化合物的制作方法與工藝
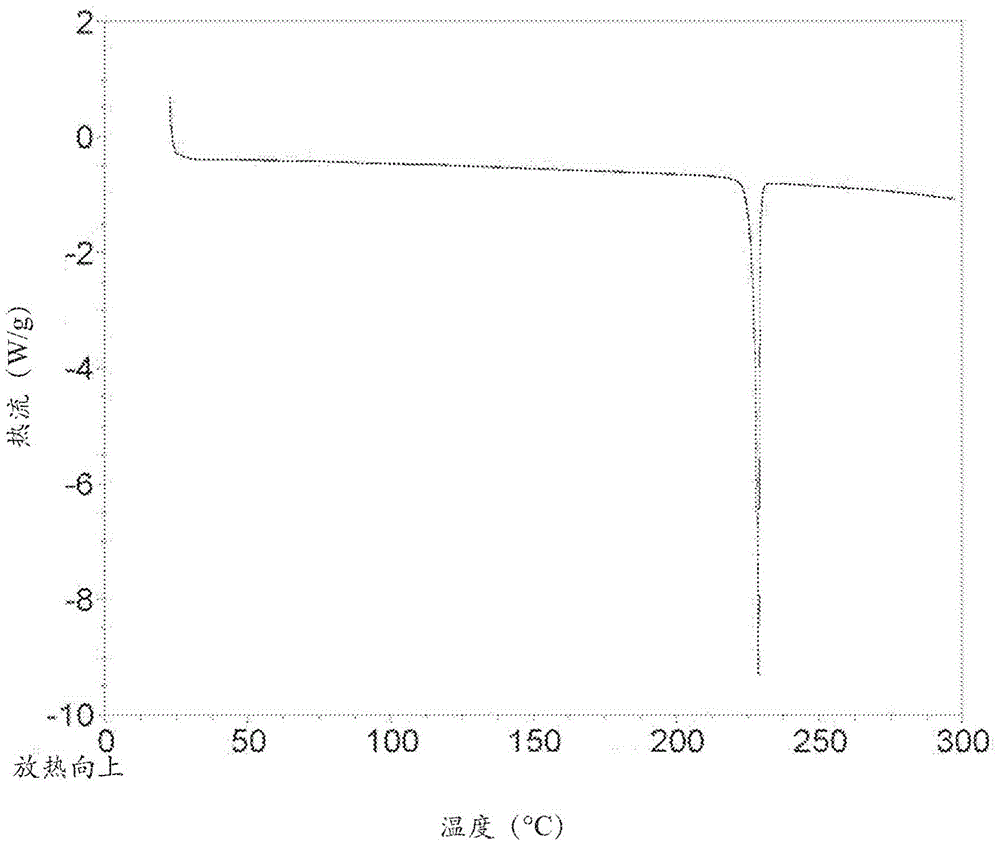
化合物本發明涉及某些新穎氨基吡嗪衍生物或其醫藥學上可接受的鹽,它們具有抗癌活性并且因此有用于治療人體或動物體的方法。本發明還涉及用于制造所述氨基吡嗪衍生物的方法、含有它們的醫藥組合物以及它們在治療方法中的用途,例如用于制造供預防或治療溫血動物(如人)的癌癥用的藥物,包括用于預防或治療癌癥。本發明還涉及氨基吡嗪衍生物,這些氨基吡嗪衍生物是酶的PI3激酶家族(它被替代性地稱為磷脂酰肌醇-3-激酶家族或PI3K家族)、尤其PI3K-α和PI3K-δ同工型的選擇性抑制劑,并且例如有用于抗腫瘤療法。在癌癥領域中,近年來已發現細胞可能由于它DNA的一部分轉化成致癌基因而變成癌性,致癌基因是一種一旦激活即導致惡性腫瘤細胞形成的基因(布拉德肖(Bradshaw),《誘變》(Mutagenesis),1986,1,91)。若干此類致癌基因引起肽的產生,這些肽是激酶,是一類能夠使它們的蛋白質或脂質底物磷酸化的酶。存在若干類激酶。首先為酪氨酸激酶,它們可以是受體酪氨酸激酶或非受體酪氨酸激酶。基于可以與不同受體酪氨酸激酶的細胞外表面結合的生長因子家族,不同類別的受體酪氨酸激酶是已知的(威爾克斯(Wilks),《癌癥研究進展》(AdvancesinCancerResearch),1993,60,43-73);作為一個實例,該分類包括I類受體酪氨酸激酶,它包括受體酪氨酸激酶的EGF家族。非受體酪氨酸激酶位于細胞內;已知不同類別的非受體酪氨酸激酶,包括Src家族,如Src、Lyn、Fyn以及Yes酪氨酸激酶。其次,某些激酶屬于也位于細胞內的絲氨酸/蘇氨酸激酶的類別。絲氨酸/蘇氨酸激酶信號傳導路徑包括Raf-MEK-ERK級聯和PI3激酶下游的那些級聯,如PDK-1、AKT以及mTOR(布盧姆-延森(Blume-Jensen)和亨特(Hunter),《自然》(Nature),2001,411,355)。還已知某些其他激酶屬于脂質激酶的類別,它們位于細胞內并且與上述激酶一樣,涉及生物化學信號的傳遞,如影響腫瘤細胞生長和侵襲性的那些生物化學信號。已知不同類別的脂質激酶,包括上述PI3激酶家族。現充分了解,致癌基因和腫瘤抑制基因的失調促成惡性腫瘤形成,例如經由增加細胞增殖或增加細胞存活。目前還已知,由PI3激酶家族介導的信號傳導路徑在多個細胞過程(包括增殖和存活)中具有重要作用,并且這些路徑的失調是廣譜人類癌癥和其他疾病的病因因素(卡索(Katso)等人,《細胞和發育生物學年鑒》(AnnualRev.CellDev.Biol.),2001,17:615-617和福斯特(Foster)等人,《細胞科學雜志》(J.CellScience),2003,116:3037-3040)。脂質激酶的PI3激酶家族是使磷脂酰肌醇(PI)的肌醇環的3-位磷酸化的一組酶。已知主要的三組PI3激酶,它們是根據其生理底物特異性分類的(凡艾斯布魯艾克(Vanhaesebroeck)等人,《生物科學趨勢》(TrendsinBiol.Sci.),1997,22,267;恩格爾曼(Engleman)等人,《自然綜述·遺傳學》(NatureReviewGenetics),2006,7,607)。III類PI3激酶僅使PI磷酸化。相比之下,II類PI3激酶使PI和PI4磷酸酯兩者磷酸化[在下文中簡稱為PI(4)P]。I類PI3激酶使PI、PI(4)P以及PI4,5-雙磷酸酯磷酸化[在下文中簡稱為PI(4,5)P2],但據相信僅PI(4,5)P2為生理細胞底物。PI(4,5)P2的磷酸化產生脂質第二信使PI3,4,5-三磷酸酯[在下文中簡稱為PI(3,4,5)P3]。與這一超家族的關系較疏遠的成員為IV類激酶,如mTOR和DNA依賴性蛋白激酶,它們使蛋白質底物內的絲氨酸/蘇氨酸殘基磷酸化。這些脂質激酶中被最多研究和了解的是I類PI3激酶。I類PI3激酶是由p110催化亞基和調節亞基組成的雜二聚體,并且基于調節搭配物和調節機制,該家族進一步分為Ia類和Ib類酶(恩格爾曼等人,《自然綜述·遺傳學》,2006,7,607)。Ia類酶由三個不同的催化亞基(p110α、p110β以及p110δ,通過命名法將PI3激酶同工型分別定義為α、β或δ)組成,這些催化亞基與五個不同的調節亞基(p85α、p55α、p50α、p85β以及p55γ)二聚,其中所有催化亞基均能夠與所有調節亞基相互作用以形成各種雜二聚體。Ia類PI3激酶總體上經由調節亞基SH2結構域與激活受體或接附蛋白的特異性磷酸化-酪氨酸殘基(如IRS-1)的相互作用,對受體酪氨酸激酶的生長因子刺激起反應得到激活。p110α和p110β均在所有細胞類型和組織中廣泛表達,而p110δ表達更受限于白細胞群體和一些上皮細胞。相比之下,單一Ib類酶由與p101調節亞基相互作用的p110γ催化亞基組成。此外,Ib類酶對G蛋白偶聯受體(GPCR)系統起反應以及通過上述機制激活。目前存在大量證據表明,在多種人類癌癥中,Ia類PI3激酶或者直接或者間接地促成腫瘤形成(維萬科(Vivanco)和索耶斯(Sawyers),《自然綜述·癌癥》(NatureReviewsCancer),2002,2,489-501)。確切地說,編碼PI3激酶的p110α催化亞基的PIK3CA基因廣泛地涉及腫瘤形成。激活點突變最常見于p110α的螺旋或催化結構域,提高全酶的PI3激酶活性并且可以轉化細胞。特別地,它們已以大頻率被報導為各式各樣腫瘤類型中的體細胞產生的突變(塞繆爾斯(Samuels)等人,《科學》(Science),2004,304,554;塞繆爾斯等人,《癌細胞》(CancerCell),2005,7,561;恩格爾曼等人,《自然綜述·遺傳學》,2006,7,607;趙L(ZhaoL)和沃格特PK(VogtPK),《致癌基因》(Oncogene)2008,275486)。也已在如卵巢癌和結腸癌的癌癥中鑒別出p85α中的腫瘤相關突變(菲利普(Philp)等人,《癌癥研究》(CancerResearch),2001,61,7426-7429)。此外,p110α亞基在如卵巢腫瘤(沙耶斯迪(Shayesteh)等人,《自然·遺傳學》(NatureGenetics),1999,21,99-102)和子宮頸腫瘤(馬(Ma)等人,《致癌基因》,2000,19,2739-2744)的一些腫瘤中擴增。除直接作用以外,據相信Ia類PI-3激酶的激活促成信號傳導路徑上游發生的腫瘤形成事件,例如經由受體酪氨酸激酶、GPCR系統或整合素的配體依賴性或非配體依賴性激活(瓦拉(Vara)等人,《癌癥治療綜述》(CancerTreatmentReviews),2004,30,193-204)。這些上游信號傳導路徑的實例包括在多種腫瘤中使得PI3-激酶介導的路徑激活的受體酪氨酸激酶Erb2的過度表達(哈拉瑞(Harari)等人,《致癌基因》,2000,19,6102-6114),以及致癌基因Ras的過度表達(考夫曼-澤(Kauffmann-Zeh)等人,《自然》,1997,385,544-548)。另外,Ia類PI3激酶可能促成由不同下游信號傳導事件造成的腫瘤形成。舉例來說,催化PI(3,4,5)P3轉化回PI(4,5)P2的PTEN腫瘤抑制因子磷酸酶的作用的缺失經由PI3激酶介導產生PI(3,4,5)P3的失調而與極寬范圍的腫瘤相關(辛普森(Simpson)和帕森斯(Parsons),《實驗細胞研究》(Exp.CellRes.),2001,264,29-41)。此外,加強其他PI3激酶介導的信號傳導事件的作用被認為促成各種癌癥,例如通過激活Akt(尼克爾森(Nicholson)和安德森(Anderson),《細胞信號傳導》(CellularSignalling),2002,14,381-395)。因此,PI3激酶的常見失調連同上游和下游信號傳導路徑的那些失調共同使PI3激酶的常見失調成為人類癌癥中最常見的失調路徑之一(亨尼西(Hennessey)等人,《自然綜述·藥物發現》(NatureReviewsDrugDiscovery),2005,4,988)。除在腫瘤細胞中介導增殖和存活信號傳導方面的作用之外,還存在充分證據表明Ia類PI3激酶還將經由其在腫瘤相關的基質細胞中的功能促成腫瘤形成。舉例來說,PI3激酶信號傳導已知在內皮細胞中對如VEGF的促血管生成因子起反應來介導血管生成事件方面起重要作用(阿比德(Abid)等人,《動脈硬化、血栓和血管生物學》(Arterioscler.Thromb.Vasc.Biol.),2004,24,294-300)。因為I類PI3激酶還涉及活動和遷移(索耶(Sawyer),《研究藥物的專家意見》(ExpertOpinionInvestig.Drugs),2004,13,1-19),所以PI3激酶抑制劑應經由抑制腫瘤細胞侵襲和轉移提供治療效益。另外,I類PI3激酶在調節具有PI3激酶活性的促成炎性細胞的促腫瘤形成作用的免疫細胞方面起重要作用(庫曾斯(Coussens)和韋布(Werb),《自然》,2002,420,860-867)。實際上,Ia類PI3激酶即PI3激酶δ尤其涉及血液科惡性疾病中的腫瘤形成,如慢性淋巴細胞白血病(CLL)、急性淋巴母細胞白血病(ALL)以及套細胞淋巴瘤(MCL)。在各式各樣的惡性淋巴細胞中報導有PI3K(主要為p110δ)的信號傳導提高(赫爾曼(Herman)等人,《血液》(Blood),2010,116.2078;池田(Ikeda)等人,《血液》,2010,116,1460;烏丁(Uddin)等人,《血液》,2006,108,4178;魯德柳斯(Rudelius)等人,《血液》2006,108,1668;加西亞-馬丁內斯(Garcia-Martinez.),《英國癌癥雜志》(BrJCancer),2011,104,1116;倫尼(Renne)等人,《白血病》(Leukemia),2007,2,780)。這已引起靶向PI3激酶δ的藥劑的開發,其中血液科惡性疾病具有有前景的初始臨床結果。(卡斯蒂略(Castillo)等人,《研究藥物的專家意見》,2012,21,15)。此等發現表明,I類PI3激酶的藥理學抑制劑應具有用于治療不同形式癌癥疾病的治療價值,該癌癥疾病包括實體腫瘤(如癌瘤和肉瘤)和白血病以及淋巴性惡性疾病。探究PI3激酶的生理學和病理學作用的早期研究(臨床前和臨床研究兩者)大量使用具有受限制的激酶抑制選擇性的藥劑,或者在更寬的激酶家族中,或者在PI3激酶家族中,或者在PI3激酶1類家族中擴展。因此,需要選擇性更高的藥物PI3激酶1類抑制劑以提供有用治療劑,它們有可能改良進入臨床的初始藥劑所遞送的治療范圍。總體而言,本發明的化合物具有針對I類PI3激酶的子組,尤其針對Ia類PI3激酶-α和-δ同工型的強力抑制活性,其中-γ和尤其-β同工型相對保守。這些化合物也選擇性針對更寬的PI3激酶家族和更寬的激酶組。這些化合物具有針對I類PI3激酶的足夠效力,它們可以按足以抑制I類PI3-激酶同工型的子組、尤其抑制Ia類PI3激酶-α和-δ的量使用,同時顯示出針對其他激酶的極小活性。人類癌癥和其他疾病中PI3激酶信號傳導的失調的理解經由被稱為個人化醫療(PHC)或個人化藥物的方法提供靶向最可能受益于這一專利中所述的藥劑的治療的患者子組的前景。關于這些藥劑,疾病取決于PI3K-α信號傳導和/或PI3K-δ信號傳導的提高或以其他方式改變的患者可能尤其受益于治療。在本領域中眾所周知,可以使用診斷學來提供反應-預測生物標記讀出。這些診斷學可以測量路徑失調的一個或多個讀出,如(但不限于)PIK3CA、PTEN或p85(PIK3R)基因的突變;PIK3CA基因的擴增或拷貝數增加;PI3K-α和/或-δ同工型的過度表達或活性提高;或在該路徑內使用磷酸化生物標記讀出,如磷酸化RTK或磷酸化AKT。另外,具有異常或失調PIK3CA或PI3K-α的腫瘤中的另外的基因(如Kras,一種潛在抗性標記)的突變狀態或激活狀態的測量(恩格爾曼(Engelman)等人,《自然·醫學》(NatureMedicine),200814,第1351-1355頁;伊爾(Ihle)等人,《癌癥研究》,2009,69,第143-160頁;揚庫(Janku)等人,《分子癌癥治療學》(MolecularCancerTherapeutics),2011,10,第558-564頁)可以有助于提高個人化藥物方法的預測。可替代地,在另一個靶向性但特異性較差的方法中,治療可以聚焦于相關PI3K同工型的失調已知最為普遍的疾病子集中。所述化合物可以用于或者單獨或者與另外的一種或多種醫藥劑組合靶向疾病。將PI3激酶抑制劑與其他療法組合可以通過克服或者先天性或者對PI3激酶藥劑起反應誘導的抗性機制來改良功效。存在大量臨床前數據來支持此類方法(考特尼(Courtney)等人,《臨床腫瘤學雜志》(JClinOncol),2010,28,1075;恩格爾曼等人,《自然綜述·遺傳學》,2006,7,607)。一種方法為與調節PI3激酶信號傳導路徑中的其他軸的藥劑(例如mTOR、AKT、RTK、其他PI3激酶藥劑)的‘路徑內’組合。第二方法為其中抑制一個以上信號傳導路徑的效益可能優于抑制單一路徑的‘路徑之間’組合(例如與MEK抑制劑、Raf抑制劑、Bcl家族調節劑、RTK抑制劑或DNA損傷信號傳導調節劑(如PARP抑制劑)組合)。其他方法包括PI3激酶抑制劑與已在臨床實踐中確定的藥劑或方案組合的方法,即所謂的護理標準(SoC)方法,或與靶向非腫瘤細胞機制(如腫瘤基質細胞或經由免疫系統)的藥劑組合。除腫瘤形成之外,有證據表明I類PI3激酶在其他疾病中起作用(懷曼(Wymann)等人,《藥理學科學趨勢》(TrendsinPharmacologicalScience),2003,24,366-376)。Ia類PI3激酶(尤其PI3K-δ)與Ib類酶(PI3K-γ)兩者和單一的Ib類酶(PI3K-γ)在免疫系統的細胞中具有重要作用(小保(Koyasu),《自然·免疫學》(NatureImmunology),2003,4,313-319),并且因此它們是炎性和過敏性適應癥的治療目標。PI3激酶的抑制也如前所述有用于經由消炎作用或直接通過影響心肌細胞來治療心血管疾病(普拉薩德(Prasad)等人,《心血管醫學趨勢》(TrendsinCardiovascularMedicine),2003,13,206-212)。因此,I類PI3激酶的抑制劑可以具有在預防和治療除癌癥以外的多種疾病方面的價值。本發明的化合物(即氨基吡嗪衍生物)已發現具有強力抗腫瘤活性,有用于抑制由惡性疾病引起的不受控制的細胞增殖。不希望僅憑借對單一生物過程的作用就暗示本發明中所披露的化合物具有藥理學活性,據相信這些化合物借助于抑制I類PI3激酶,尤其借助于抑制Ia類PI3激酶的子組,更確切地說借助于抑制PI3K-α和-δ同工型來提供抗腫瘤作用。本發明化合物也可以有用于抑制由不同非惡性疾病引起的不受控制的細胞增殖,這些疾病為如炎性疾病(例如類風濕性關節炎和炎性腸病)、纖維化疾病(例如肝硬化和肺纖維化)、絲球體腎炎、多發性硬化癥、牛皮癬、良性前列腺肥大(BPH)、皮膚超敏反應、血管疾病(例如動脈粥樣硬化和再狹窄)、過敏性哮喘、胰島素依賴性糖尿病、糖尿病性視網膜病變以及糖尿病性腎病變。脯氨酸酰胺已由諾華(Novartis)在國際專利申請WO2009/080705、WO2010/029082以及WO2011/000905中披露為選擇性PI3K-α選擇性藥劑。包含ATR激酶抑制劑的氨基吡嗪已披露于WO2011/143426和WO2010/071837(威泰克斯(Vertex))中。根據本發明的一個方面,提供一種具有化學式(I)的化合物其中:R1為甲基或乙基;并且R2為經羥基取代的(C2-3)烷基;或其醫藥學上可接受的鹽。在本發明的另一個方面中,提供一種如上文所定義的具有化學式(I)的化合物。應了解,術語“經羥基取代的(C2-3)烷基”包括直鏈和分支鏈烷基兩者,例如圖示為以下基團(i)到(xi)的那些基團:應了解,就以上所定義的某些具有化學式(I)的化合物可憑借一個或多個不對稱碳原子而以光學活性或外消旋形式存在來說,本發明在其定義中包括具有PI3K-α和-δ抑制活性的任何此類光學活性或外消旋形式。光學活性形式的合成可通過本領域中熟知的有機化學的標準技術,例如通過從光學活性起始物質合成或通過拆分外消旋形式來進行。類似地,可以使用標準實驗室技術評估上文所提及的活性。在此所述的化合物的特定對映異構體的活性可以高于同一化合物的其他對映異構體。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,它為對映異構體過量(ee%)≥95%、≥98%或≥99%的單一對映異構體。