環氧酮中間體的新鹽的制作方法
本發明涉及環氧酮化合物的新鹽,其是卡非佐米(Carfilzomib)制備中的關鍵中間體。
背景技術:
卡非佐米是國際公認的(S)-4-甲基-N-((S)-1-(((S)-4-甲基-1-((R)-2-甲基環氧乙烷-2-基)-1-戊酮-2-基)氨基)-1-氧代-3-苯基丙烷-2-基)-2-((S)-2-(2-嗎啉乙酰胺基)-4-苯基丁酰胺基)戊酰胺的非專有名稱,并具有C40H57N5O7的經驗式和719.91的分子量。
已知卡非佐米可治療性地用于患有多發性骨髓瘤的患者,并且以KyprolisTM名稱在美國銷售。
卡非佐米首次在EP1781688B1中描述。卡非佐米的合成方法以流程圖總結于該'688專利中(參見方案1)。
方案1
(2S)-2-氨基-4-甲基-1-[(2R)-2-甲基環氧乙烷-2-基]戊-1-酮(在下文中稱為式(I)化合物)及其鹽是卡非佐米制備中的關鍵中間體。
EP1781688B1中的方法涉及三氟乙酸鹽形式的式(I)的化合物,在方案1中稱為化合物E并在下文中稱為TFA-(I),其根據Biorg.Med.Chem.Lett.,1999,9,2283-2288制備。
WO2009045497A1公開了TFA-(I)的具體晶體形式、制備所述TFA-(I)和使用該TFA-(I)制備卡非佐米的方法。WO2009045497A1的實施例21還公開了制備式(I)的環氧酮的甲酸鹽,其以油形式獲得并隨后用于制備卡非佐米。
技術實現要素:
本發明提供了式(I)化合物的穩定鹽及其制備方法和其用于制備卡非佐米的用途。
本發明的一個方面涉及式MSA-(I)的甲磺酸鹽。
本發明的甲磺酸鹽MSA-(I)表現出優異的溶解性和穩定性性質。具體地,本發明的甲磺酸鹽MSA-(I)與文獻中公開的三氟乙酸鹽TFA-(I)相比更穩定。因此,本發明的甲磺酸鹽在溫度方面不需要任何特殊的儲存條件,以使得其可以在用于制備卡非佐米之前儲存較長時間。
在某些實施方式中,本發明的甲磺酸鹽MSA-(I)是結晶的。
在某些實施方式中,本發明的結晶甲磺酸鹽MSA-(I)示出了包含至少以下峰(2-θ,2θ)的XRPD圖譜:在5.4和10.7(±0.2度)處,更優選在5.4、10.7、20.3、22.3和27.5(±0.2度)度,甚至更優選在5.4、8.2、10.7、12.3、17.2、20.3、21.4、22.3、26.8、27.5、29.6和32.9(±0.2度)處。圖1示出了本文中公開的結晶甲磺酸鹽MSA-(I)的X射線粉末衍射圖譜。
本文中使用的術語結晶是指本發明的甲磺酸鹽MSA-(I)的至少20%(重量/重量)是結晶的,優選本發明的甲磺酸鹽MSA-(I)的超過50%(重量/重量)是結晶的,更優選本發明的甲磺酸鹽MSA-(I)的超過70%(重量/重量)是結晶的,且甚至更優選本發明的甲磺酸鹽MSA-(I)的超過90%(重量/重量)是結晶的。
本發明的另一方面提供了通過包括式(II)的化合物與甲磺酸在合適的溶劑中的反應的方法來制備本發明的甲磺酸鹽MSA-(I)的方法,其中PG為合適的保護基團(參見方案2)。
方案2
在某些實施方式中,在式(II)的化合物到甲磺酸鹽MSA-(I)的直接轉化中使用的溶劑是選自二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、甲苯、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(MeTHF)及其任意組合的有機溶劑。優選地,所述溶劑包含二氯甲烷、甲苯、乙腈或其任意組合。
