纖維增強樹脂組合物以及纖維增強復合材料的制造方法與工藝
本發明涉及兼具力學特性與生產性和成型加工性的纖維增強樹脂組合物。此外,本發明涉及具有優異的力學特性的纖維增強復合材料。
背景技術:
包含熱塑性樹脂和增強纖維的纖維增強樹脂組合物具有下述特征:發揮熱塑性樹脂的特性而成型加工容易,或者不需要熱固性樹脂那樣的儲存的負擔,再循環性優異。作為這樣的纖維增強樹脂組合物,公知的是使增強纖維片狀地排列的熱塑性預浸料、使增強纖維無規分散了的顆粒等多種多樣的形態。纖維增強樹脂組合物由于輕量性與力學特性的平衡優異,因此廣泛用作航空器、汽車、船舶等的結構用構件、電子設備殼體、體育用途、建材等的工業材料。在熱塑性樹脂中,聚芳撐硫醚是耐熱性、耐化學性特別優異,可以期待將所得的纖維增強樹脂組合物適合用作金屬材料的替代的樹脂。然而,作為金屬材料的替代,在發展使用了聚芳撐硫醚的纖維增強樹脂組合物方面,纖維增強樹脂組合物的力學特性,特別是抗拉強度和伸長率的進一步提高是問題。這是因為,一般的聚芳撐硫醚的拉伸伸長率比增強纖維的拉伸伸長率(例如如果是碳纖維則為2%左右)低,因此在纖維增強樹脂組合物中,未充分地發揮盡增強纖維的增強效果。作為使纖維增強樹脂組合物的抗拉強度和伸長率提高的通常的方法,可舉出所用的聚芳撐硫醚的高伸長率化。然而,聚芳撐硫醚的拉伸伸長率與其分子量、進而熔融粘度有相關關系,如果使聚芳撐硫醚的拉伸伸長率提高,則有熔融粘度也會增加,大幅損傷纖維增強樹脂組合物的成型加工性的風險。此外,廣泛已知聚芳撐硫醚的熔融粘度越高,則與增強纖維的復合化越困難。在這樣的情況下,由于工藝溫度也需要更高溫,因此對于容易生產性良好地制造纖維增強樹脂組合物而言是不適合的。基于這些理由,對于使用了聚芳撐硫醚的纖維增強樹脂組合物而言,兼具抗拉強度、伸長率的提高與生產性和成型加工性被認為是重要的技術課題。專利文獻1中公開了,對包含聚芳撐硫醚預聚物和連續的增強纖維的復合體,以相接的方式配置高分子量的熱塑性樹脂而成的成型材料。對于該成型材料,通過對連續的增強纖維束的含浸時使用低分子量體、基體樹脂使用高分子量體進行分開使用,從而謀求兼具力學特性和生產性。聚芳撐硫醚預聚物是下述材料:由于容易含浸于增強纖維束,因此提高成型材料的生產性,進一步在成型工序中與基體樹脂容易分散或相容,從而提高增強纖維在成型品中的分散的優異的材料。然而,由于聚芳撐硫醚預聚物為低分子量,因此有其添加量與獲得的成型品的力學特性會處于抵換的關系的問題。另一方面,對于對聚芳撐硫醚的添加劑也研究了各種物質。專利文獻2中公開了,通過將聚芳撐硫醚、環狀聚烯烴樹脂和聚碳二亞胺進行組合,從而提高與金屬材料、有機材料的密合性的組合物。然而,對于該組合物,雖然說明書中記載了增強纖維,但是是以對組合物賦予剛性為目的,而不是以纖維增強樹脂組合物的抗拉強度、伸長率這樣的力學特性的提高為目的,進一步并不是以兼具這些力學特性與生產性和成型加工性為目的。專利文獻3中公開了,包含聚芳撐硫醚、芳香族聚碳二亞胺系樹脂和無機填充劑的組合物。然而,該組合物以力學特性不大幅降低且提高耐濕性和耐化學性為目的,雖然公開了組合物的力學特性,但是并沒有想到其抗拉強度、伸長率飛躍性提高的現象。進一步并不是以兼具這些力學特性與生產性和成型加工性為目的。此外,纖維增強復合材料(FRP)輕量且具有優異的力學特性,廣泛用于電氣/電子設備用途、土木/建筑用途、機械/機構部件用途、遙控設備用途、二輪車/汽車用途、宇宙/航空用途等。這些FRP所使用的增強纖維中,使用了鋁纖維、不銹鋼纖維等金屬纖維、芳族聚酰胺纖維、PBO纖維等有機纖維、和碳化硅纖維等無機纖維、碳纖維等,但是從比強度、比剛性特別優異,獲得超群的輕量性的觀點出發,適合使用碳纖維。這里,作為碳纖維增強復合材料(CFRP)等FRP的代表性形態,可舉出將預浸料或由預浸料疊層而得的預成型品進行了壓制成型(在加壓力下進行脫泡而賦形的成型方法)的成型品。關于該預浸料,在使連續的增強纖維沿一個方向排列或進行了織物加工的增強纖維基材中,含浸樹脂而制造的方法是通常的方法,但也可使用不連續的增強纖維。將使用了連續的增強纖維的預浸料成型而得的成型品獲得優異的力學特性,另一方面,由于增強纖維以連續體直接使用,因此不適合成型復雜的形狀,并且由于預浸料的疊層角度對特性的影響大,因此從疊層工序的經濟負擔出發,使用用途受到限制。