適宜地,單一對映異構體以對映異構體過量(ee%)≥99%存在。根據本發明的另一個方面,提供一種醫藥組合物,它包括具有化學式(I)的化合物,所述化合物為對映異構體過量(ee%)≥95%、≥98%或≥99%的單一對映異構體;或其醫藥學上可接受的鹽,以及醫藥學上可接受的稀釋劑或載劑。適宜地,單一對映異構體以對映異構體過量(ee%)≥99%存在。一些具有化學式(I)的化合物可以為結晶并且可以具有一種以上晶形。應了解,本發明涵蓋任何晶形或非晶形或其混合物,該形式具有有用于抑制PI3K-α和-δ活性的性質,在本領域中熟知如何通過下文所述的標準測試測定晶形或非晶形對于抑制PI3K-α和/或-δ活性的功效。總體上已知可以使用常規技術分析結晶物質,如X射線粉末衍射(下文稱為XRPD)分析、差示掃描熱量測定(下文稱為DSC)、熱解重量分析(下文稱為TGA)、漫反射紅外傅里葉變換(DRIFT)光譜法、近紅外(NIR)光譜法、溶液和/或固態核磁共振光譜法。這些結晶物質的水含量可以通過卡爾費歇爾分析(KarlFischeranalysis)測定。作為一個實例,實例1的化合物展現出結晶性并且已鑒別出一種晶形。因此,本發明的另一個方面為1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在約2θ=5.1°處的特定峰。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在約2θ=18.0°處的特定峰。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少兩個在約2θ=5.1°和18.0°處的特定峰。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有在約2θ=5.1°、18.0°、10.2°、11.7°、19.4°、18.5°、14.8°、26.7°、26.6°、17.8°處的特定峰。根據本發明,提供晶形,即X射線粉末衍射圖與圖1中所示的X射線粉末衍射圖實質上相同的形式A。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在約2θ=5.1°±0.2°2θ處的特定峰。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在約2θ=18.0°±0.2°2θ處的特定峰。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少兩個在約2θ=5.1°和18.0°±0.2°2θ處的特定峰。根據本發明的另一個方面,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有在約2θ=5.1°、18.0°、10.2°、11.7°、19.4°、18.5°、14.8°、26.7°、26.6°、17.8°±0.2°2θ處的特定峰。實例3也為結晶并且在此描述三種形式(A、B以及C)。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在約2θ=4.8°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在約2θ=10.0°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少兩個在約2θ=4.8°和10.0°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有在約2θ=4.8°、10.0°、14.6°、5.2°、19.9°、10.4°、25.4°、23.6°、24.4°、16.2°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖與圖3中所示的X射線粉末衍射圖實質上相同。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在2θ=4.8°±0.2°2θ處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少一個在2θ=10.0°±0.2°2θ處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有至少兩個在2θ=4.8°和10.0°處的特定峰,其中所述值可以±0.2°2θ。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式A,它的X射線粉末衍射圖具有在2θ=4.8°、10.0°、14.6°、5.2°、19.9°、10.4°、25.4°、23.6°、24.4°、16.2°處的特定峰,其中所述值可以±0.2°2θ。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有至少一個在約2θ=5.8°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有至少一個在約2θ=10.9°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有至少兩個在約2θ=5.8°和10.9°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有在約2θ=5.8°、10.9°、11.5°、25.9°、17.3°、24.0°、19.1°、12.9°、24.7°、27.2°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖與圖5中所示的X射線粉末衍射圖實質上相同。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有至少一個在2θ=5.8°±0.2°2θ處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有至少一個在2θ=10.9°±0.2°2θ處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有至少兩個在2θ=5.8°和10.9°處的特定峰,其中所述值可以±0.2°2θ。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式B,它的X射線粉末衍射圖具有在2θ=5.8°、10.9°、11.5°、25.9°、17.3°、24.0°、19.1°、12.9°、24.7°、27.2°處的特定峰,其中所述值可以±0.2°2θ。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有至少一個在約2θ=6.9°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有至少一個在約2θ=12.3°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有至少兩個在約2θ=6.9°和12.3°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有在約2θ=6.9°、12.3°、10.5°、21.0°、24.6°、13.6°、16.4°、19.6°、20.2°、22.5°處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖與圖7中所示的X射線粉末衍射圖實質上相同。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有至少一個在2θ=6.9°±0.2°2θ處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有至少一個在2θ=12.3°±0.2°2θ處的特定峰。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有至少兩個在2θ=6.9°和12.3°處的特定峰,其中所述值可以±0.2°2θ。根據本發明,提供一種晶形,即1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮的形式C,它的X射線粉末衍射圖具有在2θ=6.9°、12.3°、10.5°、21.0°、24.6°、13.6°、16.4°、19.6°、20.2°、22.5°處的特定峰,其中所述值可以±0.2°2θ。當據陳述本發明涉及本發明化合物的晶形時,如實例1或實例3,結晶度宜大于約60%,更宜大于約80%,優選大于約90%并且更優選大于約95%。最優選地,結晶度大于約98%。當據陳述本發明涉及本發明化合物的晶形時,如實例1或實例3,該晶形優選實質上不含同一化合物的其他晶形或非晶形。在此上下文中,“實質上不含”意指宜大于約60%,更宜大于約80%,優選大于約90%,更優選大于約95%,再更優選大于約98%并且甚至更優選大于約99%純單一晶形。舉例來說,實例3可以呈形式A的形式并且實質上不含形式B和C;可替代地,實例3可以呈形式B的形式并且實質上不含形式A和C;可替代地,實例3可以呈形式C的形式并且實質上不含形式A和B。類似地,實例3可以呈形式B的形式并且實質上不含替代性晶形或非晶形。應了解,X射線粉末衍射圖的2θ值可能因機器不同或因樣品不同而略有變化,并且因此所引用的值不應理解為絕對的。已知可以獲得取決于測量條件(如,所用設備或機器)而具有一個或多個測量誤差的X射線粉末衍射圖。具體來說,總體上已知X射線粉末衍射圖的強度可以波動,取決于測量條件。因此應了解,除非另行說明,否則上述本發明的晶形不限于所提供的X射線粉末衍射圖與圖1、3、5中所示的X射線粉末衍射圖一致的晶體并且所提供的X射線粉末衍射圖與這些圖中所示的那些X射線粉末衍射圖實質上相同的任何晶體均落入本發明的范圍內。X射線粉末衍射領域的普通技術人員能夠判斷X射線粉末衍射圖的實質一致性。X射線粉末衍射領域的普通技術人員也應認識到,相對峰強度可能受例如尺寸大于30微米的晶粒和非單一縱橫比影響,這些因素可能影響樣品分析。本領域普通技術人員也將認識到,反射位置可以受樣品在衍射計中所處的確切高度和衍射計的零點校正影響。樣品的表面平坦度也可能具有細微影響。因此,所呈現的衍射圖數據不應視為絕對值(參見詹金斯R(Jenkins,R)和辛德爾R.L.(Snyder,R.L.)《X射線粉末衍射測定法的介紹》(‘IntroductiontoX-RayPowderDiffractometry’),約翰·威利父子公司(JohnWiley&Sons)1996;邦C.W.(Bunn,C.W.)(1948),《化學晶體學》(ChemicalCrystallography),倫敦克拉倫登出版社(ClarendonPress,London);克盧格H.P.(Klug,H.P.)和亞歷山大L.E.(Alexander,L.E.)(1974),《X射線衍射程序》(X-RayDiffractionProcedures))。總體而言,X射線粉末衍射圖中衍射角的測量誤差為約±0.2°2θ,并且當考慮X射線粉末衍射數據時,應將該測量誤差度考慮在內。此外,應了解強度可能波動,取決于實驗條件和樣品制備(優選取向)。特定本發明化合物為實例中的每一者,它們各自提供本發明的另一個獨立方面。另外的特定本發明化合物為這些實例中的每一者的醫藥學上可接受的鹽(多種),它們各自提供本發明的另一個獨立方面。根據本發明的另一個方面,提供一種具有化學式(I)的化合物,它通過遵循如在此所披露的實例中的任一者可獲得。另一個特征為在此所限定的范圍中的任一者,其限制條件為如實例1、3、4等的特定實例單獨地宣布棄權。本領域普通技術人員應了解,某些具有化學式(I)的化合物包含經不對稱取代的碳原子,并且因此可以按光學活性和外消旋形式存在并且分離。一些具有化學式(I)的化合物可以展現多晶型。應了解,本發明涵蓋任何外消旋、光學活性、多晶型或立體異構形式或其混合物,該形式具有有用于抑制PI3K-α和-δ活性的性質,在本領域中熟知如何制備光學活性形式(例如通過用再結晶技術拆分外消旋形式、通過從光學活性起始物質合成、通過手性合成、通過酶拆分、通過生物轉化或者通過使用手性固定相進行色譜分離)以及如何通過下文所述的標準測試測定用于抑制PI3K-α和-δ活性的功效。應了解,上文定義的某些具有化學式(I)的化合物可以展現互變異構現象。應了解,本發明在其定義中包括任何此類互變異構形式或其混合物,它具有PI3K抑制活性并且不僅僅限于在化學式圖中所用或在實例中所命名的任何一種互變異構形式。總體來講,任何此類互變異構形式中僅一者在下文的實例中命名或在下文的任何相關化學式圖中呈現。本發明打算包括存在于本發明化合物中的原子的所有同位素。同位素應理解為包括具有相同原子數但具有不同質量數的那些原子。舉例來說,氫的同位素包括氚和氘。碳的同位素包括13C和14C。具有化學式(I)的化合物的適合醫藥學上可接受的鹽為例如具有化學式(I)的化合物的酸加成鹽,例如與強無機或有機酸(如鹽酸、氫溴酸、硫酸或三氟乙酸)的酸加成鹽。具有化學式(I)的化合物的另一個適合醫藥學上可接受的鹽為例如在給予具有化學式(I)的化合物之后人體或動物體內所形成的鹽。應進一步了解,具有化學式(I)的化合物的適合醫藥學上可接受的溶劑合物也形成本發明的一個方面。適合醫藥學上可接受的溶劑合物為例如水合物,如半水合物、單水合物、二水合物或三水合物或其替代量。應進一步了解,具有化學式(I)的化合物的適合醫藥學上可接受的前藥也形成本發明的一個方面。因此,本發明化合物可以按前藥形式給予,該前藥為在人體或動物體內分解以釋放本發明化合物的化合物。前藥可以用于改變本發明化合物的物理性質和/或藥物動力學性質。當本發明化合物包含可能連接修飾基團的適合基團或取代基時,可能形成前藥。前藥的實例包括體內可裂解的酯衍生物,這些酯衍生物可以形成于具有化學式(I)的化合物的羥基處;和體內可裂解的酰胺衍生物,這些酰胺衍生物可以形成于具有化學式(I)的化合物的氨基處。因此,本發明包括當通過有機合成可獲得時以及當經由前藥的裂解而在人體或動物體內可獲得時,如上文所定義的那些具有化學式(I)的化合物。因此,本發明包括那些由有機合成方法產生的具有化學式(I)的化合物并且也包括在人體或動物體內經由前體化合物代謝而產生的這些化合物,即具有化學式(I)的化合物可以為合成產生的化合物或代謝產生的化合物。具有化學式(I)的化合物的適合醫藥學上可接受的前藥為基于合理醫學判斷,適合給予人體或動物體而無所不希望的藥理學活性且無不當毒性者。例如在以下文獻中已描述不同形式的前藥:-a)《酶學方法》(MethodsinEnzymology),第42卷,第309-396頁,由K.韋德爾(K.Widder)等人編(學術出版社(AcademicPress),1985);b)《前藥設計》(DesignofPro-drugs),由H.邦加爾德(H.Bundgaard)編,(埃爾塞維爾(Elsevier),1985);c)《藥物設計和開發教科書》(ATextbookofDrugDesignandDevelopment),由克洛格斯加德-拉森(Krogsgaard-Larsen)和H.邦加爾德編,第5章“前藥的設計和應用(DesignandApplicationofPro-drugs)”,H.邦加爾德第113-191頁(1991);d)H.邦加爾德,《高級藥物遞送綜述》(AdvancedDrugDeliveryReviews),8,1-38(1992);e)H.邦加爾德,等人,《醫藥科學雜志》(JournalofPharmaceuticalSciences),77,285(1988);f)N.掛谷(N.Kakeya)等人,《化學與藥學通報》(Chem.Pharm.Bull.),32,692(1984);g)T.樋口(T.Higuchi)和V.斯黛拉(V.Stella),《依新穎遞送系統的前藥》(“Pro-DrugsasNovelDeliverySystems”),ACS研討會系列(A.C.S.SymposiumSeries),第14卷;以及h)E.羅奇(E.Roche)(編者),《藥物設計中的生物可逆載體》(“BioreversibleCarriersinDrugDesign”),培格曼出版社(PergamonPress),1987。具有羥基的具有化學式(I)的化合物的適合的醫藥學上可接受的前藥為例如其體內可裂解的酯或醚。