EDOT官能化的共軛聚合物和包含其的光電探測器的制作方法
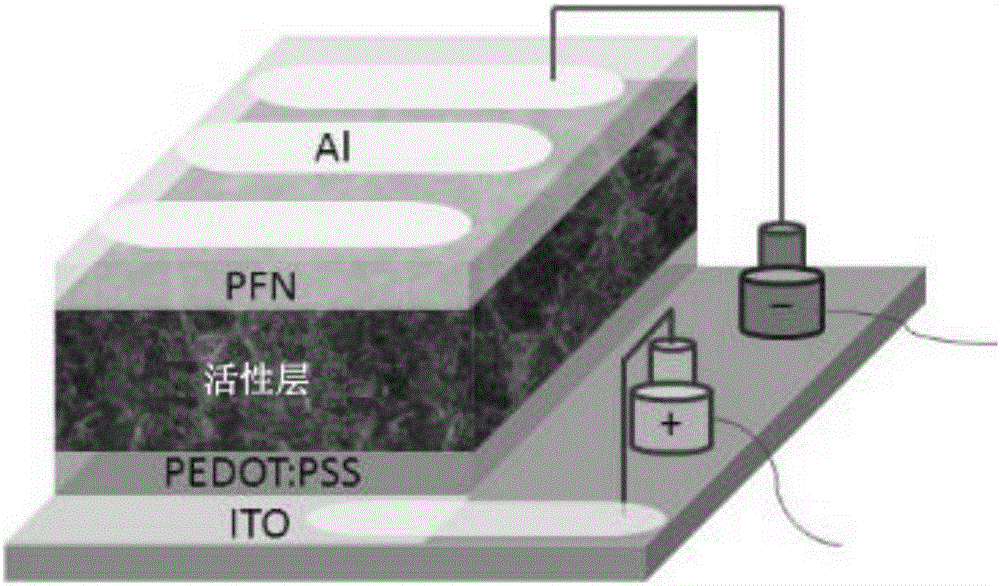
本發明一般涉及有機半導體材料領域,特別地涉及含有作為側鏈的3,4-乙撐二氧噻吩(3-4-ethylenedioxythiophene:EDOT)的給體-受體型共軛聚合物,和含有其的光電探測器。
背景技術:
作為一類可將所檢測到的光轉換成電信號的半導體器件,光電探測器對于各種各樣的應用具有重大的意義,如圖像傳感、數據通信、遠程控制等。目前,商業上可以獲得的用于光電探測器的活性材料主要是基于無機半導體材料,如氧化鋅、硅、GaAs和PbS。這些材料的探測率通常大于1012Jones,但其價格較昂貴以及對工作環境要求相對苛刻(例如,GaAs探測器需要在非常低的溫度下工作)。此外,這些無機材料是脆弱的、非可折疊的并且延展性低,這限制了其在新型高需求領域的應用。因此,這種商業材料具有在低成本、溫和工作條件和新特性(諸如柔性和半透明性)方面的優點的替代材料是被期望并且需要的。
共軛聚合物作為一類新型半導體材料,由于其具有成本低、加工簡單、柔性和半透明性的優勢,引起了學術界和工業界的廣泛關注。已經證明了聚合物光電探測器中的高外量子效率、快速的響應以及近紅外(NIR)范圍(1450nm)的檢測。此外,聚合物光電探測器適用于大面積檢測,并且可以在室溫下工作,還適用于柔性襯底,這些給傳感或檢測技術提供了新的機會。與聚合物太陽能電池相似,采用本體異質結結構構建聚合物光電檢測器,其中共軛聚合物作為電子給體,而富勒烯衍生物作為電子受體。這樣的結構有利于光子吸收和電荷分離,獲得高的外量子效率。然而,這種結構通常會導致在負偏壓下相對高的暗電流密度,以及在正向/反向偏壓下低的整流比,難以從紫外(UV)到近紅外(NIR)的寬光譜范圍內實現高的探測率。開發具有高探測率的新型聚合物光電探測器有很多種方法,包括界面工程、活性層形貌的控制以及改進結構,然而所報道的工作幾乎沒有與分子結構設計相關的。
技術實現要素:
技術問題
本發明的目的是提供一種具有重復的給體-受體單元的、高探測率有機共軛聚合物,其包含作為側鏈的EDOT。
本發明的另外一個目的是提供一種通過將3,4-乙撐二氧噻吩(EDOT)作為側鏈接入主鏈來提高給體-受體聚合物的探測率的方法。
本發明的另一個目的是提供一種光電探測器器件,其包含含有有機共軛聚合物的光活性層。
技術方案
根據本發明的實施方案,提供了一種給體-受體型共軛聚合物,其包含作為側鏈連接到聚合物的主鏈上的式(I)的3,4-乙撐二氧噻吩(EDOT),
其中R1選自由C1-C20烷基、C1-C20烷氧基和C1-C20烷基硫醇基團組成的組。
根據優選的實施方案,R1選自由CH3、C2H5、C4H9、C6H13、C8H17、C12H25、2-乙基己基、2-丁基辛基、2-己基癸基和2-辛基十二烷基組成的組。
根據本發明的另一實施方案,提供了一種光活性層,其包含與富勒烯衍生物共混的共軛聚合物,其中共軛聚合物在側鏈中具有上述式(I)的EDOT功能單元。
具體地,根據實施方案的富勒烯衍生物選自由PC61BM和PC71BM組成的組。
根據本發明的實施方案,式(I)的3,4-乙撐二氧噻吩(EDOT)環連接到聚合物的主鏈上一側或兩側的每個給體單元,即共軛共聚物中EDOT與給體單元的摩爾比為1:1或2:1。