另一方面,使用了不連續的增強纖維的片成型復合物(SMC)、玻璃氈基材(GMT)為適于壓制成型的材料,但因為比強度、比剛性等力學特性低,對預浸料那樣的薄壁成型品的應對困難,此外成型時樹脂大幅流動因此得不到各向同性的力學特性,并且特性的偏差大等課題,因而使用用途受到限制。此外,增強纖維與基體樹脂的粘接性對纖維增強復合材料的抗拉強度等力學特性帶來影響,因此增強纖維與基體樹脂的界面設計在開發纖維增強復合材料方面是極其重要的課題。專利文獻4中提出了碳二亞胺試劑附著于表面的碳纖維,記載了對成為基體樹脂的熱塑性樹脂的粘接性優異、抗彎強度提高,但在增強纖維與基體樹脂的界面強度方面留有問題,期望進一步的強度提高。此外,專利文獻5中也公開了有關于包含分子內具有2個以上碳二亞胺鍵的有機化合物的碳纖維用表面改性劑和用該改性劑改性了的碳纖維的技術,但同樣地在增強纖維與基體樹脂的界面強度方面留有問題,期望成型品的進一步強度提高。進一步專利文獻6中也公開了在聚芳撐硫醚樹脂中脂肪族聚碳二亞胺系樹脂和作為無機填充劑的纖維的樹脂組合物,但成型品的抗拉強度也不充分,期望進一步的強度提高。現有技術文獻專利文獻專利文獻1:日本特開2008-231291號公報專利文獻2:日本特開平10-168290號公報專利文獻3:日本特開2004-91504號公報專利文獻4:日本特開平5-106163號公報專利文獻5:日本特開平5-311069號公報專利文獻6:日本特開2005-239917號公報
技術實現要素:
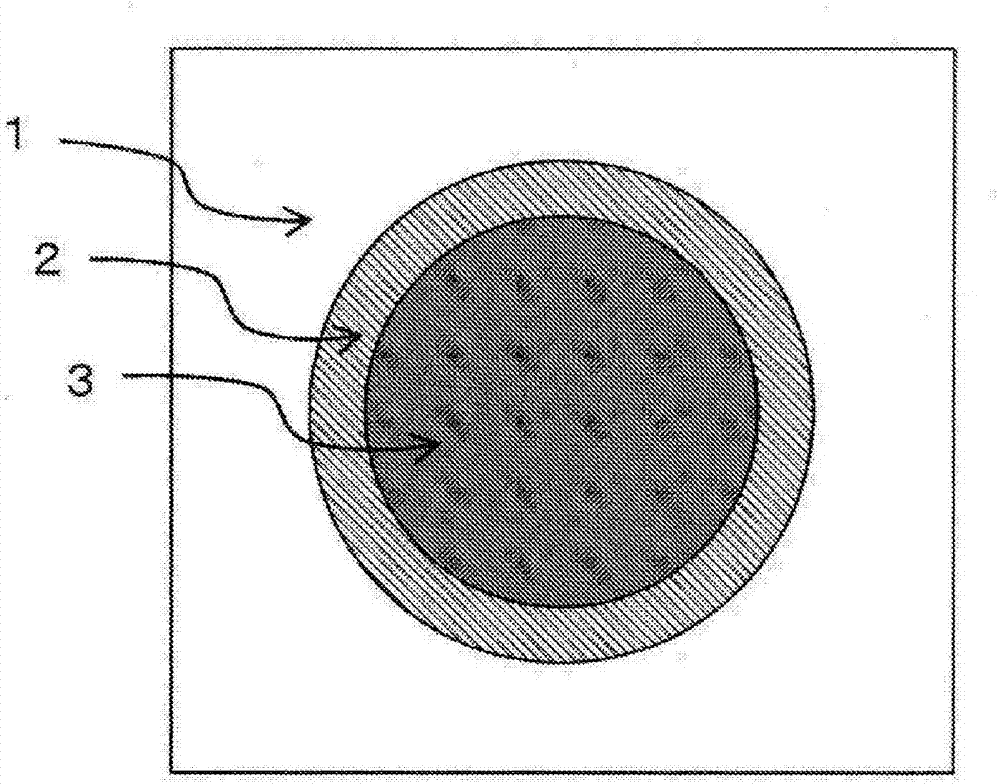
發明所要解決的課題本發明的課題在于改善這樣的現有技術的問題,提供兼具抗拉強度、伸長率這樣的力學特性與生產性和成型加工性的纖維增強樹脂組合物。此外,本發明的課題在于提供抗拉強度這樣的力學特性優異的纖維增強復合材料。用于解決課題的方法發明人等為了解決上述課題而進行了深入研究,結果發現,通過成為特定的組成,從而在將低分子量的聚芳撐硫醚用于基體樹脂的情況下,所得的纖維增強樹脂組合物的抗拉強度、伸長率飛躍性地提高,由此完成本發明。此外,發明人等為了解決上述課題而進行了深入研究,結果發現,通過使粘接性化合物在增強纖維界面局部存在,從而控制增強纖維周邊的粘接性化合物的存在比,提高與增強纖維的粘接性,所得的纖維增強復合材料的抗拉強度飛躍性地提高,由此完成本發明。用于解決這樣的問題的本發明的纖維增強樹脂組合物包含如下構成。即,一種纖維增強樹脂組合物,其含有聚芳撐硫醚(A)、碳二亞胺化合物(B)和碳纖維(C),并且上述碳纖維(C)用上漿劑(D)進行了表面處理,上述碳二亞胺化合物(B)為脂肪族碳二亞胺化合物,上述上漿劑(D)為1分子中具有3個以上選自羧基、氨基、羥基和環氧基中的至少1種官能團的化合物,相對于成分(A)100質量份,含有成分(B)0.1~10質量份、成分(C)10~300質量份,相對于成分(C)100質量份,含有成分(D)0.01~5質量份。此外,用于解決上述問題的本發明的成型品包含如下構成。即,是上述纖維增強樹脂組合物成型而成的成型品。進一步,用于解決上述問題的本發明的纖維增強復合材料包含如下任一構成。·一種纖維增強復合材料,其是使用了上述纖維增強樹脂組合物的纖維增強復合材料,式(1)所示的碳二亞胺化合物(B)的存在比Rb為1.2以上。Rb=R(≤500nm)/R(>500nm)式(1)R(≤500nm):碳纖維(C)周邊500nm以內的碳二亞胺化合物(B)的存在量R(>500nm):與碳纖維(C)周邊500nm相比靠外側的碳二亞胺化合物(B)的存在量·一種纖維增強復合材料,其含有熱塑性樹脂(A’)、粘接性化合物(B’)和增強纖維(C’),并且上述粘接性化合物(B’)為在1分子內具有2個以上選自碳二亞胺結構、脲結構和氨基甲酸酯結構中的至少1種結構的化合物,熱塑性樹脂(A’)為在主鏈的重復單元結構中包含碳以外的元素的熱塑性樹脂,式(1’)所示的粘接性化合物(B’)的存在比Rb’為1.2以上。Rb’=R’(≤500nm)/R’(>500nm)式(1’)R’(≤500nm):增強纖維(C’)周邊500nm以內的粘接性化合物(B’)的存在量R’(>500nm):與增強纖維(C’)周邊500nm相比靠外側的粘接性化合物(B’)的存在量發明的效果本發明的纖維增強樹脂組合物能夠使抗拉強度、伸長率這樣的力學特性飛躍性地提高。