包含羥基的具有化學式(I)的化合物的體內可裂解的酯或醚為例如在人體或動物體內裂解以產生母體羥基化合物的醫藥學上可接受的酯或醚。對于羥基的適合的醫藥學上可接受的酯形成基團包括無機酯,如磷酸酯(包括磷酰胺環酯)。對于羥基的另外的適合的醫藥學上可接受的酯形成基團包括(1-10C)烷酰基,如乙酰基、苯甲酰基、苯乙酰基以及經取代的苯甲酰基和苯乙酰基;(1-10C)烷氧羰基,如乙氧羰基、N,N-[二(1-4C)烷基]氨甲酰基、2-二烷基氨基乙酰基以及2-羧基乙酰基。苯乙酰基和苯甲酰基上的環取代基的實例包括氨甲基、N-烷基氨甲基、N,N-二烷基氨甲基、N-嗎啉基甲基、哌嗪-1-基甲基以及4-(1-4C)烷基哌嗪-1-基甲基。對于羥基的適合的醫藥學上可接受的醚形成基團包括α-酰氧基烷基,如乙酰氧基甲基和特戊酰氧基甲基。具有氨基的具有化學式(I)的化合物的適合的醫藥學上可接受的前藥為例如其體內可裂解的酰胺衍生物。由氨基形成的適合的醫藥學上可接受的酰胺包括例如與(1-10C)烷酰基(如乙酰基、苯甲酰基、苯乙酰基以及經取代的苯甲酰基和苯乙酰基)形成的酰胺。苯乙酰基和苯甲酰基上的環取代基的實例包括氨甲基、N-烷基氨甲基、N,N-二烷基氨甲基、N-嗎啉基甲基、哌嗪-1-基甲基以及4-(1-4C)烷基哌嗪-1-基甲基。具有化學式(I)的化合物的體內作用可以部分地由在給予具有化學式(I)的化合物之后在人體或動物體內形成的一種或多種代謝物來發揮。如上所述,具有化學式(I)的化合物的體內作用也可以經由前體化合物(前藥)代謝來發揮。具有化學式(I)的化合物包含經-C(O)R2取代的哌啶子單元,其中R2為經羥基取代的(C2-3)烷基。這些化合物的一種可能的代謝途徑為通過氧化這一基團上的羥基取代基。這些經氧化的化合物總體上保留一些PI3K-α和-δ抑制活性。因此,根據本發明的另一個方面,提供一種具有化學式(A)的化合物:其中:R1A為甲基或乙基;并且R2A為經羧基取代的(C1-2)烷基;或其醫藥學上可接受的鹽。具有化學式(A)的化合物的實例包括實例8,它為實例1的經鑒別的代謝物。和實例9,它為實例3的經鑒別的代謝物:實例3的另外的可能的代謝物為兩種替代性氧化產物,顯示如下并且進一步描述在實例10和11中:具有化學式(A)的化合物的適合的醫藥學上可接受的鹽包括例如堿金屬鹽或堿土金屬鹽,如鈣鹽或鎂鹽;或銨鹽;或與如甲胺、二甲胺、三甲胺、哌啶、嗎啉或三-(2-羥乙基)胺的有機堿形成的鹽。為避免疑義,應了解,如果在本說明書中某一基團經‘上文所定義(hereinbeforedefined/definedhereinbefore)’限定,那么所述基團涵蓋首次出現且最廣泛的定義以及對該基團的每一和所有特定定義。本發明的特定新穎化合物包括例如具有化學式(I)的化合物或其醫藥學上可接受的鹽,其中除非另行說明,否則R1和R2各自具有上文或以下陳述中所定義的含義中的任一者:R1為甲基。R1為乙基。R2為如上文所定義的基團(i)到(xi)中的任一者。R2為如上文所定義的基團(i)到(vi)。R2為基團(i)。一組特定的本發明化合物為以上具有化學式(I)的化合物,其中:-R1為甲基或乙基,R2為基團(i):或其醫藥學上可接受的鹽。特定的本發明化合物為例如在下文中所陳述的實例中所披露的具有化學式(I)的化合物。舉例來說,特定的本發明化合物為選自以下任一者的具有化學式(I)的化合物:-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-丙-1-酮(實例1和2);1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-乙基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-丙-1-酮(實例3);(3R)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-丁-1-酮(實例4);(3S)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-丁-1-酮(實例5);(2R)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-2-甲基-丙-1-酮(實例6);1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-2-羥基-2-甲基-丙-1-酮(實例7)。本發明的另一個方面提供一種制備具有化學式(I)的化合物或其醫藥學上可接受的鹽的方法。適合方法通過以下代表性方法變化形式來說明,其中除非另行說明,否則R1、R2具有上文所定義的含義中的任一者。通過有機化學的標準程序可以獲得必要的起始物質。這些起始物質的制備結合以下代表性方法變化形式并在隨附實例中描述。可替代地,通過于有機化學家的普通技術中說明的那些程序的類似程序可獲得必要的起始物質。適合的方法變化形式包括例如以下:(a)具有化學式II的化合物與羧酸R2-COOH宜在適合的激活試劑存在下進行的反應其中R1具有上文所定義的含義中的任一者,除了必要時在適合的堿存在下對任何官能團進行保護,隨后移除所存在的任何保護基。用于這一反應的適合的偶合劑包括例如在2-羥基-吡啶N-氧化物存在下的六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲基脲、TBTU(四氟硼酸2-(1H-苯并[d][1,2,3]三唑-1-基)-1,1,3,3-四甲基異脲)或1-(3-二甲氨基丙基)-3-乙基碳化二亞胺鹽酸鹽離子。該反應宜在適合堿存在下進行。適合堿為例如有機胺堿,如吡啶、2,6-二甲基吡啶、三甲基吡啶、4-二甲氨基吡啶、三乙胺、N-甲基嗎啉、二氮雜雙環[5.4.0]十一-7-烯、二異丙基乙基胺;或例如堿金屬或堿土金屬碳酸鹽或氫氧化物,例如碳酸鈉、碳酸鉀、碳酸鈣、氫氧化鈉或氫氧化鉀;優選為N-乙基-N,N-二異丙胺。該反應宜在適合的惰性溶劑存在下且在例如-50℃到100℃范圍內,優選在0℃到30℃范圍內的溫度下進行,該惰性溶劑如乙腈、N,N-二甲基甲酰胺、N-甲基吡咯烷酮、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯、甲醇、乙醇、鹵化溶劑(如二氯甲烷、氯仿或四氯化碳)。可替代地,羧酸R2-COOH可以轉化成活性物質,該活性物質可以接著與具有化學式II的化合物在本領域中熟知的條件下反應。羥基的適合保護基為四氫吡喃保護基,如實例2和3中所述。移除這一基團的適合條件包括弱酸性條件,在作為溶劑的醇(如甲醇或乙醇)存在下,在20℃到70℃之間的溫度下。所用的典型弱酸為對甲苯磺酸吡啶。具有化學式II的化合物可以由具有化學式III的化合物:其中P為保護基,如叔丁氧羰基,與具有化學式R1-L的化合物(其中L為適合的離去基,如鹵基,如溴、碘基(宜為碘))在適合堿存在下進行反應,隨后移除所存在的任何保護基而獲得。適合堿為例如有機胺堿,如1,8-二氮雜雙環[5.4.0]十一-7-烯。該反應宜在適合惰性溶劑存在下且在例如-50℃到60℃范圍內,優選在-10℃到0℃范圍內的溫度下進行,該惰性溶劑如2-甲基四氫呋喃、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯。脫除叔丁氧羰基的保護基的適合條件包括酸性條件,如三氟乙酸于如二氯甲烷的惰性溶劑中,在接近室溫(20℃-25℃)下。化合物III可以由具有化學式IV的化合物與具有化學式V的化合物在適合的激活試劑存在下,優選在適合堿存在下進行偶合反應,隨后在弱酸存在下進行環化反應而獲得。偶合反應可以在適合偶合劑存在下進行,該偶合劑如六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲基脲或TBTU(四氟硼酸2-(1H-苯并[d][1,2,3]三唑-1-基)-1,1,3,3-四甲基異脲)。該偶合反應宜在適合堿存在下進行。適合堿為例如有機胺堿,如吡啶、2,6-二甲基吡啶、三甲基吡啶、4-二甲氨基吡啶、三乙胺、N-甲基嗎啉、二氮雜雙環[5.4.0]十一-7-烯、二異丙基乙基胺;或例如堿金屬或堿土金屬碳酸鹽或氫氧化物,例如碳酸鈉、碳酸鉀、碳酸鈣、氫氧化鈉或氫氧化鉀;優選為N-乙基-N,N-二異丙胺。該偶合反應宜在適合的惰性溶劑存在下且在例如-50℃到100℃范圍內,優選在0℃到30℃范圍內的溫度下進行,該惰性溶劑如N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、N-甲基吡咯烷酮、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯、甲醇、乙醇、鹵化溶劑(如二氯甲烷、氯仿或四氯化碳)。環化條件是在弱酸(典型地為乙酸)存在下進行。該反應宜在適合的惰性溶劑存在下且在例如50℃到150℃范圍內,優選在80℃到100℃范圍內的溫度下進行,該惰性溶劑如N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、N-甲基吡咯烷酮、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯。化合物IV可以由具有化學式VI的化合物與肼的反應獲得。這一反應宜在適合惰性溶劑存在下,在例如20℃到70℃范圍內的溫度,優選約50℃下進行,該惰性溶劑如四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯或醇(如乙醇或異丙醇)。化合物VI可以由具有化學式VII的化合物與氰化物源(如二氰化鋅(II))的金屬催化反應獲得。該反應的適合催化劑包括例如金屬催化劑,如鈀(0),例如四(三苯基膦)鈀(0);或由鈀(II)鹽原位形成的催化劑,例如乙酸鈀(II)、氯化鈀(II)、溴化鈀(II)、氯化雙(三苯基膦)鈀(II)、[1,1′-雙(二苯基膦基)二茂鐵]二氯化鈀(II)或三(二苯亞甲基丙酮)二鈀以及膦配體,例如二環己基(2′,4′,6′-三異丙基聯苯-2-基)膦。該反應宜在適合溶劑中,且在例如20℃到150℃范圍內,優選在60℃到120℃范圍內的溫度下進行,該溶劑如N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯或二甲苯。該反應也宜在如鋅的另外金屬存在下進行。適合的這種類型反應描述于《金屬催化的交叉偶合反應》(‘Metal-CatalyzedCross-CouplingReactions’),第二版,由阿民·邁耶雷(ArminMeijere),弗朗索瓦·迪德里希(FrancoisDiederich)編,威利出版公司(Wiley-VCH),2004)中。化合物VII的合成已描述于實例1和2中。可替代地,具有化學式II的化合物可以通過化合物VIII(其中R為低碳烷基)與化合物IX(其中P為保護基,如叔丁氧羰基)的金屬催化反應獲得,該反應的適合催化劑包括例如金屬催化劑,如鈀(0),例如四(三苯基膦)鈀(0);或由鈀(II)鹽原位形成的催化劑,例如乙酸鈀(II)、氯化鈀(II)、溴化鈀(II)、氯化雙(三苯基膦)鈀(II)、[1,1′-雙(二苯基膦基)二茂鐵]二氯化鈀(II)或三(二苯亞甲基丙酮)二鈀以及膦配體,例如二環己基(2′,4′,6′-三異丙基聯苯-2-基)膦。該反應宜在適合溶劑中,在例如在50℃到180℃范圍內,優選在120℃到150℃范圍內的溫度下進行,該溶劑如N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯或二甲苯或醇(如4-甲基-2-戊醇)。該反應也宜在如氯化鋰的另外鹽存在下進行。適合的這種類型反應描述于《金屬催化的交叉偶合反應》,第二版,由阿民·邁耶雷,弗朗索瓦·迪德里希編,威利出版公司,2004)中。具有化學式VIII的化合物可以由化合物VII與適合的六烷基二錫烷的金屬催化反應獲得。該反應的適合催化劑包括例如金屬催化劑,如鈀(0),例如四(三苯基膦)鈀(0);或由鈀(II)鹽原位形成的催化劑,例如乙酸鈀(II)、氯化鈀(II)、溴化鈀(II)、氯化雙(三苯基膦)鈀(II)、[1,1′-雙(二苯基膦基)二茂鐵]二氯化鈀(II)或三(二苯亞甲基丙酮)二鈀以及膦配體,例如二環己基(2′,4′,6′-三異丙基聯苯-2-基)膦。該反應宜在適合溶劑中,在例如在50℃到100℃范圍內,優選在70℃到80℃范圍內的溫度下進行,該溶劑如N,N-二甲基乙酰胺、N,N-二甲基甲酰胺、四氫呋喃、1,4-二噁烷、1,2-二甲氧基乙烷、苯、甲苯、二甲苯或醇(如4-甲基-2-戊醇)。具有化學式IX的化合物可以由可商購的物質以若干步驟獲得,如實例1所示(其中R1=Me且P=叔丁氧羰基)。應了解,上述方法變化形式中的方法步驟的其他排列也有可能。應了解,通過上文所述方法中的任一者獲得的任何具有化學式(I)的化合物必要時均可以轉化成另一個具有化學式(I)的化合物。當需要具有化學式(I)的化合物的醫藥學上可接受的鹽,例如酸加成鹽時,它可以通過例如所述化合物與適合酸的反應獲得。當需要具有化學式(I)的化合物的醫藥學上可接受的前藥時,它可以使用常規程序獲得。舉例來說,具有化學式(I)的化合物的體內可裂解的酯可以通過例如包含羥基的具有化學式(I)的化合物與醫藥學上可接受的羧酸的反應獲得。關于前藥的另外的信息已在上文中提供。也應了解,在上文提及的一些反應中,可能必需或所希望的是保護化合物中的任何敏感性基團。必需或所希望保護的情況和適用于保護的方法為本領域普通技術人員所知。常規保護基可以根據標準規范來使用(關于說明,參見T.W.格林(T.W.Green),《有機合成中的保護基》(ProtectiveGroupsinOrganicSynthesis),約翰威利父子公司(JohnWileyandSons),1991)。因此,如果反應物包括如氨基、羧基或羥基的基團,那么在此提及的一些反應中可能所希望的是保護該基團。適合的氨基或烷基氨基保護基為例如酰基,例如烷酰基,如乙酰基;烷氧羰基,例如甲氧羰基、乙氧羰基或叔丁氧羰基;芳基甲氧羰基,例如苯甲氧羰基;或芳酰基,例如苯甲酰基。關于以上保護基的脫除保護基條件必然隨保護基的選擇而變化。因此,舉例來說,如烷酰基或烷氧羰基或芳酰基的酰基可以例如通過用如堿金屬氫氧化物(例如,氫氧化鋰或氫氧化鈉)的適合堿進行水解來移除。可替代地,如叔丁氧羰基的酰基可以例如通過用如鹽酸、硫酸或磷酸或三氟乙酸的適合酸處理來移除,且如苯甲氧羰基的芳基甲氧羰基可以例如通過經如鈀/碳的催化劑氫化或通過用路易斯酸(Lewisacid)(例如三(三氟乙酸)硼)處理來移除。用于伯氨基的適合的替代保護基為例如酞酰基,它可以通過用例如二甲氨基丙胺的烷基胺或用肼處理來移除。適合的羥基保護基為例如酰基(例如烷酰基,如乙酰基)、芳酰基(例如苯甲酰基)或芳甲基(例如苯甲基)。關于以上保護基的脫除保護基條件將必然隨保護基的選擇而變化。因此,舉例來說,如烷酰基或芳酰基的酰基可以例如通過用如堿金屬氫氧化物(例如,氫氧化鋰或氫氧化鈉)的適合堿進行水解來移除。可替代地,如苯甲基的芳基甲基可以例如通過經如鈀/碳的催化劑氫化來移除。適合的羧基保護基為例如酯化基團,例如甲基或乙基,它可以例如通過用如氫氧化鈉的堿進行水解來移除;或例如叔丁基,它可以例如通過用酸(例如有機酸,如三氟乙酸)進行處理來移除;或例如苯甲基,它可以例如通過經如鈀/碳的催化劑進行氫化來移除。可以使用于化學領域中熟知的常規技術于合成的任何便利階段移除這些保護基。在此所定義的某些中間體(例如具有化學式II、III、IV、VI、VII、VIII的化合物)為新穎的且提供這些中間體作為本發明的另一個特征。生物分析-使用以下分析來測量本發明化合物作為以下的作用:a)生物化學分析中PI3激酶的抑制劑;b)生物化學分析中其他激酶的抑制劑;c)BT474細胞中磷酸化AKT(Thr308)的體外抑制劑;d)MDA-MB-468細胞中磷酸化AKT(Ser473)的體外抑制劑;e)JEKO細胞中磷酸化AKT(Ser473)的體外抑制劑;f)HT29細胞中磷酸化Chk1(Ser345)的體外抑制劑;g)腫瘤細胞系池中細胞增殖的抑制劑;h和i)分別作為用人類乳癌細胞系MCF7移植的SCID小鼠中磷酸化AKT(Ser473)的體內抑制劑或腫瘤生長的體內抑制劑。