在本發明的另一方面中,甲磺酸鹽MSA-(I)通過包括如下步驟的方法制備:(a)將式(II)的化合物與合適的酸在合適的溶劑中反應以獲得式(I)的化合物或其鹽;和(b)將式(I)的化合物或其鹽與甲磺酸在合適的溶劑中反應以得到甲磺酸鹽MSA-(I),其中PG為合適的保護基團(參見方案3)。
方案3
在某些實施方式中,步驟(a)中的式(II)化合物可溶解于選自二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、甲苯、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(MeTHF)及其任意組合的有機溶劑。優選地,式(II)的化合物溶解于二氯甲烷、甲苯或其任意組合。
在某些實施方式中,步驟(a)中的合適酸選自氫溴酸、鹽酸、硫酸、磷酸、硝酸、三氟乙酸、甲磺酸、對甲苯磺酸、2-羥基乙基磺酸 等。優選地,所述酸是三氟乙酸。
在優選的實施方式中,在步驟(a)中獲得的式(I)化合物是鹽的形式,更優選是式TFA-(I)的三氟乙酸鹽形式。
在優選的實施方式中,在步驟(a)中獲得的三氟乙酸鹽TFA-(I)與甲磺酸在合適的溶劑中反應以得到甲磺酸鹽MSA-(I)。
在某個實施方式中,鹽形式的、優選三氟乙酸鹽TFA-(I)形式的式(I)化合物然后用堿中和以形成式(I)的化合物,其然后與甲磺酸在合適的溶劑中反應以得到甲磺酸鹽MSA-(I)。
在某些實施方式中,在步驟(b)中使用的溶劑是選自二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、甲苯、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(MeTHF)及其任意組合的有機溶劑。優選地,在步驟(b)中使用的溶劑包含二氯甲烷、甲苯或其任意組合。
可選地,甲磺酸鹽MSA-(I)可以通過包括以下步驟的方法制備:(a)將式(II)的化合物與合適的酸在合適的溶劑中反應,并用堿中和所形成的鹽以形成式(I)的化合物;(b)將式(I)的化合物與草酸在合適的溶劑中反應以得到草酸鹽,在下文中稱為OX-(I);和(c)將草酸鹽OX-(I)在合適的溶劑中轉化為甲磺酸鹽MSA-(I);其中PG為合適的保護基團(參見方案4)。
方案4
主要在式(II)的化合物顯示出低色譜純度(HPLC純度低于95%)的情況中,草酸鹽OX-(I)的制備和分離可以是MSA-(I)制備中的關鍵步驟。因此,該草酸鹽OX-(I)的分離允許工藝雜質的有效純化,以使得可以進行從異亮氨酸制備MSA-(I)的整個方法而不需要執行任何色譜純化步驟,從而使本發明的方法適合于工業規模。
在某些實施方式中,本發明的草酸鹽OX-(I)是結晶的。
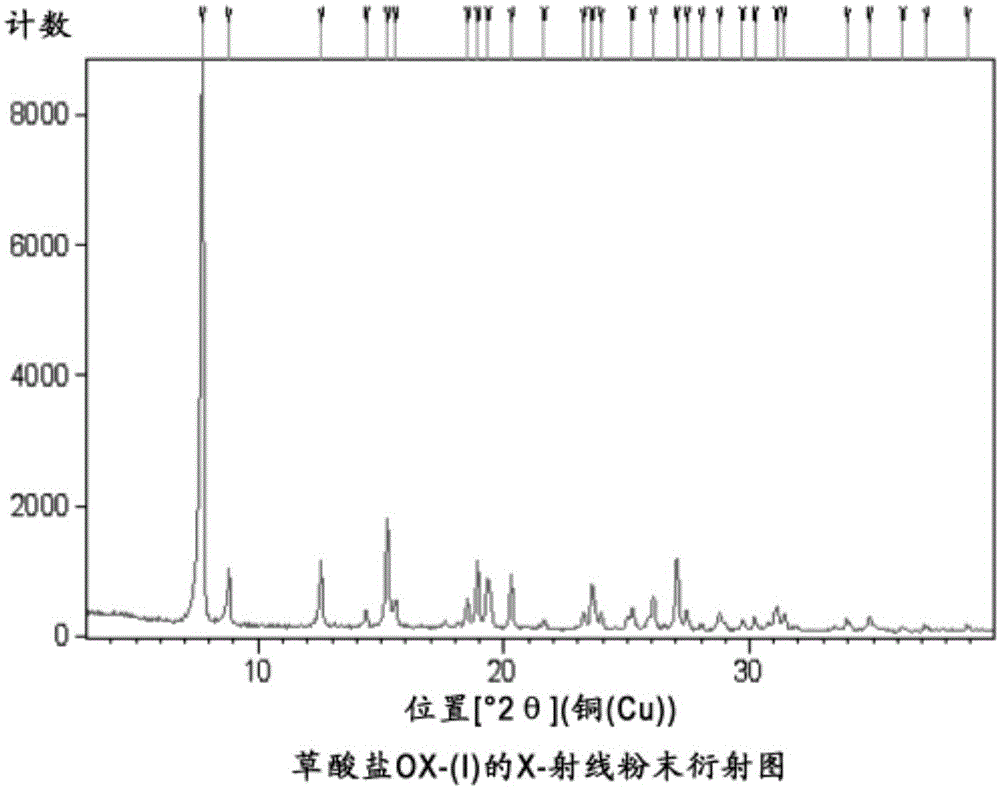
在某些實施方式中,本發明的結晶草酸鹽OX-(I)顯示了包含至少以下峰(2θ)的XRPD圖譜:在7.7和8.8(±0.2度)處,更優選在7.7、8.8、12.6、15.2、18.9和27.0(±0.2度)處,甚至更優選在7.7、8.8、12.6、14.4、15.2、15.6、18.5、18.9、19.3、20.3、23.2、23.6、24.0、25.2、26.1、27.0、27.4、28.8、31.1、31.4、34.0、34.9(±0.2度)處。圖2示出了本文中公開的結晶草酸鹽OX-(I)的X射線粉末衍射圖譜。