進一步在本發明中,在將低分子量的聚芳撐硫醚作為基體樹脂的情況下,力學特性提高,因此可以制成兼具力學特性與生產性和成型加工性的纖維增強樹脂組合物。進一步本發明的成型品的抗拉強度、伸長率這樣的力學特性飛躍性地提高,因此可以適合用于各種制品。此外,本發明的纖維增強復合材料中,增強纖維與基體樹脂的界面強度高,因此能夠使抗拉強度這樣的力學特性飛躍性地提高。附圖說明圖1為顯示在與碳纖維(C)的單絲的軸心方向正交的截面中觀察到的、本發明的纖維增強樹脂組合物的形態的一例的概略圖。圖2為顯示在與碳纖維(C)的單絲的軸心方向正交的截面中觀察到的、本發明的纖維增強樹脂組合物中的區域(T1)與區域(T2)的一例的概略圖。圖3為顯示在與碳纖維(C)的單絲的軸心方向正交的截面中觀察到的、本發明的纖維增強樹脂組合物中的區域(T1)與區域(T2)的一例的概略圖。圖4為顯示本發明的纖維增強復合材料中的與增強纖維(C’)的單絲的軸心方向正交的截面的一例的示意圖。具體實施方式本發明的纖維增強樹脂組合物包含聚芳撐硫醚(A)、碳二亞胺化合物(B)、碳纖維(C)和上漿劑(D)作為成分。首先對各成分進行說明。<聚芳撐硫醚(A)>本發明的纖維增強樹脂組合物中的聚芳撐硫醚(A)(以下,有時將聚芳撐硫醚簡稱為PAS)是以式-(Ar-S)-的重復單元作為主要構成單元的、含有該重復單元優選為80摩爾%以上、更優選為90摩爾%以上、進一步優選為95摩爾%以上的均聚物或共聚物。作為Ar,有以下式(a)~式(k)等所示的單元等,其中特別優選為式(a)所示的單元。(R1、R2為選自氫、碳原子數1~12的烷基、碳原子數1~12的烷氧基、碳原子數6~24的亞芳基、鹵基中的取代基,R1與R2可以相同也可以不同。)只要將該重復單元作為主要構成單元,就可以包含以下式(l)~式(n)等所示的少量的支鏈單元或交聯單元。這些支鏈單元或交聯單元的共聚量相對于-(Ar-S)-的單元1摩爾優選為0~1摩爾%的范圍。此外,聚芳撐硫醚(A)可以為包含上述重復單元的無規共聚物、嵌段共聚物和它們的混合物的任一種。作為它們的代表性物質,可舉出聚苯硫醚(上述式(a)、式(b)、式(f)~式(k))、聚苯硫醚砜(上述式(d))、聚苯硫醚酮(上述式(c))、聚苯硫醚醚(上述式(e))、它們的無規共聚物、嵌段共聚物和它們的混合物等。作為特別優選的聚芳撐硫醚(A),可舉出作為聚合物的主要構成單元含有對苯硫醚單元80摩爾%以上,特別是90摩爾%以上的聚苯硫醚(以下,有時將聚苯硫醚簡稱為PPS)PAS(A)的質均分子量優選為10,000~80,000,更優選為10,000~60,000,進一步優選為10,000~40,000。在質均分子量小的PAS(A)的情況下,熔融粘度低,所得的纖維增強樹脂組合物的成型加工性優異,因此優選。此外,關于本發明的纖維增強樹脂組合物,在質均分子量小的PAS(A)的情況下,有所得的纖維增強樹脂組合物的抗拉強度、伸長率這樣的力學特性提高的傾向。其原因推測是,PAS(A)所具有的官能團與碳二亞胺化合物(B)的碳二亞胺基進行化學反應,在質均分子量小的PAS(A)的情況下,末端所存在的官能團增加,與碳二亞胺化合物(B)的反應位點增加。從這些理由出發,本發明的纖維增強樹脂組合物中,使PAS(A)的質均分子量為10,000~40,000的范圍因為可以以高水平實現兼具所得的纖維增強樹脂組合物的力學特性與生產性和成型加工性,因此特別優選。另外,PAS(A)的質均分子量可以通過尺寸排阻色譜(SEC)來測定。通過洗脫液使用1-氯萘,將柱溫度設為210℃,算出聚苯乙烯換算的質均分子量,從而可以求出。PAS(A)優選在主鏈和/或側鏈的末端具有官能團。這里所謂主鏈,是指高分子結構中最長的鏈狀結構部分,從主鏈分支而構成的部分稱為側鏈。所謂高分子結構,是指單一的結構單元重復連接的部分、或多個結構單元規則地或無規地連接的部分,所謂末端,是指連接停止的最后的結構單元。PAS(A)所具有的官能團優選為在高分子結構的主鏈和/或側鏈的末端的任一者存在1處以上,具有這樣的官能團的PAS在PAS(A)中所占的比例優選為50質量%以上,更優選為60質量%以上,進一步優選為80質量%以上。通過滿足這樣的條件,從而獲得力學特性更優異的纖維增強樹脂組合物。作為PAS(A)所具有的官能團,可以使用聚合時所使用的單體的官能團殘留了的官能團;聚合時的催化劑、助劑、溶劑被引入到末端而形成的官能團;高分子結構通過熱分解、水解等被切斷而形成的官能團;和將這些官能團氧化、還原和用改性劑進行了改性的官能團。