根據本發明的實施方案,提供了一種光電探測器器件,其包括與富勒烯衍生物共混、作為光活性層的半導體共軛聚合物。半導體共軛聚合物在側鏈上具有上述式(I)的EDOT功能單元,它可以有效降低暗電流,而對光電流沒有明顯的影響。
根據本發明的另一實施方案,提供了一種通過將作為側鏈的上述式(I)的3,4-乙撐二氧噻吩接入聚合物的主鏈的給體單元來提高給體-受體聚合物的探測率的方法。
有益效果
根據本發明的實施方案,與不含EDOT側鏈的聚合物制成的參考器件相比,EDOT或其衍生物的引入大大降低了光電探測器件的暗電流約2個數量級,并且提高了光電探測率超過1個數量級。此方法可以應用到各種給體-受體型半導體聚合物,其光響應覆蓋從UV到NIR。
附圖說明
將附圖并入本文并且構成說明書一部分,其顯示出本發明的實施方案。
圖1給出了根據本申請一個實施方案的光電器件的結構示意圖。
圖2給出了根據本申請一個實施方案的聚合物在溶液(a)和固體膜(b)中的吸收光譜。
圖3給出了根據本申請一個實施方案的電子給體、電子受體、界面層和電極的能級水平。
圖4-8給出了根據本申請一個實施方案的器件的J-V(電流-電壓)測量結果。
具體實施方式
以下僅通過舉例的方式對優選實施方案進行描述,而不限于實施本發明所需特征的組合。
本申請提供了一種給體-受體型共軛聚合物,其包含連接到主鏈的給體單元的下式(I)的3,4-乙撐二氧噻吩(EDOT)單元,
其中R1選自由C1-C20烷基、C1-C20烷氧基和C1-C20烷基硫醇基團組成的組。優選地,R1選自由CH3、C2H5、C4H9、C6H13、C8H17、C12H25、2-乙基己基、2-丁基辛基、2-己基癸基和2-辛基十二烷基組成的組。更優選地,R1為2-乙基己基基團。
根據本申請的實施方案,可以有一個或兩個EDOT單元連接到聚合物的主鏈上的每個給體單元。優選地,有兩個EDOT單元連接到聚合物的主鏈上的每個給體單元的兩側。
根據本申請的實施方案,共軛共聚物具有給體-受體結構的主鏈,其中主鏈中給體單元與受體單元的摩爾比(D/A比值)是1:1,如下所示。
根據本申請的實施方案,主鏈中重復的給體-受體單元的數目為10到100。
根據本申請實施方案的共軛聚合物中的給體單元可以是有機光電材料領域的常用的給體單元。優選地,給體單元可以選自由苯并[3,4-b]二噻吩、噻吩、苯及其衍生物組成的組。
根據本申請的優選實施方案,共軛共聚物的給體單元可以是以下結構的任何一種,
其中R1為如式(I)中所描述的相同基團。更優選地,該共聚物的給體單元是苯并[3,4-b]二噻吩或其衍生物。
根據本申請的實施方案,共聚物的主鏈上的受體單元可以是下列結構表示的環的任一種,
特別地,共聚物的主鏈上的受體單元可以選自由苯并[c][1,2,5]噻二唑、苯并[c][1,2,5]噁二唑、異靛-1,3-二酮、喹喔啉、苯并[d][1,2,3]三唑、噻吩并[3,4-c][1,2,5]噻二唑、噻吩并[3,4-b]吡嗪類、噻吩并[3,4-b]噻吩、苯并[1,2-c:4,5-c']-雙([1,2,5]噻二唑)、含噻二唑環[3,4-g][1,2,5]喹喔啉、吡嗪[2,3-g]喹喔啉、[3,3'-雙吲哚滿叉]2,2'-二酮及其衍生物組成的組、其中R選自由H、CH3、C2H5、C4H9、C6H13、C8H17、C12H25、2-乙基己基、2-丁基辛基、2-己基癸基和辛基十二烷基組成的組。
優選地,共軛聚合物中受體單元是含有二酮吡咯并吡咯(DPP)或其衍生物的化合物。