本文中使用的術語結晶是指本發明的草酸鹽OX-(I)的至少20%(重量/重量)是結晶的,優選本發明的草酸鹽OX-(I)的超過50%(重量/重量)是結晶的,更優選本發明的草酸鹽OX-(I)的超過70%(重量/重量)是結晶的,且甚至更優選本發明的草酸鹽的OX-(I)超過90%(重量/重量)是結晶的。
在某些實施方式中,步驟(a)中的式(II)化合物可溶解于選自二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、甲苯、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(Me THF)及其任意組合的有機溶劑。優選地,式(II)的化合物溶解于二氯甲烷、甲苯或其混合物。
在某些實施方式中,步驟(a)中的合適酸選自氫溴酸、鹽酸、硫酸、磷酸、硝酸、三氟乙酸、甲磺酸、對甲苯磺酸、2-羥基乙磺酸等。優選地,所述酸是三氟乙酸。
在某些實施方式中,在步驟(b)中使用的溶劑是選自二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、甲苯、乙腈、四氫呋喃(THF)、 2-甲基四氫呋喃(MeTHF)及其任意組合的有機溶劑。優選地,在步驟(b)中使用的溶劑是二氯甲烷、甲基叔丁基醚(MTBE)、甲苯或其混合物。
在某些實施方式中,在步驟(b)中得到的草酸鹽OX-(I)可以通過在合適的溶劑中重結晶進一步純化。用于草酸鹽OX-(I)的重結晶的合適溶劑的非限制性實例包含二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙基醚、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(MeTHF)或其任意組合。優選地,草酸鹽OX-(I)可以在乙腈中重結晶。
在某些實施方式中,步驟(c)包括草酸鹽OX-(I)與甲磺酸在合適的溶劑中的反應以直接得到甲磺酸鹽MSA-(I)。可選地,在步驟(b)中得到的草酸鹽OX-(I)首先用堿中和以形成式(I)的化合物,其隨后與甲磺酸在合適的溶劑中反應以形成甲磺酸鹽MSA-(I)。
在某些實施方式中,在步驟(c)中使用的溶劑是選自二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(MeTHF)及其任意組合的有機溶劑。優選地,步驟(c)中使用的溶劑是甲基叔丁基醚(MTBE)、乙腈、甲苯或其混合物。
在某些實施方式中,所獲得的甲磺酸鹽MSA-(I)可以進一步從合適的溶劑分離。從其中可以分離MSA-(I)的溶劑的非限制性實例是二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙醚、甲苯、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(MeTHF)或其任意組合。優選地,從其中可以分離MSA-(I)的溶劑是甲基叔丁基醚(MTBE)、乙腈、甲苯或其混合物。