作為上述改性劑,可以例示表氯醇、多官能環氧樹脂、酸酐等。其中,從對高分子結構的破壞少,容易控制分子量考慮,優選使用聚合時所使用的單體的官能團殘留了的官能團;聚合時的催化劑、助劑、溶劑被引入到末端而形成的官能團。作為PAS(A)所具有的官能團,可以例示硫醇基、環氧基、羧基、羧基的金屬鹽、氨基、羥基、異氰酸酯基、唑啉基、磺酸基等。其中,從與碳二亞胺基的反應性方面出發,優選為硫醇基、環氧基、羧基、羧基的金屬鹽、氨基、羥基,特別優選為硫醇基、羧基、氨基、羥基。PAS(A)優選基于氯仿的低聚物提取量為2質量%以下,更優選為1質量%以下。這里的所謂基于氯仿的低聚物提取量,是有機低聚合成分(低聚物)量的指標,可以由使用氯仿200mL對測定的PAS(A)10g索格利特提取5小時處理時的殘差量算出。通過減少PAS(A)中的低聚物成分,從而PAS(A)的聚合物成分的官能團與碳二亞胺化合物(B)的碳二亞胺基能夠更加選擇性反應,因此獲得力學特性優異的纖維增強樹脂組合物。PAS(A)可以通過從使多鹵代芳香族化合物與硫化劑在極性有機溶劑中反應而得的聚合反應物,將PAS(A)進行回收、后處理,從而以高收率進行制造。所謂多鹵代芳香族化合物,是指1分子中具有2個以上鹵原子的化合物。作為具體例,可舉出對二氯苯、間二氯苯、鄰二氯苯、1,3,5-三氯苯、1,2,4-三氯苯、1,2,4,5-四氯苯、六氯苯、2,5-二氯甲苯、2,5-二氯-對二甲苯、1,4-二溴苯、1,4-二碘苯、1-甲氧基-2,5-二氯苯等。其中,優選使用對二氯苯。此外,也能夠將不同的2種以上多鹵代芳香族化合物組合而制成共聚物,優選將對二鹵代芳香族化合物作為主要成分。關于多鹵代芳香族化合物的使用量,從獲得適于加工的質均分子量的PAS(A)方面出發,可以例示硫化劑每1摩爾為0.9~2.0摩爾、優選為0.95~1.5摩爾、進一步優選為1.005~1.2摩爾的范圍。作為硫化劑,可舉出堿金屬硫化物、堿金屬氫硫化物和硫化氫。作為堿金屬硫化物的具體例,可舉出例如硫化鋰、硫化鈉、硫化鉀、硫化銣、硫化銫和它們2種以上的混合物,其中優選使用硫化鈉。這些堿金屬硫化物可以作為水合物或水性混合物、或以無水物的形式使用。作為堿金屬氫硫化物的具體例,可舉出例如氫硫化鈉、氫硫化鉀、氫硫化鋰、氫硫化銣、氫硫化銫和它們2種以上的混合物,其中優選使用氫硫化鈉。這些堿金屬氫硫化物可以作為水合物或水性混合物,或以無水物的形式使用。在PAS(A)的制造時,關于加入硫化劑的量,在由于脫水操作等因而在聚合反應開始前硫化劑的一部分發生損失的情況下,是指由實際的加入量減去該損失部分后的殘存量。另外,也能夠與硫化劑一起,并用堿金屬氫氧化物和/或堿土金屬氫氧化物。作為堿金屬氫氧化物的具體例,可舉出例如氫氧化鈉、氫氧化鉀、氫氧化鋰、氫氧化銣、氫氧化銫和它們2種以上的混合物作為優選的具體例。作為堿土金屬氫氧化物,具體例可舉出例如氫氧化鈣、氫氧化鍶、氫氧化鋇等。在這些堿金屬氫氧化物、堿土金屬氫氧化物中,優選使用氫氧化鈉。作為硫化劑,在使用堿金屬氫硫化物的情況下,特別優選同時使用堿金屬氫氧化物,其使用量可以例示相對于堿金屬氫硫化物1摩爾為0.95~1.20摩爾,優選為1.00~1.15摩爾,進一步優選為1.005~1.100摩爾的范圍。以下,對于PAS(A)的制造方法的一例,將前工序、聚合反應工序、回收工序和后處理工序依次具體地說明。首先對前工序進行說明。硫化劑通常以水合物的形式使用,優選在添加多鹵代芳香族化合物之前,將包含有機極性溶劑和硫化劑的混合物進行升溫,將過剩量的水除去至體系外。另外,在通過該操作而過度地除去水的情況下,優選添加不足部分的水進行補充。此外,作為硫化劑,也可以使用由堿金屬氫硫化物與堿金屬氫氧化物,在反應體系內就地或利用與聚合槽不同的槽調制的堿金屬硫化物。調制堿金屬硫化物的優選條件是在非活性氣體氣氛下,在常溫~150℃,更優選為常溫~100℃的溫度范圍內,在有機極性溶劑中添加堿金屬氫硫化物和堿金屬氫氧化物,在常壓或減壓下,升溫至150℃以上,更優選為180~260℃,使水分蒸餾除去。在該階段可以添加聚合助劑。此外,為了促進水分的蒸餾除去,可以添加甲苯等來進行反應。聚合反應中的聚合體系內的水分量是加入硫化劑每1摩爾優選為0.5~10.0摩爾。