根據本申請的優選實施方案,將DPP和噻吩并吡咯二酮(TPD)用作受體單元,因為它們可以賦予所獲得的共聚物良好的光電性能。
以下示例對本申請進行了詳細說明,但不限于其范圍。在以下的示例中僅對含有BDT(苯并[1,2-b:4,5-b']二噻吩)的共聚物作為給體單元而DPP、TPD或噻吩并異靛(TIIDG)作為受體單元進行了說明,然而含有以上列出的其它給體單元和受體單元的共聚物可以通過與示例1-3中方法相似的方法合成。
以下描述將EDOT(EH)作為側鏈接入D-A共聚物的一些示例,然而本領域技術人員應該理解式(I)所示的EDOT的其它衍生物都適合連接到共聚物的主鏈上的給體單元。
示例1 BDT-EDOT-DPP的合成
1.作為給體單元的Sn2-BDT-EDOT(EH)的制備
按照以下步驟進行Sn2-BDT-EDOT(EH)的合成:
首先利用2-乙基己基鏈使3,4-乙撐二氧噻吩(EDOT)官能化,得到5-(2-乙基己基)-2,3-二氫噻吩并[3,4-b][1,4]二噁英(EDOT(EH))。然后將此EDOT(EH)與苯并[1,2-b:4,5-b']雙噻吩-4,8-二酮反應,得到EDOT取代的苯并[1,2-b:4,5-b']雙噻吩(BDT-EDOT(EH))。通過與二異丙胺鋰(LDA)進行鋰化繼而以Me3SnCl進行錫化而進一步對得到的BDT-EDOT(EH)進行官能化,得到標題化合物。
5-(2-乙基己基)-2,3-二氫噻吩并[3,4-b][1,4]二噁英(EDOT(EH))的合成:將EDOT(8.17克,57.46毫摩爾)放在預先干燥好的250毫升瓶中,加入無水THF(115毫升)配制成溶液,將該溶液冷卻到-78℃。然后將溶解在正己烷(37.7毫升)中的正丁基鋰(1.6摩爾)的溶液在氬氣保護下逐滴加入,將得到的混合物在-78℃的溫度下保持1.5小時,然后將2-乙基己基溴化銨(22.2克,114.95毫摩爾)緩緩加入。該反應持續過夜,然后用水淬取,用二氯甲烷提取,并用水洗滌3次。得到的有機層用硫酸鎂干燥,然后進行減壓濃縮。在(0.1兆帕,80-85℃)蒸餾后,得到目標產物(5.49克),產率為37.55%。1H核磁共振譜(500MHz,CDCl3,δ):6.09(s,1H)、4.14(s,4H),2.54(d,2H)、1.53(m,1H),1.35-1.25(m,8H),0.87(m,6H)。
BDT-EDOT(EH)的合成:將EDOT(EH)(5.49克,21.58毫摩爾)放入預干燥的250毫升瓶中,加入無水THF(87毫升)配制成溶液,將該溶液冷卻到0℃,然后將溶解在正己烷(14.8毫升)中的正丁基鋰(1.6摩爾)在氬氣保護下逐滴加入。將得到的混合物在室溫下保持1.5小時,再冷卻至0℃,然后在一部分中加入苯并[1,2-b:4,5-b']雙噻吩-4,8-二酮(1.95克,8.85毫摩爾)。該反應在80℃進行1.5小時。再次冷卻到0℃后,引入溶解在10%鹽酸(50毫升)中的SnCl2·2H2O(12.17g,53.93毫摩爾)溶液,將得到的混合物在80℃另外攪拌2個小時。冷卻到環境溫度后,將混合物浸入到冰水中。用EtOAc提取有機層、并且水沖洗至少3次。用二氯甲烷/正己烷(v/v,3:7)作為洗脫劑進行柱層析進行進一步純化。最終產產物(5.04克)通過在乙醇中重結晶獲得,經減壓干燥,產率為81.94%。1H核磁共振譜(500MHz,CDCl3,δ):7.43(s,4H)、4.23(q,8H),2.68(d,4H)、1.65(m,2H),1.48-1.32(m,16H),0.90(m,12H)。