在某些實施方式中,本發明的甲磺酸鹽MSA-(I)可以通過在合適的溶劑中重結晶的方法進一步純化。對甲磺酸鹽MSA-(I)的重結晶來 說,合適的溶劑的非限制性實例是二氯甲烷、甲苯、乙酸乙酯、乙酸異丙酯、乙酸異丁酯、乙酸丁酯、乙酸丙酯、乙醚、甲基叔丁基醚(MTBE)、異丙基醚、乙腈、四氫呋喃(THF)、2-甲基四氫呋喃(甲基THF)或其任意組合。優選地,甲磺酸鹽MSA-(I)可在乙腈中重結晶。
在本發明中使用的式(II)的化合物可以根據在Bioorg.Med.Chem.Lett.1999,9,2283-2288或US20050256324中公開的合成策略來產生,其中PG為合適的保護基團。
在某個實施方式中,式(II)的化合物根據包括以下步驟的方法獲得:(a)將式(VII)的化合物的活化衍生物與N,O-二甲基羥胺反應以得到式(VI)的化合物;(b)將式(VI)的化合物與2-溴代異丙烯基溴化鎂反應以得到式(V)的化合物;和(c)將式(V)的化合物氧化以得到式(II)的化合物;或者,可選地,(c-i)將式(V)的化合物還原以得到式(IV)的化合物;(c-ii)將式(IV)的化合物氧化以得到式(III)的化合物;和(c-ⅲ)將式(III)的化合物氧化以得到式(II)的化合物,其中PG為合適的保護基團(參見方案5)。
方案5
在某些實施方式中,PG選自叔丁氧基羰基(Boc)、芴-9-基甲氧基羰基(Fmoc)、三氯乙氧基羰基(Troc)和芐氧基羰基(Cbz)。 優選地,PG是叔丁氧基羰基(Boc)。
在本發明的另一個方面,甲磺酸鹽MSA-(I)作為中間體用于制備卡非佐米。
在本發明的某個實施方式中,甲磺酸鹽MSA-(I)進一步與式A的化合物的活化衍生物偶聯以獲得卡非佐米(參見方案6):
方案6
在本發明的另一個方面,草酸鹽OX-(I)作為中間體用于制備卡非佐米。
實驗實施例
以下實施例進一步說明本發明,但其當然不應被解釋為以任何方式限制本發明的范圍。
一般實驗條件:
HPLC方法
使用Waters對稱C18,5μm,4.6×250毫米柱,在30℃下進行色譜分離。
通過在4L蒸餾水中溶解1.42g Na2HPO4和1.2g NaH2PO4制備流動相A。pH為7.0-7.1。
流動相B為乙腈。
色譜法編程如下:初始0-4分鐘。等度80%流動相A,4-8分鐘。線性梯度至70%流動相A,8-30分鐘。線性梯度至20%流動相A。30-35分鐘。等度20%流動相A,35-35.1分鐘。線性梯度至80%流 動相A,35.1-45分鐘。等度80%流動相A。
該色譜儀配備了205nm UV檢測器。流速為1.0ml/分鐘。
通過在10.0ml乙腈/水(50/50)中溶解約100mg樣品來制備MSA-(I)和OX-(I)的測試樣品。注射5μl測試樣品。
X射線粉末衍射(XRPD):
在Si單晶支架上使用PANalyticalXPERT-3XRPD進行XRPD。在實驗中使用的XRPD方法的細節在以下列出。
實施例1:制備式(VI)的化合物,其中PG為Boc
將1050g Boc-L-亮氨酸(式(VII)的化合物,其中PG為Boc)和7.0L二氯甲烷裝入反應器中,加入936g N-甲基嗎啉。攪拌混合物直至完全溶解并冷卻至-20℃。加入575g氯碳酸異丁酯。將反應混合物在-20℃下攪拌30分鐘。分批加入411g N,O-二甲基羥胺鹽酸鹽,并將該混合物加熱至15-25℃。16小時后,加水直至混合物完全溶解。收集有機相,并用10%檸檬酸水溶液洗滌兩次。收集有機層,隨后用飽和NaHCO3水溶液、水和飽和NaCl水溶液洗滌。通過蒸餾除去溶劑,以產出1057g油狀的式(VI)化合物,其中PG為Boc。