這里所謂聚合體系內的水分量,為從加入至聚合體系的水分量減去了除去至聚合體系外的水分量而得的量。此外,加入的水可以為水、水溶液、結晶水等的任一形態。水分量的更優選的范圍是硫化劑每1摩爾為0.75~2.5摩爾,更優選為1.0~1.25摩爾的范圍。為了將水分調整至這樣的范圍,也能夠在聚合前或聚合中途添加水分。在聚合反應工序中,在N-甲基-2-吡咯烷酮等有機極性溶劑中使硫化劑與多鹵代芳香族化合物在200℃以上290℃以下的溫度范圍內進行反應,從而生成PAS(A)。開始聚合反應工序時,優選在非活性氣體氣氛下,在常溫~220℃,優選為100~220℃的溫度范圍,在有機極性溶劑中添加硫化劑和多鹵代芳香族化合物。在該階段可以添加乙酸鈉等聚合助劑。這里,所謂聚合助劑,是指具有調整所得的PAS(A)的粘度的作用的物質。這些原料的加入順序可以順序不同,也可以同時。將這樣的混合物升溫至通常200℃~290℃的范圍。升溫速度沒有特別限制,通常選擇0.01~5℃/分鐘的速度,更優選為0.1~3℃/分鐘的范圍。最終,升溫至250~290℃的溫度,在該溫度反應0.25~50小時,優選為0.5~20小時。在達到最終溫度之前的階段,例如在200℃~245℃反應一定時間,然后升溫至250~290℃的方法在獲得更高的聚合度的情況下是有效的。此時,作為在200℃~245℃的反應時間,通常在0.25小時~20小時的范圍內選擇,優選在0.25~10小時的范圍內選擇。聚合結束后,在回收工序中,從包含聚合物、溶劑等的聚合反應物回收固體物質。作為回收方法,可舉出例如閃蒸法,即,使聚合反應物從高溫高壓(通常245℃以上,0.8MPa以上)的狀態閃蒸至常壓或減壓的氣氛中,與溶劑回收同時將聚合物制成粉粒狀而進行回收的方法;驟冷法,即,將聚合反應物從高溫高壓的狀態逐漸地冷卻而使反應體系內的PAS成分析出,并且在70℃以上,優選為100℃以上的狀態進行過濾分離,從而將包含PAS成分的固體制成顆粒狀而進行回收的方法等。PAS(A)的回收方法不限定于驟冷法、閃蒸法的任一者,優選為通過驟冷法獲得的PAS(A),因為以氯仿提取成分為代表那樣的低聚物成分少,所得的纖維增強樹脂組合物的抗拉強度、伸長率特別優異。作為通過驟冷法獲得的PAS的基于氯仿的低聚物提取量,可以例示2質量%以下,可以例示更優選為1質量%以下。PAS(A)是在經由上述聚合、回收工序而獲得的固體物質中,然后,作為后處理工序,通過熱水處理、有機溶劑實施洗滌來使用。經由上述回收工序而獲得的固體物質除了PAS(A)以外,還包含作為聚合副產物的堿金屬鹵化物、堿金屬有機物等離子性雜質,因此進行洗滌是慣例。可舉出作為洗滌液使用例如水、有機溶劑進行洗滌的方法,從簡便并且便宜地獲得PAS(A)方面出發,可以例示使用了水的洗滌作為優選的方法。作為使用的水的種類,優選使用離子交換水、蒸餾水。將經由回收工序而獲得的固體物質進行洗滌時的洗滌溫度優選為50℃以上200℃以下,更優選為150℃以上200℃以下,進一步優選為180℃以上200℃以下。利用100℃以上的液體的處理的操作通常在規定量的液體中,規定量投入經由回收工序而獲得的固體物質,在常壓或壓力容器內進行加熱、攪拌來進行。洗滌可以進行多次,各洗滌時的洗滌溫度可以不同,為了獲得離子性雜質少的PAS(A),可以在150℃以上的溫度進行至少1次,優選進行2次以上洗滌,在各洗滌之間經由將聚合物與洗滌液進行分離的過濾工序是更優選的方法。在獲得PAS(A)時,在進行洗滌的情況下,可以使用洗滌添加劑,作為這樣的洗滌添加劑,可以例示酸、堿金屬鹽或堿土金屬鹽。在使用酸的情況下,優選在洗滌所使用的水中添加有機酸或無機酸等而制成酸性的水溶液中,使被洗滌的PAS浸漬,以使加熱洗滌后的水溶液的pH成為2~8。作為有機酸、無機酸,可以例示乙酸、丙酸、鹽酸、硫酸、磷酸、甲酸等,但不限定于此,優選為乙酸、鹽酸。在本發明中,使用酸作為洗滌添加劑而獲得的PAS(A)被稱為酸末端品。在使用堿金屬鹽或堿土金屬鹽作為洗滌添加劑的情況下,可以例示在洗滌所使用的水中添加了堿金屬鹽或堿土金屬鹽的水溶液中,使要洗滌的PAS浸漬的方法,這樣的堿金屬鹽或堿土金屬鹽的量相對于PAS(A)優選為0.01~5質量%,進一步優選為0.1~0.7質量%。作為堿金屬鹽、堿土金屬鹽,可以例示上述有機酸或無機酸的鈣鹽、鉀鹽、鈉鹽、鎂鹽等,但不限定于此。