分析方案中所用的縮寫:PIP2:PI(4,5)P2,磷脂酰肌醇4,5-雙磷酸酯s.c.:皮下ATP:三磷酸腺苷DMSO:二甲亞砜TRIS:三(羥甲基)氨基甲烷CHAPS:3-[(3-膽酰胺基丙基)二甲基銨基]-1-丙烷磺酸鹽DTT:二硫蘇糖醇FBS:胎牛血清DMEM:杜爾貝科氏改良伊格爾培養基(Dulbecco′sModifiedEagleMedium)EDTA:乙二胺四乙酸EGTA:乙二醇四乙酸BSA:牛血清白蛋白PBS:磷酸鹽緩沖鹽水HRP:辣根過氧化酶RPMI:洛斯維公園紀念研究所(RoswellParkMemorialInstitute)1640培養基4NQO:4-硝基喹啉N-氧化物EMEM:伊格爾氏最低必需培養基(Eagle′sMinimalEssentialmedium)CO2:二氧化碳PBST:磷酸鹽緩沖鹽水/TweenAb:抗體MTS試劑:[3-(4,5-二甲基噻唑-2-基)-5-(3-羧基甲氧基苯基)-2-(4-磺酸基苯基)-2H-四唑鎓,內鹽;MTS]和電子偶合試劑(甲基硫酸吩嗪)PMS。(a)體外酶抑制分析使用人類重組酶,在基于KinaseGlo的酶活性分析中評估PI3K-β、PI3K-α、PI3K-γ以及PI3K-δ的抑制作用。該分析平臺間接測量在用酶、PIP2底物、ATP以及化合物孵育之后ATP的耗盡。在酶反應完成之后,剩余ATP用于第二酶促反應,其中熒光素酶在光的發射下將甲蟲熒光素轉化成氧化熒光素。所測量的發光與已完成的激酶反應中所剩余ATP之間存在直接關系。因此,發光與激酶活性呈逆相關。典型地,測試十二種不同的化合物濃度并且繪制PI3K-β、PI3K-α、PI3K-γ或PI3K-δ的抑制作用的原始數據對比抑制劑濃度的曲線。方法細節:通過聲學分配將于100%DMSO中的化合物添加到分析板中。在Tris緩沖液(50mMTrispH7.4,0.05%CHAPS,2.1mMDTT以及10mM氯化鎂)中添加PI3K酶且使其與化合物預孵育20分鐘,隨后添加包含PIP2和ATP的底物溶液。80分鐘后通過添加包含熒光素和熒光素酶(來自KinaseGlo(R)增強版發光激酶分析試劑盒(PlusLuminecentKinaseAssaykit)(普洛麥格(Promega)#V3772)的KinaseGlo檢測溶液來停止酶反應。使板在室溫下靜置30分鐘,接著在具有標準發光過濾塊的Pherastar儀器上讀取。在該分析中DMSO、ATP以及PIP2的最終濃度分別為1%、8μM以及80μM。數據分析使用擬合為非線性回歸擬合的對數曲線來計算IC50值。IC50值為抑制50%酶活性的測試化合物的濃度。(b)除PI3激酶1類酶以外的激酶選擇性評估由一系列商業供應商(如密理博(Millipore)、英杰(Invitrogen)以及普洛秋內斯(ProQinase))提供龐大激酶分析池。這些池允許評估既定化合物的整體激酶選擇性。確切方法/技術將變化,取決于供應商。使用在英國鄧迪的MRC蛋白質磷酸化單位的MRC-信號轉導療法部門(DSTT)(MRC-DivisionofSignalTransductionTherapy(DSTT),MRCProteinPhosphorylationUnit,Dundee,UK)所進行的酶分析來產生一些在此所述化合物的選擇性數據。使用放射化學形式進行蛋白激酶分析。在多點384孔板中在室溫下以25.5μl的總分析體積進行分析。在酶和肽/蛋白質底物存在下使化合物預孵育5分鐘,隨后通過添加10μlATP起始反應(針對每一激酶所選擇的最終濃度為5μM、20μM或50μM)。在室溫下進行分析,隨后通過添加5μl正磷酸終止。接著通過帕卡德收集器(PackardHarvester)(洗滌緩沖液為50mM正磷酸)將分析板內容物收集到Whatman-P81-Unifilter板上且在空氣中干燥。接著添加MicroScintO來密封干Unifilter板且在帕卡德TopcountNXT閃爍計數器中計數。這一方案擷取適用于池中大部分激酶的通用形式,但如本領域普通技術人員應熟悉,對于少數激酶需要對這些方案進行修改。針對約18種脂質激酶的脂質激酶分析也在DSTT進行。在384孔板中在室溫下以40μl的總分析體積進行所有脂質激酶分析。根據由ADP-GLO分析(普洛麥格,#V9101)提供的方案進行該分析。這一方案擷取適用于池中大部分激酶的通用形式,但如本領域普通技術人員應熟悉,對于少數激酶需要對這些方案進行修改。也使用經由迪斯科韋爾克斯(DiscoverX)可獲得的KINOMEscanTM篩選平臺評估激酶選擇性。這一篩選平臺采用活性位點定位的競爭結合分析來定量地測量在測試化合物和超過450種人類激酶與疾病相關突變體變異體之間的相互作用。KINOMEscanTM分析不需要ATP并且因此報導真實的熱力學相互相用親和力,與IC50值形成對照,IC50值可以取決于ATP濃度。該方法是基于以下化合物:它們結合激酶活性位點并且直接(空間)或間接(異位)防止激酶結合到固定配體,由此減少固體支撐物上所捕獲的激酶的量。反之,不結合激酶的測試分子對固體支撐物上所捕獲的激酶的量無影響。通過使用檢測相關的DNA標記的定量qPCR方法測量在測試對比對照樣品中所捕獲的激酶的量來鑒別篩選“命中”。以類似方式,通過測量隨測試化合物濃度而變的固體支撐物上所捕獲的激酶的量來計算測試化合物-激酶相互作用的解離常數(Kd)。(C)測量BT474細胞中磷酸化AKT(Tyr308)的分析方案這一分析用于測量細胞中的PI3K-α抑制。將BT474細胞(人類乳腺管癌,ATCCHTB-20)以每孔5600個細胞的密度接種于黑色384孔板(科斯塔(Costar),#3712)中包含10%FBS和1%谷氨酰胺的DMEM中并且使其粘附過夜。次日早晨通過聲學分配將于100%DMSO中化合物的添加到分析板中。在37℃和5%CO2下孵育2小時之后,抽吸培養基并且用包含25mMTris、3mMEDTA、3mMEGTA、50mM氟化鈉、2mM原釩酸鈉、0.27M蔗糖、10mMβ-甘油磷酸鹽、5mM焦磷酸鈉、0.5%TritonX-100以及康普利特(complete)蛋白酶抑制劑混合片劑(羅氏(Roche)#04693116001,每50ml溶解緩沖液使用1片)的緩沖液溶解這些細胞。20分鐘后,將細胞溶解物轉移到已預涂布于PBS緩沖液中的抗所有AKT抗體的ELISA板(葛萊娜(Greiner)#781077)中,并且用于包含0.05%Tween20的PBS中的1%BSA阻斷非特異性結合。在4℃下將板孵育過夜。次日用包含0.05%Tween20的PBS緩沖液洗滌這些板并且再與小鼠單克隆抗磷酸化AKTT308一起孵育2小時。再次如上洗滌板,隨后添加馬抗小鼠HRP結合的二級抗體。在室溫下孵育2小時后,洗滌板并且向每一孔中添加QuantaBlu底物工作溶液(賽默科技(ThermoScientific)#15169,根據提供者指令制備)。60分鐘后通過向孔中添加停止溶液使已發展的熒光產物停止。使用帝肯(Tecan)Safire讀板儀分別使用325nm激發波長和420nm發射波長讀取板。除非有所說明,否則在這一ELISA分析中使用來自細胞信號傳導(CellSignalling)(#7144)的PathScan磷酸化AKT(Thr308)夾心ELISA試劑盒中所含的試劑。(d)用于檢測MDA-MB-468細胞中的磷酸化AKT(Ser473)作為PI3激酶-β抑制的量度的方案.這一分析用于測量細胞中的PI3K-β抑制并且與以上分析(c)聯合用于測定細胞中的α對比β選擇性。將MDA-MB-468細胞(人類乳腺癌#ATCCHTB132)以每孔1500個細胞接種于葛萊娜384孔黑色平底板中的包含10%FBS和1%谷氨酰胺的40μlDMEM中。在37℃孵育箱中將細胞板孵育18小時,隨后使用聲學分配給予于100%DMSO中的化合物。在12點濃度范圍中將化合物給予到隨機板圖中。或者通過給予100%DMSO(最大信號)或者添加完全消除pAKT信號的參考化合物(PI3K-β抑制劑)(最小對照)來產生對照孔。在37℃下將板孵育2小時,接著通過添加10μl3.7%甲醛溶液固定細胞。30分鐘后,使用帝肯PW384洗板機用PBS洗滌板。阻斷各孔并且通過添加40μl包含0.5%Tween20和1%MarvelTM(經干燥的奶粉)的PBS滲透細胞且使細胞在室溫下孵育60分鐘。用包含0.5%(v/v)Tween20的PBS洗滌板且添加于相同PBS-Tween+1%MarvelTM中的20μl兔抗磷酸化AKTSer473(細胞信號傳導技術(CellSignallingTechnologies),#3787)并在4℃下孵育過夜。使用帝肯PW384,用PBS+0.05%Tween20將板洗滌3次。向每一孔中添加于PBS+0.05%包含1%MarvelTM的Tween20中稀釋的20μl二級抗體AlexaFluor488抗兔(分子探針(MolecularProbes),#A11008)且在室溫下孵育1小時。如之前般將板洗滌三次,接著向每一孔中添加20μlPBS且用黑色板密封物對板進行密封。在用488nm激光激發之后,盡可能快地在Acumen讀板儀上讀取這些板,測量綠色熒光。使用這一系統產生IC50值且通過對照孔測定板的質量。每次均操作參考化合物以監測分析性能。(e)檢測Jeko細胞中的磷酸化AKT(Ser473)的方案這一分析用于測量細胞中的PI3K-δ抑制。將化合物以×10最終濃度添加到葛萊娜V形底96孔板(西格瑪(Sigma)#M9686)的孔中的10μl1%(v/v)DMSO中。以1μM或10μM最高劑量的10點濃度范圍給予化合物,在一個板上給予8種化合物。每一給予有抗IgM(AffiniPureF(ab′)2片段山羊抗人類IgM(斯特拉泰克(Stratech),#109-006-129)和媒劑的板存在8個最大信號對照孔,且給予有抗IgM和參考PI3K-δ抑制劑的對照孔存在8個最小信號。最終媒劑濃度為0.1%DMSO。每一操作中均包括PI3K-δ選擇性化合物的完整劑量反應曲線。將JekoB細胞(人類套細胞淋巴瘤,ATCC#CRL-3006)接種于包含化合物的葛萊娜96孔V形底板中。以每孔100,000個細胞將細胞接種于包含1%谷氨酰胺的70μlRPMI中。使細胞板與化合物一起在37℃孵育箱中孵育1小時。在此化合物預孵育時間之后,以20μl分析緩沖液(包含1%谷氨酰胺的RPMI)中的×5最終濃度將上述抗IgM添加到這些板。最終抗IgM濃度為0.06μg/ml或等效的EC90劑量。在37℃下將板孵育10min,接著立即將板置放于冰上且在12000rpm下離心4min。在冰上,用手動移液管小心地移除上清液且添加40μl溶解緩沖液。將板在冰上孵育5min且儲存在-80℃下直到根據制造商的說明書(麥索斯蓋爾診斷(MesoscaleDiagnostics),#K11100D-3)在磷光體(Ser473)/總Akt全細胞溶解物試劑盒中分析。(f)用于檢測HT29細胞中的磷酸化Chk1(Ser345)的方案ATR(共濟失調毛細管擴張+Rad3相關激酶)為對DNA損傷或復制阻斷起反應而使絲氨酸或蘇氨酸殘基上的多個底物磷酸化的PI3激酶相關的激酶。Chk1(ATR的下游蛋白激酶)在DNA損傷檢查點控制中起重要作用。Chk1的激活涉及Ser317和Ser345的磷酸化(后者視為通過ATR磷酸化/激活的優先目標)。這是基于細胞的分析,用于通過在用化合物和UV模擬劑4NQO(西格瑪#N8141)處理之后測量HT29細胞中Chk1(Ser345)的磷酸化減少,來測量ATR激酶的抑制。將HT29細胞(ECACC#85061109)以每孔6000個細胞的密度接種于384孔分析板(科斯塔#3712)中包含1%L-谷氨酰胺和10%FBS的40μlEMEM培養基中且使其粘附過夜。次日早晨通過聲學分配將于100%DMSO中化合物的添加到分析板中。在37℃和5%CO2下孵育1小時之后,通過聲學分配向所有孔中添加40nl于100%DMSO中的3mM4NQO,除了未用4NQO處理以產生空反應對照的最小對照孔。將板返回到孵育箱,再持續1小時。接著通過添加20μl于PBS溶液中的3.7%甲醛且在室溫下孵育20min來固定細胞。隨后添加20μl于PBS中的0.1%TritonX100且在室溫下孵育10分鐘以滲透細胞。接著使用伯騰(Biotek)EL405洗板機,每孔用50μlPBS洗滌這些板一次。在包含0.05%聚山梨醇酯/Tween的PBS中將磷酸化Chk1Ser345抗體(細胞信號傳導技術#2348)稀釋150倍且向每一孔中添加15μl并在室溫下孵育過夜。次日早晨使用伯騰EL405洗板機,每孔用50μlPBS洗滌板三次,且接著添加20μl于PBST中的二級抗體溶液(包含500倍稀釋的AlexaFluor488山羊抗兔IgG(分子探針#A-11008)和0.002mg/mlHoeschst染料(分子探針#H-3570))。在室溫下孵育2小時后,使用伯騰EL405洗板機,每孔用50μlPBS洗滌板三次,且接著用黑色板密封物對板進行密封直到讀取。使用ArrayScanVTI儀器,使用具有10×物鏡的XF53濾光器來讀取板。使用雙激光設置來分析細胞核的Hoeschst(405nM)染色和二級抗體的pChk1(488nM)染色。(g)腫瘤細胞系中的細胞增殖分析(用于證實個人化藥物假設)在標準增殖分析中測定人類癌細胞系池對化合物作用的靈敏度。經由阿斯特拉捷利康(AstraZeneca)細胞庫獲得細胞系。大部分細胞系經由本領域普通技術人員已知的細胞庫貯藏地(CellBankRepositories)也可獲得,例如ATCC、ECACC、DMSZ、RIKEN、KCLB、JCRB(HSRRB)、LBNL、CLS以及ICLC。將細胞以每孔1000-6000個細胞的密度涂在96孔板中包含10%FBS的RPMI培養基中。在37℃下孵育16小時之后,向分析板添加不同濃度的化合物。在再孵育72h之后,通過向每一孔中添加MTS試劑(普洛麥格#3582)并持續2h來測定活細胞。MTS為四唑鎓鹽,它在電子偶合試劑存在下通過代謝活性細胞生物還原成甲躦。接著通過490nm下的吸光度對甲躦產物進行定量,作為活細胞的相對數目的指標。為測定GI50(細胞生長受到50%抑制時所處的濃度),通過與添加藥物前的MTS讀數進行比較來測定藥物添加時所存在的細胞的相對數目,且從未經處理的細胞的72小時值扣除這一值作為分析期間細胞生長的量度。這一數據的分析描述在下文‘個人化醫療/個人化藥物實例’下,說明可以如何分析這一數據以揭露PI3Kα抑制劑展示出具有PIK3CA基因突變的細胞系的選擇性生長抑制。這說明個人化醫療(PHC)或個人化藥物機會,其中反應預測生物標記讀數可以用于鑒別患有包含PIK3CA基因突變的腫瘤的患者以及更可能對在此所述的化合物起反應的患者。對在此所述的化合物起反應的其他可能標記包括(但不限于)增加的拷貝數、PIK3CA基因的擴增或易位以及提供PI33激酶路徑激活或依賴性的量度的其他遺傳、基因組或蛋白質組變化;例如(但不限于)一種或多種受體酪氨酸激酶的激活或編碼PI3激酶的調節亞基(p85)的PIK3R基因中的突變或易位,或如pAKT、pS6或FOXO狀態的下游信號傳導標記的磷酸化。另外,另外的基因和/或它們的蛋白質產物(例如Kras)的信號傳導的分析可以幫助改良個人化藥物方法的預測。(h)用于檢測生長于雄性SCID小鼠中的MCF-7腫瘤的磷酸化AKT(Ser473)的方案這是在動物模型中提供PI3K-α抑制的量度的藥效學分析。使雄性SCID小鼠(英國AZ公司(AZUK),從英國查爾斯河公司(CharlesRiver,UK)也可獲得)皮下(s.c)移植人類乳腺癌細胞系MCF7(倫敦帝國癌癥研究基金會(ICRFLondon),從ATCC#HTB-22也可獲得)),來測定PI3激酶抑制劑對AKT的磷酸化的抑制。在細胞植入前24小時,使小鼠植入0.5mg21天雌激素丸劑(美國創新研究公司(InnovativeResearchofAmerica),#E121)。將50%基質膠(碧迪生物科學(BDBioscience))中的5×106個細胞皮下注射在這些動物的左側腹上。當腫瘤達到400mm3體積時將動物隨機分成8只對照動物的組和4只處理動物的組且在次日開始給藥。在所選擇的時間點獲取腫瘤,此時也獲取血液樣品進行PK測量。將從小鼠切除的腫瘤置放于FastPrep管(2ml包含溶解基質A的脊形管,MP生物醫學(MPBiomedicals)#6910-500)中并且立即急凍。向每一管中添加1ml溶解緩沖液(25mMTris、3mMEDTA、3mMEGTA、50mM氟化鈉、2mM原釩酸鹽、0.27M蔗糖、10mMβ-甘油磷酸鹽、5mM焦磷酸鹽、0.5%Tritonx-100)加磷酸酶抑制劑(西格瑪#P2850和西格瑪#P5726,1∶100稀釋)和蛋白酶抑制劑(西格瑪#P8340,1∶200稀釋)。