Sn2-BDT-EDOT(EH)的合成:將先前步驟中獲得的BDT-EDOT(EH)(1.5克,2.16毫摩爾)溶解在裝有無水THF(33毫升)的100ml充氬氣的瓶中,然后在-78℃下加入二異丙基氨基鋰(2摩爾,3.24毫升)。該反應混合物在該溫度(-78℃)下攪拌1.5小時。隨后,加入溶解在四氫呋喃(7.12毫升)中的三甲基甲錫烷基氯化銨(1.0M)溶液,將得到的混合物在室溫下攪拌過夜。有機層用乙醚提取,用水洗滌2次,濃縮得到粗產物。標題化合物(1.35克)是通過將粗產物在異丙醇重結晶而獲得,產率達到60.94%。1H核磁共振譜(500MHz,CDCl3,δ):7.46(s,2H)、4.24(dd,8H),2.70(m,4H),1.68(m,2H),1.57-1.35(m,16H),0.92(m,12H),0.39ppm(s,18H)。
2.作為受體單元的Br2-DPP(BO)的制備
根據E.J.Zhou等人(E.J.Zhou,S.P.Yamakawa,K.Tajima,C.H.Yang,K.Hashimoto,Chem.Mater.2009,21,4055)報道的方法合成出3,6-雙(5-溴噻吩-2-基)-二(2-丁基辛基)吡咯并[3,4-c]吡咯-1,4(2H、5H)-二酮(Br2-DPP(BO))。
3.給體-受體共聚物的制備
以Pd(PPh3)4為催化劑而以甲苯/DMF混合物作為反應溶劑通過雙錫烷化BDT基給體單元與二溴化受體單元之間的Stille縮聚反應制備出給體-受體共聚物。
BDT-EDOT-DPP的制備方法總結如下:
向25毫升的預干燥瓶子里裝上Sn2-BDT-EDOT(EH)(250毫克,0.245毫摩爾),Br2-DPP(BO)(194.68毫克,0.245毫摩爾)以及Pd(PPh3)4(11.3毫克,0.010毫摩爾)。瓶子先抽真空然后充進氬氣,這個過程重復了3次。然后加入甲苯(9.8毫升)與DMF(0.98毫升)。將得到的混合物在120℃保溫24小時。粗產物經硅藻土過濾,和丙酮沉淀收集。然后將得到的固體在索氏提取器中先后經MeOH、丙酮、正己烷和氯仿漂洗。將在氯仿中的產物濃縮,然后在正己烷中沉淀。減壓干燥后,得到標題共聚物(301毫克),產率為92.55%。
對比例1 BDT-T-DPP的合成
1.作為給體單元的Sn2-BDT-T(EH)制備
按照L.Huo等人(L.Huo,S.Zhang,X.Guo,F.Xu,Y.Li,J.Hou,Angew.Chem.Int.Ed.2011,50,9697)報道的步驟進行(4,8-雙(5-(2-乙基己基)噻吩-2-基)苯并[1,2-b:4,5-b']雙噻吩-2,6-二基)雙(三甲基錫)(Sn2-BDT-T(EH))的合成。
2.作為受體單元的Br2-DPP(BO)的制備
以與示例1中相同的方法制備出Br2-DPP(BO)。
3.給體-受體共聚物的制備
以與示例1中制備BDT-EDOT-DPP的相同步驟進行由Sn2-BDT-T(EH)和Br2-DPP(BO)合成BDT-T-DPP。獲得了標題共聚物,其產率為82.36%。
示例2 BDT-EDOT-TPD的合成
1.作為給體單元的Sn2-BDT-EDOT(EH)的制備