洗滌添加劑可以在洗滌工序的任一階段使用,為了利用少量的添加劑有效率地進行洗滌,優選將利用上述回收工序回收的固體物質用水進行數次洗滌,然后在添加有洗滌添加劑的水溶液中,使要洗滌的PAS含浸,在150℃以上進行處理的方法。洗滌時的PAS與洗滌液的比例優選洗滌液多,通常,相對于洗滌液1升,優選選擇PAS(A)10~500g的浴比,進一步優選為50~200g。這樣獲得的PAS(A)在常壓下和/或減壓下進行干燥。作為這樣的干燥溫度,優選為120~280℃的范圍,更優選為140~250℃的范圍。干燥氣氛為氮氣、氦氣、減壓下等非活性氣氛,也可以為氧氣、空氣等氧化性氣氛,空氣與氮氣的混合氣氛中的任一種,從熔融粘度的關系出發,優選為非活性氣氛。干燥時間優選為0.5~50小時,更優選為1~30小時,進一步優選為1~20小時。<碳二亞胺化合物(B)>本發明中的碳二亞胺化合物(B)為脂肪族碳二亞胺化合物。在碳二亞胺化合物(B)中使用了不是脂肪族碳二亞胺化合物,例如芳香族碳二亞胺化合物的情況下,不能獲得本發明的纖維增強樹脂組合物。推測這是因為與脂肪族碳二亞胺化合物具有的碳二亞胺基相比,芳香族碳二亞胺化合物所具有的碳二亞胺基與PAS(A)所具有的官能團的反應性低。所謂脂肪族碳二亞胺化合物,為以通式-N=C=N-R3-(式中,R3表示環亞己基等脂環式化合物的2價有機基團、或亞甲基、亞乙基、亞丙基、甲基亞乙基等脂肪族化合物的2價有機基團)所示的重復單元作為主要構成單元的、含有該重復單元優選70摩爾%以上、更優選為90摩爾%以上、進一步優選為95摩爾%以上的均聚物或共聚物。脂肪族碳二亞胺化合物的合成法沒有特別限定,可以通過例如使有機多異氰酸酯在促進異氰酸酯基的碳二亞胺化反應的催化劑(以下也稱為“碳二亞胺化催化劑”)的存在下進行反應,從而合成脂肪族碳二亞胺化合物。作為該脂肪族碳二亞胺化合物的合成所使用的有機多異氰酸酯,優選為有機二異氰酸酯。作為這樣的有機二異氰酸酯,可舉出例如,環亞丁基-1,3-二異氰酸酯、環亞戊基-1,3-二異氰酸酯、環亞己基-1,3-二異氰酸酯、環亞己基-1,4-二異氰酸酯、1-甲基環亞己基-2,4-二異氰酸酯、1-甲基環亞己基-2,6-二異氰酸酯、1-異氰酸酯-3,3,5-三甲基-5-異氰酸酯甲基環己烷、環己烷-1,3-雙(甲基異氰酸酯)、環己烷-1,4-雙(甲基異氰酸酯)、二環己基甲烷-2,4’-二異氰酸酯、二環己基甲烷-4,4’-二異氰酸酯、亞乙基二異氰酸酯、四亞甲基-1,4-二異氰酸酯、六亞甲基-1,6-二異氰酸酯、十二亞甲基-1,12-二異氰酸酯、賴氨酸二異氰酸酯甲基酯等、通過這些有機二異氰酸酯的化學計量的過剩量與2官能性含活性氫化合物的反應而獲得的兩末端異氰酸酯預聚物等。這些有機二異氰酸酯可以1種單獨使用,或也可以將2種以上混合使用。此外,作為根據情況與有機二異氰酸酯一起使用的其它有機多異氰酸酯,可舉出例如,環己烷-1,3,5-三異氰酸酯、環己烷-1,3,5-三(甲基異氰酸酯)、3,5-二甲基環己烷-1,3,5-三(甲基異氰酸酯)、1,3,5-三甲基環己烷-1,3,5-三(甲基異氰酸酯)、二環己基甲烷-2,4,2’-三異氰酸酯、二環己基甲烷-2,4,4’-三異氰酸酯等3官能以上的有機多異氰酸酯、通過這些3官能以上的有機多異氰酸酯的化學計量的過剩量與2官能以上多官能性含活性氫化合物的反應而獲得的末端異氰酸酯預聚物等。上述其它有機多異氰酸酯可以1種單獨使用,或也可以將2種以上混合使用,其使用量為有機二異氰酸酯每100質量份,優選為0~40質量份,更優選為0~20質量份。進一步,在脂肪族碳二亞胺化合物的合成時,可以根據需要添加有機單異氰酸酯,從而將所得的脂肪族碳二亞胺化合物的分子量進行適當地控制。作為這樣的有機單異氰酸酯,可舉出例如甲基異氰酸酯、乙基異氰酸酯、正丙基異氰酸酯、正丁基異氰酸酯、月桂基異氰酸酯、硬脂基異氰酸酯等烷基單異氰酸酯類、環己基異氰酸酯、4-甲基環己基異氰酸酯、2,5-二甲基環己基異氰酸酯等環烷基單異氰酸酯類。這些有機單異氰酸酯可以1種單獨使用,或也可以將2種以上混合使用,其使用量根據脂肪族碳二亞胺化合物的所期望的分子量等而變化,有機多異氰酸酯成分每100質量份,優選為0~40質量份,更優選為0~20質量份。