使腫瘤在FastPrep)-TM機(MP生物醫學#116004500)上均質化1分鐘,并且剩余在冰上靜置5分鐘,再繼之以兩個均質化步驟,每一均質化步驟后在冰上孵育5分鐘。使樣品在13,000rpm下在冷凍離心機中離心10分鐘。接著將澄清溶解物放入新鮮管中且使用10μl進行蛋白質測定分析。使用MSD多點分析試劑盒(麥索斯蓋爾發現(MesoScaleDiscovery)#K1510OD-3)進行總AKT和磷酸化AKT(Ser473)的檢測。板的每一孔包含4個點;這些點中的兩個點用由該試劑盒提供的小鼠單克隆抗體涂布;一個用針對總AKT的捕捉抗體涂布并且一個用針對磷酸化AKT(Ser473)的抗體涂布。在冷室中在振蕩器上每孔用150μl阻斷溶液來阻斷這些板過夜,該阻斷溶液是使用20ml具有洗滌溶液加600mg由該試劑盒供應的BlockerA的1×溶液制造。每孔用0.3ml洗滌溶液洗滌板三次。從每一腫瘤獲取溶解物的等分試樣且用溶解緩沖液將其稀釋到2mg/ml的濃度,接著向每一孔中添加25μl經稀釋的溶解物,得到每孔50μg的總量。將這些板在室溫下置放于振蕩器中,持續一小時,隨后洗滌板三次。使用阻斷溶液和洗滌溶液加1∶50稀釋的50×SULFO-TAG-TM抗總AKT抗體的混合物制備檢測抗體溶液。將這些板在室溫下置放于振蕩器中,持續一小時,隨后洗滌板三次。用去離子水1∶4稀釋150μl由該試劑盒供應的讀取緩沖液且將其添加到每一孔,并且接著在MSD板分析儀上讀取這些板。讀取緩沖液提供電化學發光的正確化學環境,因此當讀板儀向板施加電壓時,板基底上的電極使得標記與檢測抗體結合而發射光。所發射的光的強度為所存在的AKT(或者總AKT或者磷酸化AKT)的定量量度。為計算磷酸化AKT與總AKT的比率,應用如由麥索斯蓋爾所推薦的計算法:兩倍磷酸化信號除以總信號加磷酸化信號,接著乘以100,得到磷蛋白%。將這些值轉化成Log10,且接著使用這些值來計算每一組的幾何平均值加標準誤差。隨后使用雙尾公式和不等變異數來應用學生T檢驗(studentTtest)以檢查顯著性。研究顯示,對照組8只動物和每個治療組4只動物足以支持該研究。(i)用于檢測移植到SCID小鼠中的人類乳腺癌細胞系MCF7中的腫瘤生長抑制的方案。這一方法提供在PI3K-α依賴性模型中體內PI3激酶抑制劑的抗腫瘤功效的評估。關于上文所指出的PD研究,使雄性SCID小鼠皮下移植人類乳腺癌細胞系MCF7。在細胞植入之前24小時,使小鼠植入0.5mg21天雌激素丸劑。將50%基質膠中的5×106個細胞皮下注射在這些動物之左側腹上。當腫瘤達到約200-300mm3體積時將動物隨機分成10-15只動物的組且開始治療。通過經口、靜脈內或腹膜內途徑向動物給予適于經由所需途徑給予且符合福利要求的于媒劑中的化合物(用于經口給藥的懸浮液,pH范圍為4-7;用于ip/iv給藥的溶液,pH范圍5.5-7.0)且以限定劑量給藥2-4周)。通常每周兩次通過測徑規來測量腫瘤,并且使用橢圓公式(π/6×寬度×寬度×長度)計算腫瘤體積。雖然具有化學式(I)的化合物的藥理學性質正如所預期的隨結構變化而變化,但總體來講,具有化學式(I)的化合物所具有的活性可以在以下濃度或劑量下在以上測試(a)和(c)中的一者或多者中得到證明:-測試(a):-在例如1nM-100nM范圍內,IC50對比PI3K-α;測試(c):-在例如10nM-1μM范圍內,BT474細胞中的IC50對比細胞磷酸化AKT(Tyr308);適宜地,特定本發明化合物在以下濃度或劑量下在以上測試(a)和(c)中的一者或多者中具有活性:-測試(a):-在例如1nM-100nM范圍內,IC50對比PI3K-α;測試(c):-在例如10nM-1μM范圍內,BT474細胞中的IC50對比細胞磷酸化AKT(Tyr308);適宜地,特定本發明化合物在以下濃度或劑量下在以上測試(a)、(c)、(h)以及(i)中的一者或多者中具有活性:-測試(a):-在例如1nM-100nM范圍內,IC50對比PI3K-α;測試(c):-在例如10nM-1μM范圍內,BT474細胞中的IC50對比細胞磷酸化AKT(Tyr308);測試(h):-在例如1-200毫克/千克/天范圍內,體內磷酸化AKT(Ser473)抑制>50%;測試(i):-在例如1-200毫克/千克/天范圍內,異種移植活性。產生這些實例的以下數據:表A*測試方案a:這些值是從許多重復測試計算的平均值。**測試方案c:這些值是從許多重復測試計算的平均值。#測試方案f:僅進行一次測試重復。組合研究材料與方法MCF7為攜帶PIKC3CA基因突變(E545K)的雌激素受體陽性乳房腫瘤細胞系。使雄性SCID小鼠(英國AZ公司)皮下(s.c)移植人類乳腺癌細胞系MCF7(倫敦帝國癌癥研究基金會)以測定PI3激酶抑制劑的抗腫瘤活性。在細胞植入前24小時,使小鼠植入0.5mg21天雌激素丸劑(美國創新研究公司)。將50%基質膠(碧迪生物科學)中的5×106個細胞皮下注射在這些動物的左側腹上。BT474為Her2表達提高的雌激素受體陽性乳房腫瘤細胞系并且攜帶PIK3CA基因突變(K111N)。使雌性瑞士無胸腺裸小鼠(swissnu/nu-AZUK)皮下移植在小鼠中傳代的人類上皮細胞乳腺管癌細胞系BT474c(以AZ形式源自BT474-ATCCHTB-20)腫瘤。在細胞植入之前24小時,使小鼠植入0.36mg60天雌激素丸粒(美國創新研究公司)。將50%基質膠(碧迪生物科學)中的5×106個細胞皮下注射在這些動物的左側腹上。HCC70為缺乏PTEN基因表達的乳房腫瘤細胞系。使雌性瑞士無胸腺裸小鼠(swissnu/nu-AZUK)皮下移植乳腺管上皮細胞腫瘤細胞系HCC70(ATCC-CRL2315)細胞。將50%基質膠(碧迪生物科學)中的1×106個細胞皮下注射在這些動物的左側腹上。當腫瘤達到約200-300mm3體積時將動物隨機分成10-15只動物的組且開始治療。通過經口途徑,以限定劑量和時程向動物給予于適合媒劑中的化合物,持續3-4周。每周二-三次通過測徑規來測量腫瘤,并且使用橢圓公式(π/6×寬度×寬度×長度)計算腫瘤體積。當單獨給藥時,AZD5363是在10%DMSO、25%Kleptose溶液中配制。(Kleptose是源自羅蓋特藥物公司(Roquette-Pharma)(商標)羥丙基β-環糊精,適于體內使用和配制)。當與實例3共同給藥時,AZD5363是在HPMC/Tween(0.5%Methocel(羥丙基甲基纖維素)/0.1%聚山梨醇酯80)中配制。使懸浮液球磨過夜。實例3是在HPMC/Tween(0.5%Methocel(羥丙基甲基纖維素)/0.1%聚山梨醇酯80)中配制。AZD8186是在HPMC/Tween(0.5%Methocel(羥丙基甲基纖維素)/0.1%聚山梨醇酯80)中配制。當與實例3共同給藥時,AZD8186是在HPMC/Tween(0.5%Methocel(羥丙基甲基纖維素)/0.1%聚山梨醇酯80)中配制。使懸浮液球磨過夜。奧拉帕尼是在10%DMSO/30%Kleptose溶液中配制。通過實例3與AKT抑制劑(AZD5363)的組合依序給藥抑制腫瘤生長在BT474異種移植模型中進行研究。實例3和AZD5363依序以每周2天給藥/5天停藥的周期,每天給予兩次(BID),相隔6-8小時,以使得AZD5363在每周周期的第1天和第2天給予且實例3在每周周期的第3天和第4天給予。實例3是以50毫克/千克BID給予且AZD5363是以170毫克/千克BID給予,分別在HPMC/Tween和DMSO/Kleptose中。腫瘤生長曲線(圖9中所示)指示,間歇地給予或者實例3或者AZD5363相對于僅用媒劑的對照(HPMC/Tween)部分地抑制腫瘤生長。實例3加AZD5363的組合誘導腫瘤消退。通過實例3與AKT抑制劑(AZD5363)的組合共同給藥抑制腫瘤生長在BT474異種移植模型中進行研究。實例3和AZD5363每天給予兩次(BID),相隔6-8小時且以每周2天給藥/5天停藥的周期伴隨給藥。實例3是以25毫克/千克BID給予且AZD5363是以100毫克/千克BID給予,均在HPMC/Tween中。腫瘤生長曲線(圖10中所示)指示,間歇地給予實例3或AZD5363相對于僅用媒劑的對照(HPMC/Tween)部分地抑制腫瘤生長。實例3加AZD5363的組合在給藥期間誘導腫瘤消退,但在不給藥期間繼之以腫瘤再生長。通過實例3與PARP抑制劑(奧拉帕尼)的組合抑制腫瘤生長在BT474異種移植模型中進行研究。實例3和奧拉帕尼在整個研究中每天給予,實例3以25毫克/千克各劑量每天給予兩次(BID),相隔6-8小時;且奧拉帕尼在實例3的第一個日劑量之后1小時以100毫克/千克每天給予一次(QD)。兩種藥劑均在HPMC/Tween中給予。腫瘤生長曲線(圖11)指示,奧拉帕尼單獨對腫瘤生長無重要作用,實例3單獨部分地抑制生長,但實例3加奧拉帕尼的組合誘導腫瘤消退。通過實例3與PARP抑制劑(奧拉帕尼)的組合抑制腫瘤生長在MCF7異種移植模型中進行研究。實例3和奧拉帕尼在整個研究中每天給予,實例3以25毫克/千克各劑量每天給予兩次,相隔6-8小時;且奧拉帕尼在實例3的第一個日劑量之后1小時以100毫克/千克每天給予一次(QD)。兩種藥劑均在HPMC/Tween中給予。腫瘤生長曲線(圖12)指示,奧拉帕尼單獨對腫瘤生長具有極少作用,實例3單獨引起一些腫瘤消退,但實例3加奧拉帕尼的組合誘導更強腫瘤消退。通過實例3與PI3K-β/δ抑制劑(AZD8186)的組合抑制腫瘤生長在HCC70異種移植模型中進行研究。實例3和AZD8186在整個研究中每天給予,每天兩次(BID),實例3以25毫克/千克各劑量給予且AZD8186以50毫克/千克各劑量給予。兩種藥劑均在HPMC/Tween中給予。腫瘤生長曲線(圖13)指示,AZD8186部分地抑制腫瘤生長,實例3單獨更強烈地抑制生長,但實例3加AZD8186的組合誘導腫瘤消退。根據本發明的另一個方面,提供一種醫藥組合物,它包括一種如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽與醫藥學上可接受的稀釋劑或載劑組合。用于片劑配制品的適合的醫藥學上可接受的賦形劑包括例如惰性稀釋劑、成粒劑和崩解劑、粘合劑、潤滑劑、防腐劑以及抗氧化劑。片劑配制品可以未包覆包衣或包覆包衣,以改變它們的崩解性和隨后活性成分在胃腸道內的吸收,或改良它們的穩定性和/或外觀,在任一種情況下,均使用在本領域中熟知的常規包衣劑和程序。經口使用的組合物可以可替代地呈硬明膠膠囊形式,其中活性成分與惰性固體稀釋劑混合;或呈軟明膠膠囊形式,其中活性成分與水或油混合。水性懸浮液總體上包含呈細粉形式的活性成分以及一種或多種懸浮劑、分散劑或潤濕劑。水性懸浮液還可以包含一種或多種防腐劑、抗氧化劑、著色劑、調味劑和/或甜味劑。油性懸浮液可以通過將活性成分懸浮于植物油中或礦物油中來配制。油性懸浮液還可以包含增稠劑。可以添加甜味劑(如上文所列的甜味劑)和調味劑,以提供可口的口服制劑。這些組合物可以通過添加抗氧化劑來保存。適于通過添加水來制備水性懸浮液的可分散性散劑和顆粒劑總體上包含活性成分以及分散劑或潤濕劑、懸浮劑以及一種或多種防腐劑。也可以存在如甜味劑、調味劑以及著色劑的額外賦形劑。本發明的醫藥組合物還可以呈水包油乳液形式。油相可以是植物油或礦物油或任何這些油的混合物。乳液也可以包含甜味劑、調味劑以及防腐劑。糖漿和酏劑可以使用甜味劑配制,并且也可以包含緩和劑、防腐劑、調味劑和/或著色劑。醫藥組合物還可以呈無菌可注射水性或油性懸浮液的形式,它可以根據已知程序,使用上述適當分散劑或潤濕劑和懸浮劑中的一者或多者來配制。無菌可注射制劑還可以是于無毒腸胃外可接受的稀釋劑或溶劑中的無菌可注射溶液或懸浮液。用于經吸入給藥的組合物可以呈常規加壓氣霧劑的形式,該加壓氣霧劑經安排以分配或者呈包含細粉狀固體的氣霧劑形式或者液滴形式的活性成分。可以使用如揮發性氟化烴或烴的常規氣霧劑推進劑,并且便利地安排氣霧劑設備以分配定量的活性成分。有關配制品的另外的信息,請讀者參考《綜合醫藥化學》(ComprehensiveMedicinalChemistry)(科溫·漢施(CorwinHansch);編輯委員會主席),培格曼出版社1990的第5卷,第25.2章。與一種或多種賦形劑組合以產生單一劑型的活性成分的量將必要地變化,取決于所治療主體和特定給藥途徑。舉例來說,向人類經口給藥將總體上需要例如從1mg到2g活性劑(更適當地從100mg到2g,例如從250mg到1.8g,如從500mg到1.8g,尤其從500mg到1.5g,宜為從500mg到1g)與適當且適宜量的賦形劑混合給予,這些賦形劑可以在總組合物的從約3重量%到約98重量%內變化。應了解,如果需要大劑量,那么可能需要多種劑型,例如兩種或更多種片劑或膠囊,其中活性成分的劑量宜在其間劃分。適宜地,單一固體劑型可以包含1mg與300mg之間的活性成分。為實現治療或預防目的的具有化學式(I)的化合物的劑量大小自然將根據疾病病況的性質和嚴重程度、動物或患者的年齡和性別以及給藥途徑,根據熟知的醫學原則而變化。在使用具有化學式(I)的化合物以實現治療或預防目的時,總體上將給予具有化學式(I)的化合物以致接受例如每千克體重1mg到每千克體重100mg的范圍內的日劑量,如果需要那么分次給藥。總體來講,當采用腸胃外途徑時,將給予更低劑量。因此,例如,對于靜脈內給予來說,總體上將使用介于例如每千克體重1mg到每千克體重25mg范圍內的劑量。類似地,對于經吸入給予來說,將使用介于例如每千克體重1mg到每千克體重25mg范圍內的劑量。然而,經口給予(確切地說以片劑形式)為優選的。典型地,單位劑型將包含約10mg到0.5g的本發明化合物。本發明化合物可以每天一次或多于每天一次給予。本發明化合物還可以按適合的給藥時程給予,例如,本發明化合物可以每天給予一次或多次(例如一天一次、兩次或三次)持續一定天數,接著是不給予劑量的數天時間。可以接著重復這一劑量周期(由給藥日和非給藥日組成)。適宜地,劑量周期是5-14天的時間,如5、7、10或14天,更適宜地是7天。在一個方面中,具有化學式(I)的化合物在劑量周期中給予1天或者連續2或3天,接著3、4、5或6天不給藥。在一個方面中,具有化學式(I)的化合物給予1天,接著持續2、3或4天不給藥。在另一個方面中,具有化學式(I)的化合物給予2天,接著4、5或6天不給藥。在另一個方面中,具有化學式(I)的化合物給予3天,接著3、4或5天不給藥。在另一個方面中,具有化學式(I)的化合物給予4天,接著2、3或4天不給藥。在另一個方面中,具有化學式(I)的化合物給予5天,接著1、2或3天不給藥。在另一個方面中,具有化學式(I)的化合物每隔一天給予。以上給藥時程適宜地在本發明化合物作為單一療法使用時應用。在下文中描述本發明化合物作為組合療法給予的可能的給藥時程的另外的實例。如上所述,已知PI3K-α和-δ酶通過以下作用中的一者或多者促成腫瘤形成:介導癌癥和其他細胞的增殖、介導血管生成事件以及介導癌細胞的活動、遷移性以及侵襲性。我們已發現,本發明化合物具有強力抗腫瘤活性,該活性據相信借助于抑制涉及信號轉導步驟的PI3K-α和-δ酶而獲得,這些信號轉導步驟引起腫瘤細胞增殖和存活以及使腫瘤細胞轉移的侵襲性和遷移能力。因此,本發明化合物具有作為抗腫瘤劑的價值,具體來說,作為哺乳動物癌細胞增殖、存活、活動、播散以及侵襲的選擇性性抑制劑,從而抑制腫瘤生長和存活并抑制轉移性腫瘤生長。確切地說,本發明化合物具有在抑制和/或治療實體腫瘤疾病方面作為抗增殖性及抗侵襲性藥劑的價值。確切地說,本發明化合物預期有用于預防或治療對PI3K-α和/或-δ酶的抑制敏感且涉及信號轉導步驟的那些腫瘤,這些信號轉導步驟引起腫瘤細胞增殖和存活以及使腫瘤細胞轉移的遷移能力和侵襲性。另外,本發明化合物預期有用于預防或治療單獨或部分地通過抑制PI3K-α和/或-δ酶來介導的那些腫瘤,即這些化合物可以用于在需要該治療的溫血動物中產生PI3K-α和/或-δ酶抑制作用。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義在如人類的溫血動物中用作藥物。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于在如人類的溫血動物中產生抗增殖性作用。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于在如人類的溫血動物中作為抑制和/或治療實體腫瘤疾病的抗侵襲性藥劑。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義用于在如人類的溫血動物中產生抗增殖性作用。