提取器,相比柱層析色譜法只需較少的溶劑即可完成抽提;提純損失少,產率在一般80%以上,比柱層析色譜法的產率高;獲取的產物純度高,普遍在97%以上;由于儀器,步驟簡單,所需溶劑少,耗費時間少等優勢,因此,此方法在工業化的大量生產中具有可行性。
實施例4
本實施例為的制備:
步驟1),制備目標產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-(反式-4-戊基環己基)苯硼酸、2,6-二氟-4-溴苯胺、催化劑Pb(dppf)Cl2和Ba(OH)2;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。4小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度97%以上產物。
索氏提取法與柱層析色譜法對比:
步驟3),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入上述步驟2)提純后的產物和溶劑二氯甲烷或氯仿,攪拌溶解,并將反應瓶置于冰浴中,待冷至冰浴溫度,緩慢滴加CSCl2,滴加完畢,移除冰浴,反應液慢慢升至室溫后,開始對反應加熱回流。12個小時后,用TLC檢測反應程度;反應完畢后,加入碳酸鈉溶液,使過量的CSCl2消耗完,然后分離出有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得紅褐色油狀物,利用柱層析色譜法可分離出純度為99%以上的最終產物。
實施例5
本實施例為的制備:
步驟1),制備目標產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入2,6-二氟-4-溴苯胺、催化劑Pd(Pph3)4和K3PO4;密封反應系統,將密封系統里的空氣置換成氮氣;注入有甲苯和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。4小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏 提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度98%以上產物。
索氏提取法與柱層析色譜法對比:
步驟3),制備最終產物:
反應瓶內放入攪拌子,加入上述步驟2)提純后的產物和稀鹽酸,并將反應置于冰浴中,緩慢滴加亞硝酸鈉水溶液;在冰浴條件下反應2小時候后,用碳酸鈉溶解將上述反應液調至中性,并向反應液滴加溶于氨與氯化銨緩沖溶液的鎳氰化鉀,然后將反應升至60℃,反應15分鐘,對反應液進行抽濾并用少量水洗濾固,收集濾液,用二氯甲烷對濾液經行萃取,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鎂干燥,最后,旋轉蒸發干有機相得淡黃色固體,利用柱層析色譜法可分離出純度為99%以上的最終產物。
實施例6
本實施例為的制備:
步驟1),制備目標產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-丙基硼酸苯、2,6- 二氟-4-溴苯胺、催化劑Pd(Pph3)2Cl2、碳酸鈉;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。2.5小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流3小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度98%以上產物。
索氏提取法與柱層析色譜法對比:
步驟3),制備最終產物:
反應瓶內放入攪拌子,加入上述步驟2)提純后的產物和稀鹽酸,并將反應置于冰浴中,緩慢滴加亞硝酸鈉水溶液;在冰浴條件下反應2小時候后,用碳酸鈉溶解將上述反應液調至中性,并向反應液滴加溶于氨與氯化銨緩沖溶液的鎳氰化鉀,然后將反應升至60℃,反應15分鐘,對反應液進行抽濾并用少量水洗濾固,收集濾液,用二氯甲烷對濾液經行萃取,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鎂干燥,最后,旋轉蒸發干有機相得淡黃色固體,利用柱層析色譜法可分離出純度為99%以上的最終產物。
實施例7
本實施例為的制備:
步驟1),制備目標產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-丙氧基環己基硼酸、2,6-二氟-4-溴苯胺、催化劑Pd(Pph3)2Cl2、碳酸鈉;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。3小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流1小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度98%以上產物。
索氏提取法與柱層析色譜法對比:
步驟3),制備最終產物:
反應瓶內放入攪拌子,加入上述步驟2)提純后的產物和稀鹽酸,并將反應置于冰浴中,緩慢滴加亞硝酸鈉水溶液;在冰浴條件下反應2小時候后,用碳酸鈉溶解將上述反應液調至中性,并向反應液滴加溶于氨與氯化銨緩沖溶液的鎳氰化鉀,然后將反應升至60℃,反應15分鐘,對反應液進行抽濾并用少量水洗濾固,收集濾液,用二氯甲烷對濾液經行萃取,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鎂干燥,最后,旋轉蒸發干有機相得淡黃色固體,利用柱層析色譜法可分離出純度為99%以上的最終產物。
實施例8
本實施例為的制備:
步驟1),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入1-氟-4-溴苯、催化劑Pd(Pph3)4和碳酸鈉;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。5小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏 提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度97%以上產物。
索氏提取法與柱層析色譜法對比:
本發明提供的聯苯類液晶材料的制備方法,操作方便,儀器裝置簡單,只需索氏提取裝置和使用抽濾裝置即可;節省溶劑,利用索氏提取器,相比柱層析色譜法只需較少的溶劑即可完成抽提;提純損失少,產率在一般80%以上,比柱層析色譜法的產率高;獲取的產物純度高,普遍在97%以上;由于儀器,步驟簡單,所需溶劑少,耗費時間少等優勢,因此,此方法在工業化的大量生產中具有可行性。
實施例9
本實施例為的制備:
步驟1),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-戊基聯苯硼酸、1,2-二氟-4-溴苯、催化劑Pd(Pph3)2Cl2、碳酸鈉;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。5小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包 含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度97%以上產物。
索氏提取法與柱層析色譜法對比:
實施例10
本實施例為的制備:
步驟1),制備最終產物:
搭建回流反應裝置,向反應瓶內放入攪拌子,加入4-戊基聯苯硼酸、1,2-二氟-4-溴苯、催化劑Pd(Pph3)4、碳酸鈉;密封反應系統,將密封系統里的空氣置換成氮氣;注入有異丙醇和水,攪拌并慢慢加熱回流,確保系統在氮氣保護下反應。5小時后,用TLC檢測反應程度。
步驟2),用索式提取器對步驟1)粗產品進行提純:
反應完畢后,將反應液倒入冰水,滴加稀鹽酸并攪拌,滴至中性,使反應液里的碳酸鈉全部反應完;用二氯甲烷萃取上述液體,收集有機相,所得有機相用鹽水洗兩次后,接著用無水硫酸鈉或無水硫酸鎂干燥,最后,旋轉蒸發干有機相得褐色固體,此時褐色固體為suzuki反應的粗產品,包含目標產物、自身耦合副產物以及其他副產物。搭建好提取裝置,在索氏提取器的提取瓶內倒入石油醚;將上述褐色固體裝于濾紙筒里面,將濾紙筒放置在索氏提取器的提取管;加熱回流2小時,將提取瓶的石油醚冷卻至室溫或更低,產物固體析出,利用抽濾裝置過濾,獲得純度97%以上產物。
索氏提取法與柱層析色譜法對比:
上述僅為本發明的優選實施例而已,并不用于限制本發明,對于本領域的技術人員來說,本發明可以有各種更改和變化。凡在本發明的精神和原則之內,所作的任何修改、等同替換、改進等,均應包含在本發明的保護范圍之內。
- 還沒有人留言評論。精彩留言會獲得點贊!