此外,作為碳二亞胺化催化劑,可舉出例如1-苯基-2-磷雜環戊烯-1-氧化物、1-苯基-3-甲基-2-磷雜環戊烯-1-氧化物、1-苯基-2-磷雜環戊烯-1-硫化物、1-苯基-3-甲基-2-磷雜環戊烯-1-硫化物、1-乙基-2-磷雜環戊烯-1-氧化物、1-乙基-3-甲基-2-磷雜環戊烯-1-氧化物、1-乙基-2-磷雜環戊烯-1-硫化物、1-乙基-3-甲基-2-磷雜環戊烯-1-硫化物、1-甲基-2-磷雜環戊烯-1-氧化物、1-甲基-3-甲基-2-磷雜環戊烯-1-氧化物、1-甲基-2-磷雜環戊烯-1-硫化物、1-甲基-3-甲基-2-磷雜環戊烯-1-硫化物、它們的3-磷雜環戊烯異構體等磷雜環戊烯化合物、五羰基鐵、九羰基二鐵、四羰基鎳、六羰基鎢、六羰基鉻等金屬羰基配位化合物、鈹、鋁、鋯、鉻、鐵等金屬的乙酰丙酮配位化合物、磷酸三甲酯、磷酸三乙酯、磷酸三異丙酯、磷酸三叔丁酯、磷酸三苯酯等磷酸酯等。上述碳二亞胺化催化劑可以1種單獨使用,或也可以將2種以上混合使用。該催化劑的使用量是有機多異氰酸酯成分每100質量份,優選為0.001~30質量份,更優選為0.01~10質量份。脂肪族碳二亞胺化合物的合成反應的溫度根據有機多異氰酸酯、有機單異氰酸酯、碳二亞胺化催化劑的種類進行適宜選定,通常為20~200℃。在脂肪族碳二亞胺化合物的合成反應時,有機多異氰酸酯和有機單異氰酸酯成分可以在反應前全部量添加,或可以將其一部分或全部在反應中,連續地或階段性地添加。此外,也可以將可以與異氰酸酯基反應的化合物在脂肪族碳二亞胺化合物的合成反應的初期至后期的適宜的反應階段進行添加,將脂肪族碳二亞胺化合物的末端異氰酸酯基進行封閉,調節所得的脂肪族碳二亞胺化合物的分子量,此外也可以在脂肪族碳二亞胺化合物的合成反應的后期添加,將所得的脂肪族碳二亞胺化合物的分子量限制為規定值。作為這樣的可以與異氰酸酯基反應的化合物,可舉出例如甲醇、乙醇、異丙醇、環己醇等醇類、二甲基胺、二乙基胺、芐基胺等胺類。本發明所使用的脂肪族碳二亞胺化合物的質均分子量優選為500~10,000,更優選為1,000~5,000。如果脂肪族碳二亞胺化合物的質均分子量在該范圍,則可以以高水平達成兼具作為本發明的效果的抗拉強度、伸長率這樣的力學特性的提高與生產性和成型加工性。另外,脂肪族碳二亞胺化合物的質均分子量可以通過SEC(尺寸排阻色譜)等分析方法而求出。<碳纖維(C)>作為本發明中的碳纖維(C),可以使用聚丙烯腈(PAN)系碳纖維、瀝青系碳纖維、人造絲系碳纖維等,也可以使這些纖維2種以上混合存在。碳纖維(C)的抗拉強度優選為2,000MPa以上,更優選為3,000MPa以上,進一步優選為4,000MPa以上。此外,碳纖維(C)的拉伸彈性模量優選為200GPa以上700GPa以下。進一步,碳纖維(C)的拉伸伸長率優選為0.5%以上,更優選為1.0%以上,進一步優選為1.8%以上,特別優選為2.0%以上。通過使用高伸長率的碳纖維(C),從而可以以高水平達成本發明的纖維增強樹脂組合物的抗拉強度、伸長率這樣的力學特性的提高,因此特別優選。從這樣的抗拉強度、拉伸彈性模量、拉伸伸長率的平衡的觀點出發,優選使用PAN系碳纖維作為碳纖維(C)。碳纖維(C)的通過X射線光電子能譜法(XPS)測得的纖維表面的氧(O)與碳(C)的原子數之比即表面氧濃度比(O/C)優選為0.05~0.50,更優選為0.08~0.40,進一步優選為0.10~0.30。表面氧濃度比(O/C)越高,則碳纖維表面的官能團量越多,越可以提高碳纖維(C)與上漿劑(D)的粘接性,另一方面,如果表面氧濃度比(O/C)過高,則擔憂碳纖維表面的晶體結構的破壞,因此表面氧濃度比(O/C)在優選的范圍內,可以獲得力學特性特別優異的纖維增強樹脂組合物。碳纖維(C)的表面氧濃度比(O/C)通過X射線光電子能譜法,按照以下步驟來求出。首先,將用溶劑除去了上漿劑(D)等的碳纖維(C)進行切割并在銅制的試樣支持臺上展開排列后,將光電子脫出角度設為90°,作為X射線源使用MgKα1,2,將試樣室中保持于1×10-8托。作為伴隨測定時的帶電的峰的校正,使C1S的主峰的動能值(K.E.)與969eV一致。C1S峰面積通過作為K.E.在958~972eV的范圍引出直線的基線而求出。O1S峰面積通過作為K.E.在714~726eV的范圍引出直線的基線而求出。這里所謂表面氧濃度比(O/C),由上述O1S峰面積與C1S峰面積的比,使用裝置固有的靈敏度校正值而作為原子數比算出。作為控制表面氧濃度比(O/C)的手段,沒有特別限定,可以采用例如,電解氧化處理、藥液氧化處理和氣相氧化處理等手法,其中優選為電解氧化處理。