實施例2:制備式(V)的化合物,其中PG為Boc
將257g鎂屑、0.5g碘和400ml MeTHF裝入反應器,并加熱至60-70℃。加入200ml預先制備的1680g 2-溴丙烯在4.06L MeTHF 中的溶液。將該混合物在50℃下攪拌1小時,然后冷卻至10-30℃。將上層澄清溶液轉移至清潔反應器中并冷卻至-10至0℃。加入根據前述實施例1制備的700g式(VI)的化合物(其中PG為Boc)在1.0L MeTHF中的溶液。將反應混合物在0-5℃下攪拌16小時。然后將反應混合物加入4.60kg 15%乙酸水溶液中,并將所得混合物在0-5℃下攪拌30分鐘。收集有機層。將水層用1.4L庚烷洗滌兩次。合并有機層,并首先用1M鹽酸水溶液,然后用水和最后用氯化鈉水溶液洗滌。通過蒸餾除去溶劑以產出649g油狀的式(V)的化合物,其中PG為Boc。
實施例3:制備式(IV)的化合物,其中PG為Boc
將根據前述實施例2制備的649g式(V)的化合物(其中PG為Boc)和1.140kg CeCl3·7H2O在0℃下加入到26.0L乙醇中。將混合物在0-5℃下攪拌至完全溶解。加入115g NaBH4,攪拌2小時。將340g乙酸加入到反應中并攪拌30分鐘。加入5.0kg水。通過減壓蒸餾除去乙醇。加入5.0L乙酸乙酯和6.8kg水,收集有機層并用水和飽和NaCl水溶液洗滌。通過減壓蒸餾除去溶劑。加入0.948kg乙腈,將混合物冷卻至-20℃到0℃,并過濾所得懸浮液。從乙腈中重結晶粗物質以得到420g式(IV)的化合物,其中PG為Boc。
實施例4:制備式(II)的化合物,其中PG為Boc
將0.5g乙酰丙酮氧釩(vanadyl acetylacetonate)加入根據前述實施例3制備的50.0g式(IV)的化合物(其中PG為Boc)在2.0L二氯甲烷中的溶液中。加入50.0g叔丁基過氧化氫,并將所得混合物在0-5℃下攪拌1小時。加熱至15℃后,將2.0g乙酰丙酮氧釩在4小時內以4份加入。將反應混合物在15-20℃下攪拌30分鐘。然后,過濾所得混合物,并將過濾的溶液冷卻至0℃。將1000g 10%Na2S2O3水溶液和600g 8%NaHCO3水溶液加入過濾的溶液中。收集有機層。將水層用二氯甲烷洗滌。合并有機層,并首先用水和然后用飽和NaCl 水溶液洗滌。將含有式(III)的化合物(其中PG為Boc)的這種有機層在0-5℃下加入200g戴斯-馬丁高碘烷在752ml二氯甲烷中的溶液中。將反應混合物在0-5℃下攪拌2小時。過濾所得懸浮液,將過濾的溶液冷卻至0-5℃,并隨后用8%NaHCO3水溶液、10%Na2S2O3水溶液、水和飽和NaCl水溶液洗滌。通過蒸餾除去溶劑以得到黃色油狀物。將產物通過柱色譜純化以得到29g式(II)的化合物,其中PG為Boc。
實施例5:制備MSA-(I)
將根據前述實施例4制備的2.0g式(II)的化合物(其中PG為Boc)與6.6ml甲苯在0-5℃下混合并攪拌。在0-5℃下加入4.2g三氟乙酸并在溫度20-30℃下維持2小時。將混合物冷卻至0-5℃,并加入0.71g甲磺酸。將混合物在0-5℃下攪拌0.5小時。通過減壓蒸餾除去溶劑。在20-30℃下將6ml乙腈加入所獲得的殘余物,并攪拌混合物直至完全溶解。將12ml MTBE加入溶液,并將混合物在20-30℃下攪拌0.5小時。將混合物冷卻至0-5℃并將溫度在0-5℃維持0.5小時。過濾固體,并用MTBE洗滌以得到1.5g MSA-(I),純度為99.5%(%面積,HPLC)。
