5?(羥甲基)?糠醛還原產物的酸催化酰化的制作方法
發明領域
本披露涉及可用作聚合物合成的單體以及各種工業化學品的前體的糖衍生的二醇。特別地,本發明涉及呋喃酯及其合成方法。
背景
常規地,商品化學品大部分地由化石或石油基烴制成。近年來,隨著對可再生資源的興趣的增長,更有效技術的進展使可再生或“綠色”原料成為可行的經濟替代品。一類可再生的生物衍生的烴資源已經衍生自糖。然而,由于糖分子的可以容易地降解(特別是在較高的溫度下)的固有復雜官能度,這些材料對于工業用途傳統上已經受到有限關注。
最近的研究工作已經集中在生產具有較小官能度的糖基原料上。呋喃是可以容易地衍生自糖的各種通用的分子平臺。除了其他工業成分外,這些分子具有在制備聚合物、藥物或溶劑中特別有用的結構特征。
引起關注的化合物是5-(羥甲基)-糠醛(hmf),果糖的主要脫水產物,一種廉價且豐富的單糖(方案a)。
方案a.果糖的酸催化脫水環化,產生hmf
hmf的雙醇和醛部分使其能夠用作許多潛在的工業材料如聚合物、溶劑、表面活性劑和藥物的前體。
直接使用hmf的缺點是其在空氣的存在下容易聚合和氧化降解的傾向,因此需要存在抗氧化劑以改善壽命。hmf可以被還原為更穩定的分子,如呋喃-2,5-二甲醇(縮寫為fdm)和雙-2,5-(羥甲基)-四氫呋喃(縮寫為bhmthf),如方案b所示。
方案b.a)fdm和bhmthf的b)順式、c)反式非對映異構體的結構
fdm由hmf的部分氫化(醛還原)產生,如方案c所描繪,而徹底的氫化產生飽和的類似物bhmthf,典型地以9:1順式與反式非對映體比例產生,如方案d中。(參見例如美國專利號7,317,116或7,393,963b2。)
方案c.來自hmf的部分氫化的fdm
方案d.來自hmf的徹底催化還原的順式/反式bhmthf
這些化合物的雙官能性質使得它們能夠容易地作為起始材料用于各種化學合成中,并且用作石油基芳香族烴的可行替代品。這些材料可能是,例如,對于聚酯、聚氨酯泡沫、增塑劑、樹脂、表面活性劑、分散劑、潤滑劑、農業化學品有價值的前體,或作為溶劑、粘合劑、或濕潤劑。在四氫呋喃環的2和5位處的羥甲基附屬物提供兩個手性中心,其允許bhmthf在不對稱有機合成的新興領域中是藥物或手性助劑的可能的支架。fdm和bhmthf的應用需要提供從hmf大規模制造這些化合物的既成本效益又流線型的方法。
使用fdm和/或bhmthf制備分子衍生物的探索受到這些原料的過高成本(例如,商業上約$200/克)的限制。為了有效地與衍生自化石基烴源的化學前體競爭,從普通農業來源制備hmf衍生物需要更好的轉化和生產所希望的衍生物和中間體化合物的方法。直到最近,呋喃化合物的大規模商業化已經是相對地成本低效的。
增強使用fdm和bhmthf作為前體或原料的起始平臺的一種方法是將其轉化為酯。已建立的酯的商業合成典型地需要通過布朗斯特酸催化的用羧酸的直接醇酰化。該方案通常被指定為費歇爾-斯皮爾酯化(fischer-speieresterification)。典型地,強無機酸如h2so4和hcl用作該催化劑。這些強酸是容易獲得的、廉價的材料但是難以再生,這增加了廢物流。此外,這些酸可以按照一種所不希望的方式通過其陰離子部分的加成進行反應,形成副產物(如硫酸酯)。