以與示例1中相同的方法得到了給體單元Sn2-BDT-EDOT(EH)。
2.作為受體單元的Br2-TPD的制備
按照Y.P.Zou等人(Y.P.Zou,A.Najari,P.Berrouard,S.Beaupre,B.R.Aich,Y.Tao,M.Leclerc,J.Am.Chem.Soc.2010,132,5330)報道的步驟合成出受體單元1,3-二溴-5-辛基-4H-噻吩并[3,4-c]吡咯-4,6(5H)-二酮(Br2-TPD)。
3.給體-受體共聚物的制備
根據以下步驟進行BDT-EDOT-TPD的合成。
BDT-EDOT-TPD的合成:向25毫升的預干燥瓶內裝入Sn2-BDT-EDOT(EH)(250毫克,0.245毫摩爾),Br2-TPD(103.64毫克,0.245毫摩爾)和Pd(PPh3)4(11.3毫克,0.010毫摩爾)。將瓶子抽真空然后充填氬氣,這個過程重復3次。然后加入甲苯(9.8毫升)與DMF(0.98毫升)。將得到的混合物在120℃保溫24小時。粗產物經硅藻土過濾,和丙酮沉淀收集。將得到的固體在索氏提取器中先后經MeOH、丙酮、正己烷和氯仿漂洗。將氯仿中的產物濃縮,然后在正己烷中沉淀。減壓干燥后,得到了標題產物,產率為98.16%。
對比例2 BDT-T-TPD的合成
以如示例2中合成BDT-EDOT-TPD的相同步驟進行由Sn2-BDT-T(EH)和Br2-TPD合成BDT-T-TPD。得到了標題共聚物,其產率為79.65%。
示例3 BDT-EDOT-TIIDG的合成
為進一步證實了EDOT單元的功能的普遍性,還合成了側鏈含EDOT單元的窄帶隙聚合物,并將它用于根據本申請聚合物光電探測器的制備。低能帶隙聚合物BDT-EDOT-TIIDG的分子結構由式(II)所示
1.作為給體單元的BDT-EDOT-TIIDG的制備
以示例1中相同的方法得到了給體單元Sn2-BDT-EDOT(EH)。
2.Br2-TIIDG的制備
按照G.W.P.Van Pruissen等人(G.W.P.Van Pruissen,F.Gholamrezaie,M.M.Wienk,R.A.J.Janssen,J.Mater.Chem.2012,22,20387)報道的步驟合成出噻吩并異靛化合物,2,2'-二溴-4,4'-雙(辛基十二烷基)-[6,6'-二噻吩并[3,2-b]吡咯亞基]-5,5'(4H,4'H)-二酮(Br2-TIIDG)。
3.給體-受體共聚物的制備
根據以下步驟進行BDT-EDOT-TIIDG的合成。
BDT-EDOT-TIIDG的合成:通過與示例1中制備BDT-EDOT-DPP相同的步驟獲得該共聚物,除了將Br2-DPP(BO)換成Br2-TIIDG。得到了標題產物,其產率為71.5%。
示例4含有BDT-EDOT-DPP的光電器件的制備與測試
制備了如圖3中所示的具有ITO/PEDOT:PSS/活性層/PFN/Al結構的光電器件,其中銦錫氧化物(ITO)是底層。該器件包括透明金屬氧化物電極(即ITO)和作為陽極的PEDOT:PSS層,共軛聚電解質PFN改性的Al為陰極,而EDOT聚合物與PCBM制備而成的光活性層則置于兩電極之間。在本示例中,EDOT聚合物為BDT-EDOT-DPP,PCBM為PC71BM;而在對比例中,EDOT聚合物為BDT-T-DPP,PCBM與本示例中相同。