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于在如人類的溫血動物中產生抗增殖性作用。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于在如人類的溫血動物中作為抑制和/或治療實體腫瘤疾病的抗侵襲性藥劑。根據本發明的這一方面的另一個特征,提供一種在需要治療的溫血動物(如人類)中產生抗增殖性作用的方法,該方法包括向所述動物給予有效量的如上定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的這一方面的另一個特征,提供一種通過在需要治療的溫血動物(如人類)中抑制和/或治療實體腫瘤疾病來產生抗侵襲性作用的方法,該方法包括向所述動物給予有效量的如上定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于在如人類的溫血動物中預防或治療癌癥。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于在如人類的溫血動物中預防或治療癌癥。根據本發明的這一方面的另一個特征,提供一種在需要治療的溫血動物(如人類)中預防或治療癌癥的方法,該方法包括向所述動物給予有效量的如上定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于在如人類的溫血動物中預防或治療實體腫瘤疾病。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于在如人類的溫血動物中預防或治療實體腫瘤疾病。根據本發明的這一方面的另一個特征,提供一種在需要治療的溫血動物(如人類)中預防或治療實體腫瘤疾病的方法,該方法包括向所述動物給予有效量的如上定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于預防或治療對抑制會涉及信號轉導步驟的PI3K-α和/或-δ酶敏感的那些腫瘤,這些信號轉導步驟引起腫瘤細胞的增殖、存活、侵襲性以及遷移性能力。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于預防或治療對抑制會涉及信號轉導步驟的PI3K-α和/或-δ酶敏感的那些腫瘤,這些信號轉導步驟引起腫瘤細胞的增殖、存活、侵襲性以及遷移性能力。根據本發明的這一方面的另一個特征,提供一種用于預防或治療對抑制會涉及信號轉導步驟的PI3K-α和/或-δ酶敏感的那些腫瘤的方法,這些信號轉導步驟引起腫瘤細胞的增殖、存活、侵襲性以及遷移性能力,該方法包括向所述動物給予有效量的如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的另一個方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于提供PI3K-α和-δ酶抑制作用。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于提供PI3K-α和-δ酶抑制作用。根據本發明的另一個方面,還提供一種用于提供PI3K-α和-δ酶抑制作用的方法,該方法包括給予有效量的如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。如上文所述,某些本發明化合物針對PI3K-α和-δ酶的效力實質上比針對其他PI3激酶或其他激酶的效力更好。這些化合物具有針對PI3K-α和-δ酶的足夠效力,它們可以按足以抑制PI3K-α和-δ酶同時針對PI3K-β酶和針對大多數其他激酶顯示極少活性的量使用。這些化合物可能有用于選擇性抑制PI3K-α和-δ酶并且可能有用于有效治療例如PI3K-α和/或-δ酶驅動的腫瘤。根據本發明的這一方面,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如上文所定義適用于提供選擇性PI3K-α和-δ酶抑制作用。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如上文所定義在藥物制造中適用于提供選擇性PI3K-α和-δ酶抑制作用。根據本發明的另一個方面,還提供一種用于提供選擇性PI3K-α和-δ酶抑制作用的方法,該方法包括給予有效量的如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。“選擇性PI3K-α和-δ酶抑制作用”意指具有化學式(I)的化合物針對PI3K-α和-δ酶的效力比針對其他1類PI3激酶的效力更強,并且總體上展示出關于更寬PI3激酶家族的其他成員和在包括酪氨酸和ser/thr激酶的更寬激酶類別中的良好選擇性。根據本發明的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如在此之前所定義適用于治療乳腺癌;胃(Stomach/Gastric)癌和食管癌;非小細胞肺癌(NSCLC),包括鱗狀細胞癌(SCC)和腺癌、頭頸部(H&N)的SCC;婦科癌癥(包括子宮內膜癌、卵巢癌以及子宮頸癌);以及血液科癌癥,如多發性骨髓瘤、淋巴瘤以及白血病(包括慢性淋巴細胞白血病(CLL)、急性淋巴母細胞白血病(ALL)以及套細胞淋巴瘤(MCL))。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如在此之前所定義適用于治療膀胱癌、腦/CNS癌、結腸直腸癌、肺癌(所有其他形式)、膽囊癌和膽管癌、以及皮膚癌。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽,如在此之前所定義適用于治療前列腺癌、骨癌、腎癌、肝癌、黑素瘤癌、胃腸組織癌、胰臟癌、睪丸癌、甲狀腺癌、陰莖癌、外陰癌以及經由突變、擴增或其他畸變具有PI3激酶依賴性的其他腫瘤類型。根據本發明的這一方面的另一個特征,提供一種用于治療需要治療的溫血動物(如人類)中的乳腺癌、胃癌和食管癌、NSCLC(包括SCC和腺癌、H&N的SCC)、婦科癌癥(包括子宮內膜癌、卵巢癌以及子宮頸癌)以及血液科癌癥(如多發性骨髓瘤、淋巴瘤以及白血病(包括CLL、ALL以及MCL))的方法,該方法包括給予有效量的如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的這一方面的另一個特征,提供一種用于治療需要治療的溫血動物(如人類)中的膀胱癌、腦/CNS癌、結腸直腸癌、肺癌(所有其他形式)、膽囊癌和膽管癌、以及皮膚癌的方法,該方法包括給予有效量的如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的這一方面的另一個特征,提供一種用于治療需要治療的溫血動物(如人類)中的前列腺癌、骨癌、腎癌、肝癌、黑素瘤癌、胃腸組織癌、胰臟癌、睪丸癌、甲狀腺癌、陰莖癌、外陰癌以及經由突變、擴增或其他畸變具有PI3激酶依賴性的其他腫瘤類型的方法,該方法包括給予有效量的如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽。根據本發明的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如在此之前所定義在藥物制造中適用于治療乳腺癌、胃癌和食管癌、NSCLC(包括SCC和腺癌、H&N的SCC)、婦科癌癥(包括子宮內膜癌、卵巢癌以及子宮頸癌)以及血液科癌癥(如多發性骨髓瘤、淋巴瘤以及白血病(包括CLL、ALL以及MCL))。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如在此之前所定義在藥物制造中適用于治療膀胱癌、腦/CNS癌、結腸直腸癌、肺癌(所有其他形式)、膽囊癌和膽管癌、以及皮膚癌。根據本發明的這一方面的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽的用途,如在此之前所定義在藥物制造中適用于治療前列腺癌、骨癌、腎癌、肝癌、黑素瘤癌、胃腸組織癌、胰腺癌、睪丸癌、甲狀腺癌、陰莖癌、外陰癌以及經由突變、擴增或其他畸變具有PI3激酶依賴性的其他腫瘤類型。在本發明的一個特征中,待治療的癌癥為乳癌。在這一特征的另一個方面中,該乳癌為雌激素受體陽性。在這一方面的一個實施例中,具有化學式(I)的化合物或其醫藥學上可接受的鹽與如在此所定義的抗激素劑組合給予。在這一方面的另一個實施例中,實例3與如在此所定義的抗激素劑組合給予。在這一方面的另一個實施例中,實例3與奧拉帕尼或其醫藥學上可接受的鹽組合,并且任選地進一步與如在此所定義的抗激素劑組合給予。在這一方面的另一個實施例中,實例3與AZD5363或其醫藥學上可接受的鹽組合,并且任選地進一步與如在此所定義的抗激素劑組合給予。在表明癌癥治療的一個方面中,應了解,這可能指預防轉移以及治療轉移(即癌癥擴散)。因此,本發明化合物可能用于治療不具有轉移的患者而終止轉移發生,或延長轉移發生前的時間段,以及針對已具有轉移的患者來治療轉移本身。此外,癌癥治療可能指治療確定的原發性腫瘤(多種)和發展中的原發性腫瘤(多種)。因此,在一個方面中,癌癥治療涉及預防轉移。在本發明的另一個方面中,癌癥治療涉及治療轉移。在本發明的另一個方面中,癌癥治療涉及治療確定的原發性腫瘤(多種)或發展中的原發性腫瘤(多種)。如上文所述,具有化學式(I)的化合物的體內作用可以部分通過在給予具有化學式(I)的化合物之后在人體或動物體內形成的一種或多種代謝物(如如上文所定義的具有化學式A的化合物)來發揮。特定本發明化合物針對PI3激酶-α和-δ的效力比優于針對其他I類PI3激酶同工型(如β和γ)的效力更好。在一個方面中,本發明化合物具有相比于PI3K-β或-γ針對PI3K-α和-δ的選擇性。因此,本發明還涵蓋一種用于抑制患者中的PI3激酶-α的方法,該方法包括向患者給予有效抑制該患者中的磷酸肌醇3-激酶-α的量的具有化學式(I)的化合物或其醫藥學上可接受的鹽。因此,本發明還涵蓋一種用于抑制患者中的PI3激酶-α和-δ的方法,該方法包括向患者給予有效抑制該患者中的PI3激酶-α和-δ的量的具有化學式(I)的化合物或其醫藥學上可接受的鹽。具有化學式(I)的化合物或其醫藥學上可接受的鹽為PI3激酶的抑制劑,在各種其他疾病病況中也具有潛在治療用途。舉例來說,PI3激酶在促進血管樹(即血管平滑肌細胞)中(蒂貝里(Thyberg),《歐洲細胞生物學雜志》(EuropeanJournalofCellBiology),1998,76(1),33-42),和肺(氣管平滑肌細胞)中(克雷姆斯卡亞V.P.(Krymskaya,V.P.),《生物藥物》(BioDrugs),2007,21(2),85-95)的平滑肌增殖方面起重要作用。血管平滑肌細胞的過度增殖在侵襲性血管程序之后在形成動脈粥樣硬化斑中和在發展新生內膜增生中起重要作用(施瓦茲(Scwartz)等人,《心血管疾病進展》(ProgressinCardiovascularDisease),1984,26,355-372;克洛斯(Clowes)等人,《實驗室研究》(LaboratoryInvestigations),1978,39,141-150。此外,氣管平滑肌細胞的過度增殖在哮喘和慢性支氣管炎背景下引起COPD的發展。因此,可以使用PI3激酶活性的抑制劑來預防血管再狹窄、動脈粥樣硬化以及COPD。PI3激酶在白細胞功能(富勒(Fuller)等人,《免疫學雜志》(TheJournalofImmunology),1999,162(11),6337-6340;埃德(Eder)等人,《生物化學雜志》(TheJournalofBiologicalChemistry),1998,273(43),28025-31)和淋巴細胞功能(韋森特-曼薩納雷斯(Vicente-Manzanares)等人,《免疫學雜志》,1999,163(7),4001-4012)中也起重要作用。舉例來說,白細胞與發炎內皮的粘附涉及通過PI3激酶依賴性信號傳導過程激活內源性白細胞整合素。此外,嗜中性白細胞中的氧化爆發(西岡(Nishioka)等人,《FEBS快報》(FEBSLetters),1998,441(1),63-66和康德利夫A.M.(Condliffe,A.M.)等人,《血液》,2005,106(4),1432-40)和細胞骨架重組(基什(Kirsch)等人,《美國國家科學院論文集》(ProceedingsNationalAcademyofSciencesUSA),1999,96(11),6211-6216)似乎涉及PI3激酶信號傳導。嗜中性白細胞遷移和定向移動也取決于PI3激酶活性(坎普斯M.(Camps,M.)等人,《自然·醫學》,2005,11(9),936-43和薩都C.(Sadhu,C.)等人,《免疫學雜志》,2003,170(5),2647-54)。因此,PI3激酶的抑制劑可以有用于減少在炎癥位點處的白細胞粘附和激活并且因此可以用于治療急性和/或慢性炎性病癥。PI3激酶在淋巴細胞增殖和激活中也起重要作用(弗魯曼(Fruman)等人,《科學》,1999,283(5400),393-397)。確切地說,PI3K-δ對B細胞發育和功能為至關重要的,包括IgM特異性抗體誘導的B細胞增殖(奧肯海于格K(OkkenhaugK)等人,《科學》,2002,297(5583),1031-1034)、B細胞受體誘導的DNA合成與增殖以及IL-4-誘導的存活(比蘭恰A(BilancioA)等人,《血液》,2006,107,642-650)。這些觀察結果表明,PI3K-δ在不能通過其他多種I類PI3K補償的B細胞功能中具有至關重要且非冗余的作用。考慮到淋巴細胞在自體免疫疾病中的重要作用,可以使用PI3激酶活性的抑制劑來治療這些病癥(隆美爾C(RommelC),坎普斯M(CampsM)以及吉H(JiH),《自然綜述·免疫學》(NatRevImmunol),2007,1038,191-201)。上文所定義的抗癌治療可以按單獨療法形式施用,或除本發明的化合物外,還可以涉及常規外科手術或放射線療法或化學療法。該化學療法可以包括以下抗腫瘤劑類別中的一者或多者:-(i)如腫瘤醫學中所用的抗增生性/抗贅生性藥物和其組合,如烷基化劑(例如順鉑、奧賽力鉑、卡鉑、環磷酰胺、氮芥子氣、美法侖、苯丁酸氮芥、白消安、替莫唑胺以及亞硝基脲);抗代謝物(例如吉西他濱和抗葉酸制劑,如氟嘧啶,如5-氟尿嘧啶和喃氟啶、拉替曲澤、甲氨喋呤、胞嘧啶阿拉伯糖苷以及羥基脲);抗腫瘤抗生素(例如蒽環霉素,如阿霉素、博萊霉素、小紅莓、道諾霉素、表柔比星、艾達霉素、絲裂霉素C、更生霉素以及光神霉素);抗有絲分裂劑(例如長春花生物堿,如長春新堿、長春堿、長春地辛以及長春瑞濱,和紫杉類,如紫杉醇和多烯紫杉醇,以及polo激酶抑制劑);以及拓撲異構酶抑制劑(例如表鬼臼毒素,如依托泊苷和替尼泊苷,安吖啶、拓樸替康以及喜樹堿);(ii)抗激素劑,如抗雌激素(例如他莫昔芬、氟維司群、托瑞米芬、雷諾昔酚、屈洛昔芬以及艾多昔芬)、抗雄激素(例如比卡魯胺、氟他胺、尼魯米特以及乙酸環丙孕酮)、LHRH拮抗劑或LHRH激動劑(例如戈舍瑞林、亮丙瑞林以及布舍瑞林)、孕激素(例如乙酸甲地孕酮)、芳香酶抑制劑(例如阿那曲唑、來曲唑、維拉唑以及依西美坦)以及5α還原酶抑制劑(如非那雄安);(iii)生長因子功能和其下游信號傳導路徑的抑制劑:包括任何生長因子或生長因子受體目標的Ab調節劑,由施特恩(Stern)等人《腫瘤學/血液學的重要綜述》(CriticalReviewsinOncology/Haematology),2005,54,第11-29頁)綜述;也包括這些目標的小分子抑制劑,例如激酶抑制劑,實例包括抗erbB2抗體曲妥珠單抗[HerceptinTM]、抗EGFR抗體帕尼單抗、抗EGFR抗體西妥昔單抗[Erbitux,C225]以及酪氨酸激酶抑制劑,包括erbB受體家族的抑制劑,如表皮生長因子家族受體(EGFR/erbB1)酪氨酸激酶抑制劑,如吉非替尼或埃羅替尼,erbB2酪氨酸激酶抑制劑,如拉帕替尼,以及混合erb1/2抑制劑,如阿法替尼;類似策略可供用于生長因子和其受體的其他類別,例如肝細胞生長因子家族或其受體(包括c-met和ron)的抑制劑;胰島素和胰島素生長因子家族或其受體(IGFR、IR)的抑制劑、血小板衍生的生長因子家族或其受體(PDGFR)的抑制劑以及通過其他受體酪氨酸激酶(如c-kit、AnLK以及CSF-1R)介導的信號傳導的抑制劑;還包括在更寬PI3激酶信號傳導路徑中靶向信號傳導蛋白質的調節劑,例如其他PI3激酶同工型(如PI3K-β)和ser/thr激酶(如AKT、mTOR、PDK、SGK、PI4K或PIP5K)的抑制劑;還包括以上未列出的絲氨酸/蘇氨酸激酶的抑制劑,舉例來說,如維羅非尼的raf抑制劑,如司美替尼(AZD6244)的MEK抑制劑,如伊馬替尼或尼羅替尼的Ab1抑制劑,如依魯替尼的Btk抑制劑,如福他替尼的Syk抑制劑,極光激酶抑制劑(例如AZD1152),如JAK、STAT以及IRAK4的其他ser/thr激酶抑制劑,以及細胞周期素依賴性激酶抑制劑;iv)DNA損傷信號傳導路徑的調節劑,例如PARP抑制劑(例如奧拉帕尼)、ATR抑制劑或ATM抑制劑;v)細胞凋亡和細胞死亡路徑的調節劑,如Bcl家族調節劑(例如ABT-263/Navitoclax、ABT-199);(vi)抗血管生成劑,如抑制血管內皮生長因子的作用的那些藥劑,[例如抗血管內皮細胞生長因子抗體貝伐單抗(AvastinTM)和例如VEGF受體酪氨酸激酶抑制劑,如索拉非尼、阿西替尼、帕佐泮尼、舒尼替尼以及凡德他尼(以及通過其他機制起作用的化合物(例如利諾胺、整合素αvβ3功能的抑制劑以及血管生長抑素))];(vii)血管破壞劑,如考布他汀A4;(viii)抗侵襲劑,例如c-Src激酶家族抑制劑,如(達沙替尼,《藥物化學雜志》(J.Med.Chem.),2004,47,6658-6661)和伯舒替尼(SKI-606),以及金屬蛋白酶抑制劑,如馬立馬司他、尿激酶纖維蛋白溶酶原激活劑受體功能的抑制劑或針對肝素酶的抗體];(ix)免疫療法方法,包括例如增加患者腫瘤細胞的免疫原性的離體和體內方法,如以如白介素2、白介素4或粒細胞-巨噬細胞集落刺激因子的細胞因子轉染,減少T細胞無變應性的方法,使用如細胞因子轉染的樹突狀細胞的經轉染免疫細胞的方法,使用細胞因子轉染的腫瘤細胞系的方法,以及使用抗個體基因型抗體的方法。