此外,碳纖維(C)的平均纖維直徑沒有特別限定,從所得的纖維增強樹脂組合物的力學特性與表面外觀的觀點出發,優選為1~20μm的范圍內,更優選為3~15μm的范圍內。關于碳纖維(C),從操作性方面出發,可以作為將多根單絲成束而成的碳纖維束使用。作為構成碳纖維束的單絲數,從操作性方面出發,優選為100根以上350,000根以下,更優選為1,000以上250,000以下,進一步優選為10,000以上100,000以下。通過使構成碳纖維束的單絲數為這樣的范圍,從而經濟性良好地獲得本發明的纖維增強樹脂組合物。<上漿劑(D)>本發明中的上漿劑(D)是在1分子中具有3個以上選自羧基、氨基、羥基和環氧基中的至少1種官能團的化合物。上述官能團可以在1分子中混合存在2種以上,可以將1分子中具有3個以上1種官能團的化合物并用2種以上。在僅使用在1分子中不足3個上述官能團的化合物的情況下,與碳纖維(C)的表面官能團、碳二亞胺化合物(B)的反應位點變得不充分,所得的纖維增強樹脂組合物的抗拉強度、伸長率這樣的力學特性降低。因此,構成上漿劑(D)的化合物所具有的官能團的數目需要在1分子中有3個以上。上述官能團以外的官能團,例如烷氧基硅烷缺乏與碳纖維表面的反應性,因此將在1分子中具有烷氧基硅烷和環氧基各1個的硅烷偶聯劑用于上漿劑(D)的情況下,得不到本發明作為目的的力學特性優異的纖維增強樹脂組合物。上漿劑(D)優選構成其的化合物為脂肪族化合物。這里所謂脂肪族化合物,是指非環式直鏈狀飽和烴、支鏈狀飽和烴、非環式直鏈狀不飽和烴、支鏈狀不飽和烴、或將這些烴的碳原子(CH3、CH2、CH、C)置換成氧原子(O)、氮原子(NH、N)、羰基原子團(CO)的鏈狀結構的化合物。即,脂肪族化合物不具有芳香環等環狀結構。通過使上漿劑(D)為脂肪族化合物,從而與作為碳二亞胺化合物(B)而使用的脂肪族碳二亞胺化合物的親和性提高,因此獲得力學特性優異的纖維增強樹脂組合物,因此優選。作為構成上漿劑(D)的化合物的具體例,可舉出多官能環氧樹脂、丙烯酸系聚合物、多元醇、聚乙烯亞胺等,特別優選為與碳纖維(C)的表面官能團、碳二亞胺化合物(B)這兩者的反應性高的多官能環氧樹脂。作為多官能環氧樹脂,可舉出3官能以上的脂肪族環氧樹脂、苯酚酚醛清漆型環氧樹脂等。其中,從與脂肪碳二亞胺化合物的親和性的觀點出發,優選為3官能以上的脂肪族環氧樹脂。另外,所謂3官能以上的脂肪族環氧樹脂,是指1分子中具有3個以上環氧基的脂肪族環氧樹脂。作為3官能以上的脂肪族環氧樹脂的具體例,可舉出例如,甘油三縮水甘油基醚、二甘油聚縮水甘油基醚、聚甘油聚縮水甘油基醚、山梨糖醇聚縮水甘油基醚、阿拉伯糖醇聚縮水甘油基醚、三羥甲基丙烷三縮水甘油基醚、季戊四醇聚縮水甘油基醚等脂肪族多元醇的聚縮水甘油基醚等。在這些脂肪族環氧樹脂中,從1分子中大量包含反應性高的環氧基,并且水溶性高,向碳纖維(C)的涂布容易出發,在本發明中優選使用甘油三縮水甘油基醚、二甘油聚縮水甘油基醚、聚甘油聚縮水甘油基醚。本發明中的所謂丙烯酸系聚合物,為丙烯酸、甲基丙烯酸和馬來酸的聚合物,是1分子中含有3個以上羧基的聚合物的總稱。具體而言,可舉出聚丙烯酸、丙烯酸與甲基丙烯酸的共聚物、丙烯酸與馬來酸的共聚物、或這些的2種以上的混合物。進一步,丙烯酸系聚合物只要上述官能團的數目在1分子中為3個以上,就可以將羧基用堿部分地中和(即,成為羧酸鹽)。作為上述堿,可舉出例如,氫氧化鈉、氫氧化鋰、氫氧化鉀等堿金...
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 纖維增強樹脂組合物以及纖維增強復合材料的制造方法與工藝
- 多巴胺改性玻璃纖維?環氧樹脂復合材料的制備方法與流程
- 一種環氧樹脂發泡材料基材及其制備工藝的制造方法與工藝
- 一種纖維增強的生物樹脂膠復合材料及其制備方法與制造工藝
- 全玄武巖纖維增強樹脂基復合材料自行車及其制備方法與制造工藝
- 高硬度環氧改性聚氨酯復合材料及其制備方法與制造工藝
- 環氧樹脂復合材料和包含使用其的絕緣層的印刷電路板的制造方法與工藝
- 環氧樹脂組合物、樹脂片、預浸料及覆金屬層疊板、印刷布線基板、半導體裝置的制造方法
- 環氧樹脂組合物、樹脂片、預浸料及覆金屬層疊板、印刷布線基板、半導體裝置的制造方法
- 碳纖維增強復合材料用環氧樹脂組合物、樹脂片、預浸料、碳纖維增強復合材料的制造方法與工藝