實施例6:制備MSA-(I)
將根據前述實施例4制備的10g式(II)的化合物(其中PG為Boc)與30.1ml二氯甲烷在0-5℃下混合并攪拌。在0-5℃下加入21.0g三氟乙酸,并將溫度在20-30℃維持2小時。通過減壓蒸餾除去溶劑。加入100g水和117ml庚烷。收集水層并用庚烷洗滌。丟棄有機層。將200g 8%NaHCO3水溶液和30g飽和NaCl水溶液加入水層。將水層用75.19ml二氯甲烷洗滌兩次。收集有機層,合并,并用飽和NaCl水溶液洗滌。加入1.77g甲磺酸。將混合物在15-25℃下攪拌30分鐘。通過減壓蒸餾除去溶劑。加入250ml MTBE,將混合物冷卻至5-10℃。過濾固體并用乙腈洗滌以得到4.1g MSA-(I),純度:98%(%面 積,HPLC)。
從乙腈中將所獲得的MSA-(I)重結晶以得到3.2g MSA-(I),純度:99.5%(%面積,HPLC)。
實施例7:制備OX-(I)
將根據前述實施例4制備但未進行色譜純化步驟的10g的式(II)粗化合物(其中PG為Boc)與30.1ml二氯甲烷在0-5℃下混合并攪拌。在0-5℃下加入21.0g三氟乙酸,并將溫度在20-30℃下維持2小時。通過減壓蒸餾除去溶劑。加入100g水和117ml庚烷。收集水層并用庚烷洗滌。丟棄有機層。將200g 8%NaHCO3水溶液和30g飽和NaCl水溶液加入水層。將水層用75.19ml二氯甲烷洗滌兩次。收集有機層,合并,并用飽和NaCl水溶液洗滌。加入44.3g草酸在MTBE中的7.5%重量/重量溶液。將混合物在15-25℃下攪拌兩小時。過濾固體并用MTBE洗滌以得到4.0g OX-(I),純度:95.1%(%面積,HPLC)。
所獲得的OX-(I)在乙腈中重結晶以得到3.2g OX-(I),純度:97.3%(%面積,HPLC)。
實施例8:從OX-(I)制備MSA-(I)
將根據前述實施例7制備的3.2g OX-(I)與32.5ml乙腈混合。加入1.20g甲磺酸并將混合物在10-20℃下攪拌1小時。加入81ml MTBE并將混合物在10-20℃下攪拌1小時。過濾固體并用MTBE洗滌以得到2.78g MSA-(I),純度:99.0%(%面積,HPLC)。
所獲得的MSA-(I)在乙腈中重結晶以得到2.33g MSA-(I),純度:99.8%(%面積,HPLC)。
實施例9:從MSA-(I)制備卡非佐米
將3.280g式A的化合物、2.012g甲磺酸鹽MSA-(I)、1.775g EDC·HCl、1.152g HOBt·H2O和100ml DMF裝入燒瓶中。用N2吹掃 燒瓶,同時將混合物劇烈攪拌并冷卻至-5℃。加入1.52ml DIPEA。
在加入完成后,將反應混合物在-5℃下攪拌1小時,并通過加入66ml飽和NaHCO3水溶液淬滅。將所獲得的漿液用乙酸乙酯萃取,并將有機層用飽和NaHCO3水溶液洗滌。減壓濃縮有機層以得到卡非佐米。
實施例10:MSA-(I)與TFA-(I)的穩定性比較
下表顯示了與現有技術中公開的三氟乙酸鹽TFA-(I)相比,本發明的甲磺酸鹽MSA-(I)的不同穩定性。
在60℃下儲存后的HPLC純度(面積%)。
在60℃下儲存后的外觀
- 還沒有人留言評論。精彩留言會獲得點贊!