雖然最近已經發展了一些穩健的方法,其中獲得了較高的純度。(總體上參見,x.tong等人,“生物質到化學品:通過催化方法將糖轉化為呋喃衍生物,”應用催化a:總論(“biomassintochemicals:conversionofsugarstofuranderivativesbycatalyticprocesses,”appliedcatalysisa:general)385(2010)1-13),開發更有效的純化的問題仍然存在。
已經嘗試使用固體樹脂催化劑克服這些問題的努力,但是對于大體積尚未成功。不幸的是,傳統使用的固體酸不是水解穩定的并且甚至痕量的水可能不利地影響催化活性。均相金屬催化劑還由于其對水解的敏感性而顯示出有限的活性,這降低了催化活性。
鑒于這些使用常規方法的缺點,仍然存在對以下方法的需要,在該方法中人們能夠在用于制備用作前體的hmf衍生的化合物的經濟的催化劑負載量下獲得較高的酯產率。
發明概述
本披露部分地涉及一種用于5-(羥甲基)-糠醛(hmf)的還原產物、特別是或者呋喃-2,5-二甲醇(fdm)或雙-2,5-(羥甲基)-四氫呋喃(bhmthf)的酸催化酰化的方法。該方法涉及在耐水性路易斯酸催化劑的存在下,在足以產生相應的酯產物混合物的反應溫度和時間下在反應中使fdm或bhmthf與過量的有機酸反應。在從約150℃至約250℃的范圍內的溫度下進行該酯化。
本發明方法的另外的特征和優點將披露于以下詳細說明中。應理解的是上述概述以及以下詳細說明和實例都僅代表本發明,并且旨在提供用于理解如所要求保護的本發明的綜述。
附圖簡要說明
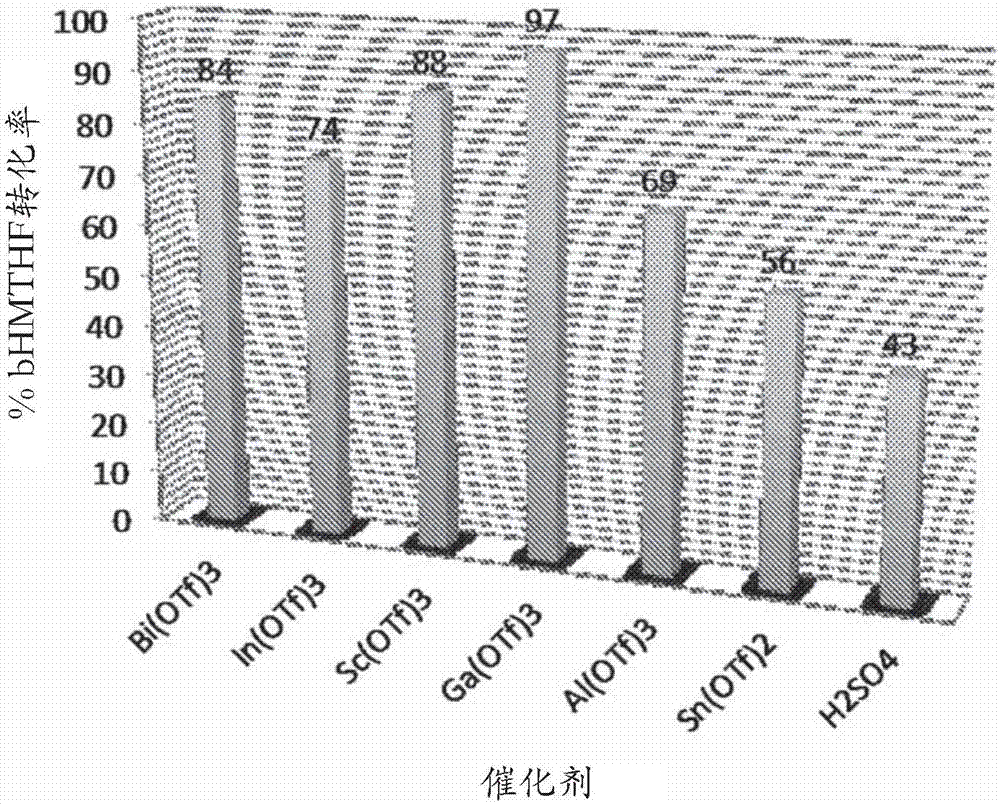
圖1顯示了使用各種耐水性路易斯酸催化劑(金屬三氟甲磺酸鹽)根據bhmthf的酸催化酰化的實例制備的幾種產物混合物的組成,各自在相對于bhmthf的量0.1wt.%的催化劑負載量下。
圖2顯示了在用圖1所示的某些路易斯酸催化劑和硫酸催化的反應中轉化成酯的bhmthf的量的比較。
圖3顯示了使用各種耐水性路易斯酸催化劑(金屬三氟甲磺酸鹽)根據bhmthf的酸催化酰化的實例制備的幾種產物混合物的組成,各自在相對于bhmthf的量0.01wt.%的催化劑負載量下。
圖4是bhmthf立體異構體的標準氣相色譜(gc)跡線。
圖5是通過0.1mol%bi(otf)3催化的bhmthf酯化的氣相色譜(gc)跡線。
圖6是通過0.1mol%in(otf)3催化的用2-乙基己酸的bhmthf酰化的氣相色譜(gc)跡線。
圖7是通過0.1mol%ga(otf)3催化的用2-乙基己酸的bhmthf酰化的氣相色譜(gc)跡線。
圖8是通過0.1mol%sn(otf)2催化的用2-乙基己酸的bhmthf酰化的氣相色譜(gc)跡線。
圖9是通過0.1mol%硫酸催化的用2-乙基己酸的bhmthf酰化的對比氣相色譜(gc)跡線。
圖10是通過0.01mol%硫酸催化的用2-乙基己酸的bhmthf酰化的對比氣相色譜(gc)跡線。
發明的詳細說明
部分i.-說明
呋喃-2,5-二甲醇(fdm)以及((2r,5s)-四氫呋喃-2,5-二基)二甲醇和((2s,5s)-四氫呋喃-2,5-二基)二甲醇(bhmthf)(從hmf的還原獲得)對于常規石油衍生的化學原料是有希望的可再生分子替代物。為了促進將這些還原的分子轉化成用于各種用途的原料,還原產物可以通過酰化轉化為單酯和二酯。所得到的hmf還原產物的單酯和二酯可以用作例如聚合物、增塑劑、潤滑劑和化學添加劑的前體。