制備光電器件的方法總結如下。ITO襯底先后經洗滌劑、去離子水、丙酮、異丙醇超聲清洗。然后,將ITO襯底在烘箱中干燥,并在紫外臭氧室處理4分鐘。PEDOT:PSS[聚(3,4-乙烯二氧噻吩):聚(苯乙烯磺酸)]水溶液經0.22μm濾膜過濾,然后在ITO電極上以2500轉進行旋涂30s,繼而將襯底在空氣150℃下烘烤10分鐘。PEDOT:PSS層厚度為約40nm。隨后,將由
ITO層和PEDOT:PSS層組成的襯底轉移到充有氮氣的手套箱中。將示例1中制備的BDT-EDOT-DPP與PC71BM(1:2,w/w)(PC71BM:[6,6]-苯基C71丁酸甲酯)混合,然后將混合物溶于氯仿/1,2-二氯苯(95:5,v/v)中。PC71BM的分子結構可以表示為式(III)所示,
將得到的溶液在手套箱內旋涂在PEDOT:PSS層上,以形成活性層(DPP膜),其厚度為約130nm。在沉積Al電極前,先在活性層上覆蓋聚[(9,9-二(3′-(N,N-二甲基氨基)丙基)-2,7-芴)-alt-2,7-(9,9-二辛基芴)](PFN)層。最后,將得到的結構轉移到真空室,然后在3×10-6mbar的基準壓力下在PFN層上熱沉積約100nm的Al。所得到的器件的光活性層面積為0.16cm2。
對所獲得器件的電流密度-電壓(J-V)的測量是在AM1.5G太陽模擬器光照下(100mWcm-2)由計算機控制的Keithley 2400源測量單元在空氣中測量得到。器件的暗導J-V特性曲線則在手套箱中由計算機控制的Keithley 236源測量單元測量的。單色外量子效率是在室溫環境下使用DSR100UV-B光譜儀與SR830鎖相放大器測量的。本測試中采用溴鎢燈作為光源。
對比例4含有BDT-T-DPP的光電器件的制備與測試
本對比例中,參考器件的制造方法與示例4中的制造方法相同,除了將活性層含有的EDOT替代為噻吩。將對比例1中制備的BDT-T-DPP與PC71BM(1:2,w/w)(PC71BM:[6,6]-苯基C71丁酸甲酯)混合,然后將混合物溶于氯仿/1,2-二氯苯(95:5,v/v)。將得到的溶液在手套箱內涂布在由ITO層和PEDOT:PSS層組成的襯底上,以形成活性層,其厚度為約130nm。
示例5含有BDT-EDOT-TPD光電器件的制備與測試
制備了如圖3所示的具有ITO/PEDOT:PSS/活性層/PFN/Al結構的光電器件,其中ITO為底層。獲得銦錫氧化物(ITO)襯底,先后經洗滌劑、去離子水、丙酮、異丙醇超聲清洗。然后,將ITO襯底在烘箱中干燥,并在紫外臭氧室處理4分鐘。將經0.22μm濾膜過濾的PEDOT:PSS水溶液以2500轉在ITO襯底上旋涂30s,然后在空氣中150℃下烘烤10分鐘。所獲得的PEDOT:PSS層厚度為約40nm。隨后,將由ITO層和PEDOT:PSS層組成的結構轉移到充有氮氣的手套箱中。將示例2中制備的的BDT-EDOT-TPD與PC61BM(1:1,w/w)(PC61BM:[6,6]-苯基C61丁酸甲酯)混合,并將混合物溶于氯仿/1,8-二碘辛烷(97:3,v/v)中。PC61BM的分子結構可以表示為式(IV),
將得到的溶液旋涂在PEDOT:PSS層上,以形成活性層,其厚度為約100nm。在活性層上覆蓋聚[(9,9-雙(3′-(N,N-二甲基氨基)丙基)-2,7-芴)-alt-2,7-(9,9-二辛基芴)](PFN)層。最后,將得到的結構轉移到真空室,然后在3×10-6mbar的基準壓力下在PFN層上熱沉積約100nm的Al。所得到的器件的光活性層面積為0.16cm2。