特定實例包括靶向PD-1(例如BMS-936558)或CTLA4(例如伊匹單抗和曲美單抗)的單克隆單抗;(x)反義或基于RNAi的療法,例如有關所列目標的那些療法。(xi)基因療法方法,包括例如置換異常基因(如異常p53或異常BRCA1或BRCA2)的方法;GDEPT(基因定向酶前藥療法)方法,如使用胞嘧啶脫氨酶、胸苷激酶或細菌性硝基還原酶的那些方法;以及增強患者對化學療法或放射線療法的耐受性的方法(如多重耐藥性基因療法)。根據本發明的這一方面,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和另一種抗腫瘤劑,尤其是在上文的(i)-(xi)下所列的抗腫瘤劑中的任一者。確切地說,以上在(i)-(xi)下所列的抗腫瘤劑為待治療的特定癌癥的護理標準;本領域普通技術人員應了解“護理標準”的含義。因此,在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與另一種抗腫瘤劑的組合,尤其是選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與另一種抗腫瘤劑的組合,尤其是選自在上文的(i)下所列的一者的抗腫瘤劑。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和在上文的(i)下所列的任一種抗腫瘤劑。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和紫杉類,如紫杉醇或紫杉德,適宜地為紫杉德。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與另一種抗腫瘤劑的組合,尤其是選自于在此上文的(ii)下所列的一者的抗腫瘤劑。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和在上文的(ii)下所列的抗激素劑中的任一者,例如在上文的(ii)中所列的抗雌激素中的任一者。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與mTOR抑制劑的組合,如WO2008/023161中所披露的那些mTOR抑制劑,例如在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和mTOR抑制劑,如WO2008/023161中所披露的那些mTOR抑制劑,例如確切地說,mTOR抑制劑為AZD2014,它具有以下結構:在一個方面中,具有化學式(I)的化合物與AZD2014的以上組合適用于治療ER陽性乳癌,任選地與護理性激素療法標準組合。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與PI3K-β的抑制劑的組合。具有化學式(I)的化合物與PI3K-β的抑制劑的組合可以尤其有用于治療腫瘤,例如前列腺腫瘤、乳房腫瘤(例如三陰性乳房腫瘤)、鱗狀細胞NSCLC以及腎癌,其中PTEN背景缺失。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和PI3K-β的抑制劑。在一個方面中,在此所述的PI3K-β的抑制劑也具有一些PI3K-δ抑制活性。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與PI3K-β的抑制劑的組合,如國際專利申請WO2011/051704中的實例中的任一者。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和PI3K-β的抑制劑,如國際專利申請WO2011/051704中的實例中的任一者。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與PI3K-β和PI3K-δ的抑制劑的組合,該抑制劑如8-((1R)-1-(3,5-二氟苯氨基)乙基)-N,N-二甲基-2-N-嗎啉基-4-氧代-4H-色烯-6-甲酰胺(國際專利申請WO2011/051704中的實例3.06b,也被稱作AZD8186)或其醫藥學上可接受的鹽:在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和PI3K-β與PI3K-δ的抑制劑,該抑制劑如8-((1R)-1-(3,5-二氟苯氨基)乙基)-N,N-二甲基-2-N-嗎啉基-4-氧代-4H-色烯-6-甲酰胺(國際專利申請WO2011/051704中的實例3.06b,也被稱作AZD8186)或其醫藥學上可接受的鹽:在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與AKT激酶的抑制劑的組合,該抑制劑如(S)-4-氨基-N-(1-(4-氯苯基)-3-羥丙基)-1-(7H-吡咯并[2,3-d]嘧啶-4-基)哌啶-4-甲酰胺(AZD5363)或其醫藥學上可接受的鹽(參見例如WO2009/047563)。具有化學式(I)的化合物與AKT抑制劑的組合可以尤其有用于治療PIK3CA基因中突變流行率更高的腫瘤,如ER陽性乳癌、子宮內膜癌、卵巢癌、鱗狀細胞NSCLC、胃癌、膀胱癌以及膽道癌。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和AKT激酶的抑制劑,該抑制劑如(S)-4-氨基-N-(1-(4-氯苯基)-3-羥丙基)-1-(7H-吡咯并[2,3-d]嘧啶-4-基)哌啶-4-甲酰胺(AZD5363)或其醫藥學上可接受的鹽(參見例如WO2009/047563)。在本發明的另一個方面中,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與奧拉帕尼(4-[3-(4-環丙烷羰基-哌嗪-1-羰基)-4-氟-苯甲基]-2H-酞嗪-1-酮)或其醫藥學上可接受的鹽的組合。具有化學式(I)的化合物與奧拉帕尼的組合可以尤其有用于或者BRCA野生型或者缺失型的三陰性乳癌和雌激素受體陽性(ER陽性)乳癌兩者中,尤其是具有PIK3CA基因突變的那些癌癥。在本發明的另一個方面中,提供一種適用于治療癌癥的組合,包括如上文所定義的具有化學式(I)的化合物或其醫藥學上可接受的鹽和奧拉帕尼(4-[3-(4-環丙烷羰基-哌嗪-1-羰基)-4-氟-苯甲基]-2H-酞嗪-1-酮)或其醫藥學上可接受的鹽。本發明的特定組合包括在此的實例化合物(或其醫藥學上可接受的鹽)和如上文所述的mTOR抑制劑、PI3Kβ抑制劑、AKT激酶的抑制劑或奧拉帕尼中的任一者。本發明的另外的特定組合包括實例3(或其醫藥學上可接受的鹽)和如上文所述的mTOR抑制劑、PI3Kβ抑制劑、AKT激酶的抑制劑或奧拉帕尼。本發明的另外的特定組合包括實例3(或其醫藥學上可接受的鹽)和如上文所述的PI3Kβ抑制劑、AKT激酶的抑制劑或奧拉帕尼(或這三者中的任一者的醫藥學上可接受的鹽)。本發明組合的再另一個特定實例包括實例3(或其醫藥學上可接受的鹽);和AZD8186、AZD5363以及奧拉帕尼(或這三者中的任一者的醫藥學上可接受的鹽)中的任一者。本發明組合的另一個實例包括實例3和AZD2014。在所有以上組合中,應了解,該組合也可以在如本領域普通技術人員所了解的護理治療標準下給予,如上文(i)到(xi)的其他治療。舉例來說,當預期使用以上組合中的任一者來治療ER陽性乳癌時,可以使用護理性激素療法標準(如在上文的(ii)下所列的那些藥劑)以及本發明的組合。在其他方面中,適當地,護理標準可以選自以上(i)。因此,在本發明的另一個方面中,提供一種適用于治療癌癥的三重組合a)一種具有化學式(I)的化合物(如實例3)或其醫藥學上可接受的鹽;b)一種mTOR抑制劑、PI3Kβ抑制劑、AKT激酶的抑制劑或奧拉帕尼或其醫藥學上可接受的鹽;以及c)用于待治療的癌癥的護理療法標準。適當地,如本領域普通技術人員所了解,護理療法標準可以根據其常見給藥方案給予。根據本發明的另一個方面,提供一種醫藥組合物,包括具有化學式(I)的化合物或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合,以及醫藥學上可接受的稀釋劑或載劑。根據本發明的另一個方面,提供一種醫藥組合物,包括實例3或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合,以及醫藥學上可接受的稀釋劑或載劑。根據本發明的另一個方面,提供一種醫藥組合物,包括實例3或其醫藥學上可接受的鹽與AZD5363、AZD8186或奧拉帕尼(或這三者中的任一者的醫藥學上可接受的鹽)的組合以及醫藥學上可接受的稀釋劑或載劑。根據本發明的另一個方面,提供一種用于治療癌癥的醫藥組合物,包括具有化學式(I)的化合物或其醫藥學上可接受的鹽與選自于在此上文的(i)-(ix)下所列的一者的抗腫瘤劑的組合,以及醫藥學上可接受的稀釋劑或載劑。根據本發明的另一個方面,提供一種用于治療癌癥的醫藥組合物,包括實例3或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合,以及醫藥學上可接受的稀釋劑或載劑。根據本發明的另一個方面,提供一種用于治療癌癥的醫藥組合物,包括實例3或其醫藥學上可接受的鹽與AZD5363、AZD8186或奧拉帕尼(或這三者中的任一者的醫藥學上可接受的鹽)的組合以及醫藥學上可接受的稀釋劑或載劑。根據本發明的另一個特征,提供一種具有化學式(I)的化合物或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合的用途,它是用于制造用于溫血動物(如人類)的癌癥的藥物。根據本發明的另一個特征,提供實例3或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合的用途,它是用于制造用于溫血動物(如人類)的癌癥的藥物。根據本發明的另一個特征,提供實例3或其醫藥學上可接受的鹽與AZD5363、AZD8186或奧拉帕尼(或這三者中的任一者的醫藥學上可接受的鹽)的組合的用途,它是用于制造用于溫血動物(如人類)的癌癥的藥物。因此,在本發明的另一個特征中,提供一種治療需要治療的溫血動物(如人類)的癌癥的方法,該方法包括向所述動物給予有效量的具有化學式(I)的化合物或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合。因此,在本發明的另一個特征中,提供一種治療需要治療的溫血動物(如人類)的癌癥的方法,該方法包括向所述動物給予有效量的實例3或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合。因此,在本發明的另一個特征中,提供一種治療需要治療的溫血動物(如人類)的癌癥的方法,該方法包括向所述動物給予有效量的實例3或其醫藥學上可接受的鹽與AZD5363、AZD8186或奧拉帕尼(或這三者中的任一者的醫藥學上可接受的鹽)的組合。根據本發明的另一個方面,提供一種試劑盒,包括具有化學式(I)的化合物或其醫藥學上可接受的鹽與選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑的組合。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的一種選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑;以及c)用于容納所述第一和所述第二劑型的容器裝置。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的一種選自于在此上文的(i)-(xi)下所列的一者的抗腫瘤劑;c)用于容納所述第一和所述第二劑型的容器裝置;以及任選的d)使用說明書。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的mTOR抑制劑,如WO2008/023161中所披露的那些mTOR抑制劑,例如以及c)用于容納所述第一和所述第二劑型的容器裝置。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的PI3K-β的抑制劑,如國際專利申請WO2011/051704中的實例中的任一者或其醫藥學上可接受的鹽;以及c)用于容納所述第一和所述第二劑型的容器裝置。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的PI3K-β的抑制劑,如國際專利申請WO2011/051704中的實例中的任一者或其醫藥學上可接受的鹽;c)用于容納所述第一和所述第二劑型的容器裝置;以及任選的d)使用說明書。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的PI3K-β和PI3K-δ的抑制劑,它為8-((1R)-1-(3,5-二氟苯氨基)乙基)-N,N-二甲基-2-N-嗎啉基-4-氧代-4H-色烯-6-甲酰胺(國際專利申請WO2011/051704中的實例3.06b,也被稱作AZD8186)或其醫藥學上可接受的鹽;以及c)用于容納所述第一和所述第二劑型的容器裝置。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的PI3K-β和PI3K-δ的抑制劑,它為8-((1R)-1-(3,5-二氟苯氨基)乙基)-N,N-二甲基-2-N-嗎啉基-4-氧代-4H-色烯-6-甲酰胺(國際專利申請WO2011/051704中的實例3.06b,也被稱作AZD8186)或其醫藥學上可接受的鹽;c)用于容納所述第一和所述第二劑型的容器裝置;以及任選的d)使用說明書。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的AKT激酶的抑制劑,如(S)-4-氨基-N-(1-(4-氯苯基)-3-羥丙基)-1-(7H-吡咯并[2,3-d]嘧啶-4-基)哌啶-4-甲酰胺或其醫藥學上可接受的鹽(AZD5363,參見例如WO2009/047563);c)用于容納所述第一和所述第二劑型的容器裝置;以及任選的d)使用說明書。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的AKT激酶的抑制劑,如(S)-4-氨基-N-(1-(4-氯苯基)-3-羥丙基)-1-(7H-吡咯并[2,3-d]嘧啶-4-基)哌啶-4-甲酰胺或其醫藥學上可接受的鹽(AZD5363,參見例如WO2009/047563);以及c)用于容納所述第一和所述第二劑型的容器裝置。