本披露描述了使用均相耐水性路易斯酸催化劑的轉化,這些催化劑可以使得實現用有機酸的容易的直接酰化以產生fdm和bhmthf的單酯和二酯。耐水性金屬催化劑在減少的催化劑負載量下產生相對高的二酯產率(例如,≥55%-60%)的能力是高度需要的,并且相對于常規的強布朗斯特酸(例如,硫酸或對甲苯磺酸)可改善方法經濟性。常規的大規模費歇爾-斯皮爾酯化(fse)典型地使用至少1mol.%的酸催化劑負載量/摩爾的醇反應物。相比之下,本發明的酯化方法可以使用兩個或三個數量級更少的催化劑來獲得相當的酯產率。路易斯酸催化劑可以例如以相對于hmf還原產物的量少至約0.01mol.%的量存在。因此,本發明方法可以顯著地幫助成本控制,同時增加整體方法效率。
耐水性路易斯酸在促進許多化學轉化上已經受到關注,并且在化學綜述(chemrev)2002,3641-3666(將其內容通過引用結合在此)中被充分地綜述。傳統上,路易斯酸喜愛其中幾乎沒有水水分存在的條件,因為它們甚至在較小或痕量的水的情況下可以快速水解并且失去它們的催化功能。如在此使用的,術語“耐水性”指的是一種具體的催化劑的金屬離子抵抗通過水水解到高程度的特征。金屬三氟甲烷磺酸根(cf3so3-),通常也稱為三氟甲磺酸根(-otf),具有這種值得注意的特征(例如,參見,美國化學會志(j.am.chem.soc.)1998,120,8287-8288,其內容通過引用結合在此)。對于在此披露的方法,表明良好反應性的耐水性路易斯酸可以包括一種或多種金屬三氟甲磺酸鹽,例如鉿(iv)、鎵(iii)、鉍(iii)、鈧(iii)、銦(iii)、釔、銅(ii)、汞、鎳、鋅、鋁、鐵、鉈或錫(ii)的三氟甲磺酸鹽)。在其他實施例中,金屬三氟甲磺酸鹽物種可以具有鑭系稀土金屬(即鈰、鐠、釹、釤、銪、釓、鋱、鏑、鈥、鉺、鐿、镥)。在某些有利的實施例中,耐水性催化劑是鎵、鈧、鉍、鉿、銦、鋁或錫的三氟甲磺酸鹽。
總體上,用于酸催化酰化的本發明的方法需要在金屬三氟甲磺酸鹽催化劑的存在下在最高達約250℃的溫度下使hmf的還原產物與有機酸接觸最高達約12小時的時期。反應溫度可以是從約150℃或160℃至約240℃或約250℃,包括在其之間的任何范圍。典型地,該反應溫度是在從約170℃或180℃至約200℃或220℃的范圍內。反應時間可以是在約30或45分鐘與6小時之間。典型地,該反應時間是在約1-4小時內,更典型地約2或3小時。
該有機酸試劑可以是具有范圍從c2-c26的碳鏈長度的鏈烷酸、鏈烯酸、鏈炔酸(alkyonoicacid)或芳香族酸。在具體實例中,該有機酸是2-乙基己酸、己酸和辛酸。所使用的有機酸的濃度是hmf還原產物的量的摩爾過量約2倍至約10倍。通常,有機酸的量是摩爾過量約3倍至約5倍。
耐水性路易斯酸的催化劑負載量可以是相對于hmf還原產物的濃度小于約1mol.%。在某些實施例中,金屬三氟甲磺酸鹽催化劑可以以低至約0.001mol.%的量存在。典型地,催化劑負載量的量是約0.5mol.%或更少。取決于所希望的產物混合物,催化劑水平通常是在從約0.1mol.%至約0.01mol.%的范圍內,更典型地約0.03mol.%或0.05mol.%。當催化劑的量是處于約0.1mol.%或更多時,人們實現hmf還原產物的完全酰化,并且二酯是以≥60%產率的主要產物。在其他實施例中,當催化劑是以在約0.01mol.%與0.1mol.%之間的量存在時,主要酰化產物是相應的單酯和二酯的混合物。在另外的實施例中,當所存在的催化劑的量是<0.01mol.%的量時,產物混合物主要包含單酯以及未反應的hmf還原產物。