以與示例4中相同的方法對所獲得的器件進行J-V測試。
對比例5含有BDT-T-TPD的光電器件的制備與測試
本示例中器件的結構和方法與示例5中是相同的,除了將活性層中含有的BDT-EDOT-TPD替代為BDT-T-TPD。該器件的制備方法總結如下:將ITO襯底先后經洗滌劑、去離子水、丙酮、異丙醇超聲清洗。然后,將ITO襯底在烘箱中干燥,并在紫外臭氧室處理4分鐘。在ITO襯底上涂布40nm的PEDOT:PSS層。將對比例2中制備的BDT-T-TPD與PC61BM(1:1,w/w)(PC61BM:[6,6]-苯基C61丁酸甲酯)混合,然后將它們溶于氯仿/1,8-二碘辛烷(97:3,v/v)中。將該溶液涂布在PEDOT:PSS層上,以形成100nm的活性層。將PFN層覆蓋在活性層上,然后在PFN層上沉積100nm的Al層。該器件的光活性面積為0.16cm2。
以與示例4中相同的方法對所獲得的器件進行J-V測試。
示例6
制備出圖3中所示的具有ITO/PEDOT:PSS/活性層/PFN/Al結構的光電器件,其中ITO為底層,而活性層包括示例3中制備的BDT-EDOT-TIIDG。本示例中器件的制備方法與示例中的方法相同,除了活性層中BDT-EDOT-TPD被替換為示例3中制備的BDT-EDOT-TIIDG。
以與示例4中相同的方法對所獲得的器件進行J-V測試。
結果
圖2給出了示例1-2與對比例1-2中制備的聚合物的吸收光譜,其中圖(2a)為聚合物在氯仿(CF)中的吸收光譜,而圖(2b)為聚合物在固體薄膜的吸收光譜。
從圖2可以看出,EDOT改性的共軛聚合物(示例1的BDT-EDOT-DPP、示例2的BDT-EDOT-TPD)與噻吩為側鏈的參考聚合物(對比例1-2)具有相似的吸收光譜。對于溶解在氯仿(CF,濃度為0.005mg·mL-1)中的DPP聚合物的結果,BDT-EDOT-DPP相比噻吩改性的BDT-T-DPP具有更高的峰(768nm)/肩(700nm)吸收比。TPD聚合物(BDT-EDOT-TPD與BDT-T-TPD比較)也有類似的趨勢。同時,EDOT較噻吩類聚合物的吸收光譜顯示出藍移,這種趨勢在TPD聚合物(BDT-EDOT-TPD)更為明顯。
示例4與對比例4中實驗性聚合物的能級水平通過循環伏安法(CV)測定,其最高占據分子軌道(HOMO)和最低未占據分子軌道(LUMO)的數據在圖3中給出。從圖中可見,將EDOT作為側鏈引入共軛聚合物導致聚合物帶隙相比于引入噻吩聚合物略有增加。圖3還表明EDOT聚合物比噻吩類聚合物具有更高的HOMO能級。
示例4-5和對比實例4-5中制備的聚合物光電探測器的電流電壓特性如圖4所示,其中圖(4a)給出BDT-EDOT-DPP器件的短路電流密度與BDT-T-DPP器件的短路電流密度是可比較的,表明載流子在這兩體系中都可以有效產生。而圖(4b)則給出了BDT-EDOT-TPD和BDT-T-TPD的測量結果。然而,在反向電壓下從BDT-EDOT-DPP器件觀察到的暗電流比從BDT-T-DPP器件觀察到的暗電流低約2個數量級,在±2V產生的整流比達到106。從BDT-EDOT-DPP器件觀察到的良好二極管特性表明在側鏈引入EDOT單元可以顯著地抑制二極管在暗導條件下的本征漏電流。這種現象在較大的帶隙TPD聚合物體系中也被觀察到。EDOT聚合物在-2V具有暗電流7.5×10-8A/cm2,而噻吩聚合物的暗電流則為3.1×10-5A/cm2(圖5b)。BDT-EDOT-TPD器件在AM1.5G光照下的短路條件時光電流密度為8.23mA/cm2,說明此EDOT改性的聚合物中電荷載流子的產生是高效的。