hmf還原產物到相應的單酯或二酯的轉化產率為≥55%或60%。典型地,轉化率是高的(例如,≥80%、85%或90%)。在足夠的催化劑濃度下,二酯可以是產物混合物的主要產物(例如,≥60%)。酯產物混合物含有≥10%產率的二酯。典型地,在酯產物混合物中,二酯的產率為從約15%至約85%(例如,60%至75%或80%)。在某些有利的實施例中,將hmf還原產物的大多數至全部(例如,約80%至100%)轉化為其相應的二酯物種。
本發明的用于制備酯的方法的另一個優點是保持至少80%的高產物可計量性的能力,如所附表中所示的。典型地,百分比可計量性是≥85%。如在此使用的“可計量性”是產物混合物的百分比的量度,該產物混合物可以被定量地鑒別為目標酯化合物和未反應的起始材料,同時排除不被鑒別為酯產物的縮聚物、顏色體或其他物種。因此,每種產物混合物的總可計量性是單酯和二酯產物和任何未反應的起始原料的組合量。
a.bhmthf酯的制備
根據在此所述的方法的實例,方案1描繪了使用如一般表示為m(otf)x的金屬三氟甲磺酸鹽催化的bhmthf與2-乙基己酸的酯化反應。該反應在175℃的溫度下未攙雜地進行3小時。所使用的金屬三氟甲磺酸鹽的具體物種是相對于所希望的催化劑反應動力學水平和所希望的單酯和二酯產物的比率確定的。(以降序排列的實例的路易斯酸活性是:hf>ga>sc>bi>in>al>sn。)該反應產生可含有單酯或二酯二者的產物混合物。鑒于bhmthf分子的手性性質,順式和反式產物的混合物以9:1的比率存在。
方案1:bhmthf的酸催化酰化。
表1總結了各自在0.1mol.%催化劑負載量下用2-乙基己酸的bhmthf的幾種耐水性路易斯酸催化酰化的數據。該數據包括每個反應的單酯和二酯產量和總體產物混合物可計量性。還顯示了使用也在0.1mol.%的催化劑負載量下的常規強布朗斯特酸催化劑,硫酸(h2so4),進行的bhmthf酯化的產物混合物。與使用硫酸催化的反應相比,所有的耐水性路易斯酸催化劑在二酯的合成中表現顯著更好。
表1
圖1以圖形形式呈現了表1中的數據。該圖沿x軸線排列不同的金屬三氟甲磺酸鹽催化劑并測量y軸線上的每個反應的從0-100%的百分比產物可計量性。用每種金屬三氟甲磺酸鹽物種催化的反應的產物混合物展現出至少80%的可計量性。這大于用硫酸催化的反應的產物組合物的值(即74%)。
表2呈現了當使用各自在0.01mol.%催化劑負載量下的耐水性路易斯酸和常規布朗斯特酸(硫酸)催化時,bhmthf到其相應酯產物的以wt.%計的對比轉化率和總體產物可計量性的總結。每種金屬三氟甲磺酸鹽路易斯酸物種將超過50%的bhmthf轉化成酯。相比之下,硫酸催化劑將43%的bhmthf轉化成酯。對于特定的路易斯酸物種如sc(otf)3、ga(otf)3和hf(otf)4,轉化率是硫酸催化劑的轉化率的至少兩倍。總體上,用路易斯酸催化劑產生的單酯和二酯的量也大于硫酸催化反應的量。圖2是轉換率的圖示。圖3呈現了對于表2的反應的未反應的bhmthf的量(wt.%)、單酯和二酯產率以及總體產物可計量性。
對于使用路易斯酸催化劑和硫酸催化劑的反應,總體產物可計量性水平是可比較的,在約5%-10%內。例如,在表2的對比實例1中,使用硫酸(0.01mol.%)催化的反應具有95%的產物可計量性。該反應產生約33wt.%的單酯,5wt.%的二酯,并且大部分(57%)的bhmthf保持未反應。相比之下在實例1中,用處于0.01mol.%的bi(otf)3催化的反應也展現出95%的產物可計量性,由約65wt.%的單酯、14wt.%的二酯和16wt.%的未反應的bhmthf構成。這兩個實例的總體產物可計量性是相同的,但是對于路易斯酸催化的反應而言,所產生的單酯或二酯的量顯著更多。
同樣在其他實例中,在相同的低催化劑負載量下來自其他金屬三氟甲磺酸鹽反應的酯產物顯示出超過硫酸的改善的轉化率和產率。總體上,路易斯酸催化的酯產物混合物展現出與來自在相同的催化劑負載量下用強布朗斯特酸催化的反應的產物混合物的至少相同水平(如果不是更好)的產物可計量性。換句話說,路易斯酸催化的反應傾向于形成更純凈的產物混合物的最少副產物。
表2.在0.01mol%負載量下用2-乙基己酸的bhmthf的耐水性路易斯酸(金屬三氟甲磺酸鹽)和布朗斯特酸催化酰化的總結。
表3說明,催化劑濃度的變化可以對bhmthf轉化率以及制成的單酯和二酯產物的類型和比率具有顯著影響。在實例1-14中,幾種不同的金屬三氟甲磺酸鹽物種以相對于bhmthf的量0.1mol.%和0.01mol.%催化劑負載量存在。比較圖1和3的結果表明了催化劑負載量的數量級可以產生的差異。對于更活性的金屬三氟甲磺酸鹽物種,用更大的催化劑量所有的bhmthf都反應。
表3的對比實例1顯示了用對甲苯磺酸(p-tsoh),另一種強布朗斯特酸,催化的反應的結果。在每個實例中使用的金屬三氟甲磺酸鹽催化劑的量比用于進行相同反應的p-tsoh催化劑的量小一或兩個數量級。來自兩種種類的酸催化劑的結果關于bhmthf轉化率以及單酯和二酯產物產率是可比較的。該特征表明,使用耐水性路易斯酸催化劑的本發明的酯化方法可以有助于方法成本的節省。
表3.bhmthf(9:1)的2eh酰化。3摩爾當量2eh/bhmthf
b.fdm酯的制備
根據本發明方法的另一個實施例的fdm的酯化類似于對于bhmthf描述的反應。根據方案2所示的實例,fdm在通過三氟甲磺酸鉿(hf(otf)4)催化的酰化中與2-乙基己酸反應。再次,該反應在175℃的溫度下未攙雜地進行3小時。
方案2:如通過三氟甲磺酸鉿催化的fdm酰化反應。
fdm酯化的兩個實例總結在表4中。一個實例是在0.1mol.%的催化劑負載量下進行,而另一個在相對于fdm的量的0.01mol.%下進行。在這兩個實例中,所有的fdm都轉化為或者單酯或二酯物種。這種有效的轉化被認為反映了三氟甲磺酸鉿催化劑的高活性。在0.1mol.%的催化劑量下,所有的fdm都完全轉化成二酯。在0.01%的催化劑量下,大約一半的fdm轉化成二酯,而約40%保持呈單酯形式。較大的催化劑負載量似乎增強了到二酯產物的完全轉化。
表4.fdm酯化
這些反應展現出對應地對于0.1mol.%和0.01mol.%的催化劑負載量,對于產物混合物的86%和91%(39mol.%單酯+52mol.%二酯)的組成可計量性。再次,這些值是與使用硫酸催化劑的常規酯化方法的產物可計量性可比較的或比其更好。
部分ii.–實例
提供以下實例作為本披露的不同方面的說明。參數和條件的變化(如,溫度、時間和試劑濃度,以及具體的起始物種和催化劑的變化)可以影響并且延伸本發明的整個實踐。
a.bhmthf酰化
實例1.