圖5給出了在示例4-5和對比例4-5中制備的聚合物光電探測器的外量子效率(EQE),參數表示其光子-電荷的轉換能力。可以看出,BDT-EDOT-DPP器件與BDT-T-DPP器件光電轉換能力很好,而BDT-EDOT-TPD器件與BDT-T-TPD器件也可以有效地捕獲相應光并有效地產生光電流。
在本領域,有幾個參數,可用于評估實驗性光電探測器的質量和有效性。其中的一個參數是噪聲等效功率(NEP),它可以計算為:
NEP=(AΔf)1/2/D*
其中A為探測器的有效面積,單位是cm2,Δf是電帶寬,單位是Hz,和D*為探測率,單位是Jones。在這里,我們使用等式:
D*=EQE×(λ/1240)/(2qJd)1/2
計算D*值,其中q是絕對的電荷量1.60×10-19C,Jd是器件的暗電流,單位為A/cm2,λ是波長。圖6給出了聚合物光電探測器的探測率,其中器件在0V下的暗電流為3.125×10-11A/cm2。從中可見,BDT-EDOT-DPP器件在波長775nm達到8×1013Jones。然而,這BDT-T-DPP器件在其響應譜范圍內最高只有1.7×1012Jones的探測率。對于TPD聚合物,該器件具有與DPP聚合物探測器類似的特征。因為側鏈中EDOT的存在,BDT-EDOT-TPD探測器在605nm具有約2.7×1013Jones。但是,BDT-T-TPD器件在響應范圍內均低于1.9×1012Jones。這些結果表明EDOT側鏈的引入能夠提供用于具有高探測率的光電探測器件的高效聚合物。
示例3中制備的BDT-EDOT-TIIDG薄膜的光學吸收光譜如圖7所示。可以看出,BDT-EDOT-TIIDG最強吸收峰處在840nm處,其吸收覆蓋從400nm到1000nm,而吸收邊延伸到1085nm。從其吸收光譜中計算出的BDT-EDOT-TIIDG的光學帶隙約為1.14eV。
圖8給出了BDT-EDOT-TIIDG基-光電探測器在光導和暗導下的J-V特性(a),以及BDT-EDOT-TIIDG基-光電探測器的外量子效率和探測率(b)。與上述兩類EDOT基-聚合物類似,BDT-EDOT-TIIDG器件也表現出很好的二極管特性,在±2V電壓下的整流比在106。在0V下,暗導下BDT-EDOT-TIIDG器件的電流密度為5.4×10-10A/cm2,表現出器件中很低的散粒噪聲(散粒噪聲是來源于電荷離散性的電子噪聲)盡管BDT-EDOT-TIIDG的帶隙小至1.14eV。BDT-EDOT-TIIDG器件的探測率從380nm到975nm超過1012Jones,在1085nm超過1011Jones。最高的探測率在波長830nm處,接近6.3×1012Jones。這樣高的探測率可與以前報道的結果相媲美。所有這些結果進一步證明,在側鏈引入EDOT單元可以有效地抑制器件中的暗電流,進而提高光電探測器的探測率。
顯而易見的是,上述的描述目的是為了說明、解釋、描述本發明。對這些具體例子的修改和改進對于那些熟悉本領域的研究人員來說是簡單易懂的,并且可以不偏離本發明的范圍或內涵。
- 還沒有人留言評論。精彩留言會獲得點贊!