通過0.1mol%bi(otf)3催化的用2-乙基己酸的bhmthf的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gbhmthf(0.076mmol)、40g2-乙基己酸(0.277mmol)和49.7mg三氟甲磺酸鉍(0.1mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器(dean-starktrap),該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過氣相色譜(gc)(硅烷化)分析等分試樣。與圖4所示的bhmthf前體的gc跡線相比,圖5顯示了bhmthf到相應的二酯的所得跡線揭露的完全轉化。
實例2.
通過0.1mol%in(otf)3催化的用2-乙基己酸的bhmthf的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gbhmthf(0.076mmol)、40g2-乙基己酸(0.277mmol)和42.7mg三氟甲磺酸銦(0.1mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。圖6顯示了bhmthf到相應的二酯的所得跡線揭露的完全轉化。
實例3.
通過0.1mol%ga(otf)3催化的用2-乙基己酸的bhmthf的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gbhmthf(0.076mmol)、40g2-乙基己酸(0.277mmol)和39.3mg三氟甲磺酸鎵(0.1mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。圖7呈現了bhmthf到相應的二酯的所得跡線揭露的完全轉化。
實例4.
通過0.1mol%sn(otf)2催化的用2-乙基己酸的bhmthf的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gbhmthf(0.076mmol)、40g2-乙基己酸(0.277mmol)和31.7mg三氟甲磺酸錫(0.1mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。圖7顯示了bhmthf到主要二酯的所得跡線揭露的完全轉化。
實例5.
通過0.1mol%h2so4催化的用2-乙基己酸的bhmthf的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gbhmthf(0.076mmol)、40g2-乙基己酸(0.277mmol)和7.45mg硫酸(0.01mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。所得跡線揭露了bhmthf到主要單-2eh酯與極少量的二-2eh的部分轉化(圖9,參見下文)
對比實例
通過0.01mol%h2so4催化的用2-乙基己酸的bhmthf的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gbhmthf(0.076mmol)、40g2-乙基己酸(0.277mmol)和0.745mg硫酸(0.01mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。所得跡線揭露了bhmthf到主要單-2eh酯與極少量的二-2eh的部分轉化(圖10)。
b.fdm酰化
實例1.通過0.1mol%hf(otf)4催化的用2-乙基己酸的fdm的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gfdm(0.078mmol)、40g2-乙基己酸(0.277mmol)和60.5mg三氟甲磺酸鉿(0.1mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。相應的色譜圖披露了所有fdm已經被轉化(在21.23min保留處沒有特征峰)成僅二酯(54.76min的特征保留時間)。
實例2.通過0.01mol%hf(otf)4催化的用2-乙基己酸的fdm的酰化。
實驗:向含有ptfe磁力攪拌棒的三頸100ml圓底燒瓶中裝入10gfdm(0.078mmol)、40g2-乙基己酸(0.277mmol)和6.1mg三氟甲磺酸鉿(0.01mol%)。然后將該燒瓶的最左邊頸部引入用橡膠隔片塞住的夾套式迪安-斯達克分水器,該橡膠隔片被三個14”針刺穿,中心頸部用磨砂玻璃套管的熱電偶套管適配器加蓋,并且最右邊是附接到氬氣管線的磨砂玻璃適配器。然后將該燒瓶浸入高溫硅油浴中。在劇烈的氬氣吹掃和伴隨攪拌下,將混合物加熱至175℃持續3小時。此后,將產物基質冷卻至室溫并通過gc(硅烷化)分析等分試樣。相應的色譜圖揭示了所有fdm已經被轉化(在21.23min保留處沒有特征峰)成主要單酯(52wt.%,特征保留時間37.34min)和二酯(36wt.%,特征保留時間54.76min)。
已總體地并借助于實例詳細地描述了本發明。本領域的普通技術人員應理解,本發明不必然限于特定披露的實施例,而是在不脫離如由以下權利要求或其等效物(包括目前已知或有待開發的其他等效組分,它們可以在本發明的范圍內使用)所定義的本發明的范圍的情況下可以作出修改和變化。因此,除非變化另外脫離本發明的范圍,否則這些變化應被解釋為被包括在此。
- 還沒有人留言評論。精彩留言會獲得點贊!