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的AKT激酶的抑制劑,如(S)-4-氨基-N-(1-(4-氯苯基)-3-羥丙基)-1-(7H-吡咯并[2,3-d]嘧啶-4-基)哌啶-4-甲酰胺或其醫藥學上可接受的鹽(AZD5363,參見例如WO2009/047563);c)用于容納所述第一和所述第二劑型的容器裝置;以及任選的d)使用說明書。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的奧拉帕尼(4-[3-(4-環丙烷羰基-哌嗪-1-羰基)-4-氟-苯甲基]-2H-酞嗪-1-酮)或其醫藥學上可接受的鹽;以及c)用于容納所述第一和所述第二劑型的容器裝置。根據本發明的另一個方面,提供一種試劑盒,包括:a)呈一個第一單位劑型的一種具有化學式(I)的化合物或其醫藥學上可接受的鹽;b)呈一個第二單位劑型的奧拉帕尼(4-[3-(4-環丙烷羰基-哌嗪-1-羰基)-4-氟-苯甲基]-2H-酞嗪-1-酮)或其醫藥學上可接受的鹽;c)用于容納所述第一和所述第二劑型的容器裝置;以及任選的d)使用說明書。在所有以上組合、用途、治療方法以及試劑盒中,AZD5363、AZD8186以及奧拉帕尼可以呈游離堿形式或呈醫藥學上可接受的鹽形式。因此,在一個實施例中,AZD5363呈游離堿形式;在另一個實施例中,AZD5363呈醫藥學上可接受的鹽形式。在另一個實施例中,AZD8186呈游離堿形式;在另一個實施例中,AZD8186呈醫藥學上可接受的鹽形式。在另一個實施例中,奧拉帕尼呈游離堿形式;在另一個實施例中,奧拉帕尼呈醫藥學上可接受的鹽形式。雖然具有化學式(I)的化合物主要具有作為用于溫血動物(包括人類)的治療劑的價值,但它們也有用在需要抑制PI3激酶-α和-δ的作用的任何時候。因此,它們有用于作為用于發展新生物測試以及搜尋新藥理學藥劑中的藥理學標準。在此,當使用術語“組合”時,應了解,這一術語是指同時、分別或依序給藥。在本發明的一個方面中,“組合”是指同時給藥。在本發明的另一個方面中,“組合”是指分別給藥。在本發明的另一個方面中,“組合”是指依序給藥。當給藥是依序或分別進行時,給予第二組分不應延遲以致失去有利的作用。在一個實施例中,依序治療涉及在11天的時間段內給予該組合的每一組分。在另一個實施例中,這一時間段為10天。在另一個實施例中,這一時間段為9天。在另一個實施例中,這一時間段為8天。在另一個實施例中,這一時間段為7天。在另一個實施例中,這一時間段為6天以內。在另一個實施例中,這一時間段為5天以內。在另一個實施例中,這一時間段為4天以內。在另一個實施例中,這一時間段為3天以內。在另一個實施例中,這一時間段為2天以內。在另一個實施例中,這一時間段為24小時以內。在另一個實施例中,這一時間段為12小時以內。依序和共同給藥均在此于BT474模型中使用實例3和AZD5363的組合實驗中舉例說明。在這個實例中,依序給藥通過給予AZD5363持續2天,繼之以實例3持續2天,接著為3天不給予任一藥劑,隨后重復該模式(“劑量周期”)來說明。共同給藥通過以下給藥方案來說明,其中AZD5363和實例3均給予2天,繼之以5天不給予任一藥劑。在這兩個實例中,依序給藥似乎在引起腫瘤消退方面更有效,展示出使方案達到最佳的潛在重要性。其他可能的共同給藥方案包括:1)AZD5363和實例3均給予2天,繼之以3天不給予任一藥劑的劑量周期;2)AZD5363和實例3均給予3天,繼之以4天不給予任一藥劑的劑量周期;3)AZD5363和實例3均給予4天,繼之以3天不給予任一藥劑的劑量周期;4)AZD5363和實例3均給予5天,繼之以2天不給予任一藥劑的劑量周期;5)AZD5363和實例3每隔一天給予的劑量周期6)AZD5363和實例3每三天給予的劑量周期7)AZD5363和實例3以每周時程給予,給藥之間間隔3和4天(例如星期一/星期四)的劑量周期8)AZD5363和實例3以每周時程給予,給藥之間間隔2和3天(例如星期一/星期三/星期五)的劑量周期具有化學式(I)的化合物、尤其實例3與mTOR抑制劑(如AZD2014或PI3K-β抑制劑(如β/δ抑制劑AZD8186))的組合可以適當地以與上文關于實例3和AZD5363的組合所述的那些方案類似的方案給予。具有化學式(I)的化合物和奧拉帕尼的組合可以根據以下方案給予,其中奧拉帕尼每天給予且具有化學式(I)的化合物根據間歇性給藥時程給予(如2天給藥,繼之以3到5天不給藥)。這些說明性給藥方案中的每一者包括本發明的另一個方面。這些說明性給藥方案中的每一者也可以與在以上(i)到(xi)中列出的其他抗腫瘤劑組合施用。在既定劑量周期內,給予該組合的一種特定組分,隨后為另一種組分(即依序給藥)可為有利的。因此,在一個實施例中,依序給藥包括在劑量周期內依序給予具有化學式(I)的化合物(尤其實例3),隨后給予在以上(i)到(xi)中所列的另一種抗腫瘤劑,尤其是選自AZD5363、AZD8186以及奧拉帕尼的抗腫瘤劑。在另一個實施例中,依序給藥包括在劑量周期內依序給予在以上(i)到(xi)中所列的抗腫瘤劑,尤其是選自AZD5363、AZD8186以及奧拉帕尼的抗腫瘤劑,隨后給予具有化學式(I)的化合物(尤其實例3)。在一個實施例中,在以上(i)到(xi)中所列的抗腫瘤劑和具有化學式(I)的化合物相隔最多2天給予。在另一個實施例中,在以上(i)到(xi)中所列的抗腫瘤劑和具有化學式(I)的化合物相隔最多1天給予。在另一個實施例中,在以上(i)到(xi)中所列的抗腫瘤劑和具有化學式(I)的化合物相隔最多18小時給予。在另一個實施例中,在以上(i)到(xi)中所列的抗腫瘤劑和具有化學式(I)的化合物相隔最多12小時給予。在另一個實施例中,在以上(i)到(xi)中所列的抗腫瘤劑和具有化學式(I)的化合物相隔最多6小時給予。在另一個實施例中,在以上(i)到(xi)中所列的抗腫瘤劑和具有化學式(I)的化合物相隔最多3小時給予。在另一個實施例中,劑量周期的長度可以為5到10天。在另一個實施例中,劑量周期的長度可以為6到10天。在另一個實施例中,劑量周期的長度可以為7到9天。在另一個實施例中,劑量周期的長度可以為6到8天。在另一個實施例中,劑量周期的長度可以為10天。在另一個實施例中,劑量周期的長度可以為9天。在另一個實施例中,劑量周期的長度可以為8天。在另一個實施例中,劑量周期的長度可以為7天。在另一個實施例中,劑量周期的長度可以為6天。在另一個實施例中,劑量周期的長度可以為5天。在其他實施例中,劑量周期可以涉及在6到9天劑量周期長度內,具有化學式(I)的化合物(尤其實例3)給予2-4個連續日且其他幾天不給予。在其他實施例中,劑量周期可以涉及在6到9天劑量周期長度(例如長度為7天)內,具有化學式(I)的化合物(尤其實例3)給予3-4個連續日且其他幾天不給予。在其他實施例中,劑量周期可以涉及在7到10天劑量周期長度內,具有化學式(I)的化合物(尤其實例3)給予3-5個連續日且其他幾天不給予。在其他實施例中,劑量周期可以涉及在6到9天劑量周期長度內,具有化學式(I)的化合物(尤其實例3)給予5個連續日且其他幾天不給予。在其他實施例中,劑量周期可以涉及在6到9天劑量周期長度(例如長度為7天)內,具有化學式(I)的化合物(尤其實例3)給予4個連續日且其他幾天不給予。在其他實施例中,劑量周期可以涉及在6到9天劑量周期長度內,具有化學式(I)的化合物(尤其實例3)給予3個連續日且其他幾天不給予。劑量周期可以相隔數天,在此期間不給予活性組合組分。可以在典型地根據常見處方給藥時程進行的護理療法標準之上添加如上所述的組合療法。個人化醫療本發明的另一個方面是基于鑒別編碼磷酸肌醇-3-激酶的基因的狀態、催化性α多肽(PIK3CA)以及用具有化學式(I)的化合物治療的易感性之間的聯系。因此,這一方面提供用于選擇用具有化學式(I)的化合物治療的患者,尤其是癌癥患者和/或避免更不可能與該治療發生治療反應的患者治療而由此避免不必要的治療以及可能與該無效治療有關的任何副作用的機會、方法以及工具。本發明涉及患者選擇工具和方法(包括個人化藥物)。該選擇是基于待治療的腫瘤細胞是具有野生型抑或突變型PIK3CA基因。PIK3CA基因狀態可以因此用作PI3K-α和-δ抑制劑的治療易感性的生物標記。明確需要將針對以下患者富集或用于選擇這些患者的生物標記:這些患者的腫瘤將對PI3K-α和-δ抑制劑(如具有化學式(I)的化合物)治療起反應。鑒別最可能對藥劑起反應的患者的患者選擇生物標記在癌癥治療中為理想的,因為它們減少具有非反應性腫瘤的患者的不必要治療,從而減少這些藥劑的潛在副作用。生物標記可以描述為“作為正常生物過程、致病過程或對治療性干預的藥理學反應的指示劑客觀地經測量且評估的特性”。生物標記為與一種特定病狀或疾病有關的任何可鑒別且可測量的指示劑,其中在生物標記的存在或含量與該病狀或疾病的一些方面(包括該病狀或疾病的存在、它的含量或變化含量、它的類型、它的階段、它的易感性或對用于治療該病狀或疾病的藥物的反應)之間存在相關性。相關性可以為定性的、定量的或定性且定量的。典型地,生物標記為化合物、化合物片段或化合物組。這些化合物可以為在生物體中發現或通過生物體產生的任何化合物,包括蛋白質(和肽)、核酸以及其他化合物。生物標記可以具有預測能力,并且因此可以用于預測或檢測特定病狀或疾病的存在、含量、類型或階段(包括特定微生物或毒素的存在或含量)、對特定病狀或疾病的易感性(包括遺傳易感性)或對特定治療(包括藥物治療)的反應。據認為在藥物發現和發展的未來,生物標記將通過改良研究與發展程序的效率而發揮日益重要的作用。生物標記可以用作診斷劑、疾病進展監測劑、治療監測劑以及臨床結果預測劑。舉例來說,不同生物標記研究項目試圖鑒別出特定癌癥和特定心血管疾病以及免疫疾病的標記。據相信新的經驗證的生物標記的發展將使醫療和藥物發展成本顯著降低,并且使多種疾病和病狀的治療顯著改善。為了最佳設計臨床試驗并且為了從這些試驗獲得最多信息,生物標記可以為需要的。該標記在替代物和腫瘤組織中可以為可測量的。這些標記理想地也將與功效相關并且由此可以最終用于患者選擇。因此,本發明的這一方面的根本技術問題為鑒別對用具有化學式(I)的化合物治療的患者分類的方法。該技術問題通過提供在此的權利要求書和/或說明書中所表征的實施例來解決。如在此的實例中所詳述,發現具有PIK3CA突變的細胞的生長總體上更易受具有化學式(I)的化合物抑制。本發明提供一種測定細胞對具有化學式(I)的化合物的敏感性的方法。該方法包括測定所述細胞中的PIK3CA基因狀態。如果這些細胞具有突變的PIK3CA基因,那么這些細胞經鑒別為可能對具有化學式I的化合物敏感。因此預測具有突變的PIK3CA基因的那些患者尤其易受具有化學式(I)的化合物治療影響。如果一個細胞在細胞生長分析中抑制細胞數目增加(或者經由抑制細胞增殖和/或者經由增加細胞死亡),那么將該細胞定義為對具有化學式(I)的化合物敏感。本發明方法有用于預測更可能通過生長抑制對具有化學式(I)的化合物起反應的細胞。本發明進一步部分地基于可以用于測定患者對具有化學式(I)的化合物的反應的方法,包括確定是否給予具有化學式(I)的化合物。確切地說,本發明方法包括測定PIK3CA的基因狀態。突變的PIK3CA基因的存在指示腫瘤細胞當與具有化學式(I)的化合物接觸時更可能通過生長抑制起反應。PIK3CA基因狀態可以因此用于選擇用具有化學式(I)的化合物治療的患者。此外,披露一種用于鑒別可能對具有化學式(I)的化合物敏感的患者的體外方法。還披露了提供能夠檢測PIK3CA基因的突變狀態的寡或聚核苷酸引物或探針的用途。還披露用于檢測PIK3CA突變的′試劑盒的用途,包括(但不限于)由診斷公司(包括凱杰(Qiagen)和羅氏分子系統(RocheMolecularSystems))出售的PIK3CA突變檢測試劑盒。在另一個實施例中,本發明涉及一種用于確定罹患癌癥的患者是否可能為使用具有化學式(I)的化合物的藥物治療的反應者的體外方法,所述方法包括以下步驟:(i)獲得代表先前從所述患者收集的腫瘤的樣品;以及(ii)確定所述樣品中的PIK3CA基因是否包含突變。PIK3CA基因突變指示對具有化學式(I)的化合物治療的反應可能性增加。作為單一基因生物標記測試,包含PIK3CA突變的腫瘤的鑒別將使對具有化學式(I)的化合物的反應增加。包含PIK3CA突變的單獨腫瘤具有對具有化學式(I)的化合物起反應的最大可能性。“代表腫瘤”的樣品可以為經分離的實際腫瘤樣品或者可以為已進一步加工的樣品,例如從腫瘤樣品進行PCR擴增的核酸樣品。定義:在這一個人化醫療部分中:“等位基因”是指遺傳基因座的特定形式,通過它的特定核苷酸或氨基酸序列與其他形式相區分。“擴增反應”為導致目標核酸優于非目標核酸進行特異性擴增的核酸反應。聚合酶鏈反應(PCR)為熟知擴增反應。“癌癥”在此用于指由于細胞轉化成贅生性表型所出現的贅生性生長。該細胞轉化通常涉及遺傳突變。“基因”為包含關于RNA產物的經調節生物合成的所有信息的DNA區段,包括啟動子、外顯子、內含子以及可以位于控制表達的5′或3′側接區域內(不在該基因的轉錄部分內)的其他序列元件。“基因狀態”是指該基因為野生型或者不是(即突變型)。“標記”是指能夠產生指示在分析樣品中存在目標聚核苷酸的可檢測信號的組合物。適合標記包括放射性同位素、核苷酸發色團、酶、底物、熒光分子、化學發光部分、磁性粒子、生物發光部分等。因此,標記為通過光譜、光化學、生物化學、免疫化學、電學、光學或化學手段可檢測的任何組合物。“非同義變異”是指基因編碼序列中或與基因編碼序列重疊的變異(變化),導致產生不同(經改變的)多肽序列。這些變異可能影響或可能不影響蛋白質功能并且包括錯義變異體(導致一個氨基酸取代另一個)、無義變異體(由于產生過早終止密碼子而產生截短多肽)以及插入/缺失變異體。“同義變異”是指不影響所編碼多肽的序列的基因編碼序列中的變異(變化)。這些變異可以間接地(例如通過改變基因表達)影響蛋白質功能,但在不存在相反證據的情況下,總體上假定為無害的。“核酸”是指單鏈或雙鏈DNA和RNA分子,包括自然界中所發現的天然核酸和/或經修飾的具有經修飾的主鏈或堿基的人工核酸,如本領域中已知。“引物”是指能夠充當用于合成與待復制的核酸鏈互補的引物延伸產物的起始點的單鏈DNA寡核苷酸序列。引物的長度和序列必須使其能夠引發延伸產物的合成。典型引物在與目標序列實質上互補的序列長度中包含至少約7個核苷酸,但略微更長的引物為優選的。通常,引物包含約15-26個核苷酸,但也可以采用更長或更短引物。“多態位點”為基因座內導致在一個群體中發現至少兩種替代性序列的位置。“多態現象”是指在個體中在多態位點處所觀察到的序列變異。多態現象包括核苷酸取代、插入、缺失以及微衛星,并且可以(但無需)引起基因表達或蛋白質功能方面可檢測的差異。在不存在對表達或蛋白質功能的影響證據的情況下,包括非同義變異體的常見多態現象總體上視為包括在野生型基因序列的定義中。人類多態現象和相關注釋的目錄包括驗證、觀察的頻率以及疾病關聯,由NCBI(dbSNP:http://www.ncbi.nlm.nih.gov/projects/SNP/)維護。請注意,術語“多態現象(polymorphism)”當用于基因序列的情形中時不應與術語“多晶型現象(polymorphism)”當用于化合物的固態形式(即化合物的結晶或非晶性質)的情形中時混淆。本領域普通技術人員將通過其背景了解所需的含義。“探針”是指具有與待檢測的等位基因的目標序列完全互補的序列的單鏈序列特異性寡核苷酸。“反應”是通過根據實體腫瘤反應評估準則(ResponseEvaluationCriteriainSolidTumours;RECIST)進行的測量來定義,涉及將患者主要分成兩個組:顯示部分反應或穩定疾病的組和顯示進行性疾病的跡象的組。“嚴格的雜交條件”是指在42℃下在包括50%甲酰胺、5×SSC(750mMNaCl、75mM檸檬酸三鈉)、50mM磷酸鈉(pH7.6)、5×鄧哈特溶液(Denhardt′ssolution)、10%硫酸葡聚糖以及20pg/mI變性剪切的鮭魚精子DNA的溶液中孵育過夜,繼之以在0.1×SSC中在約65℃下洗滌過濾物。“存活”涵蓋患者的整體存活和無進展存活。“整體存活(OS)”定義為從給藥開始到由于任何原因死亡的時間。“無進展存活(PFS)”定義為從給藥開始到第一次出現進行性疾病或由于任何原因死亡的時間。根據本發明的一個方面,提供一種用于選擇用具有化學式(I)的化合物治療的患者的方法,該方法包括從患者提供包含腫瘤細胞的樣品;確定包含患者的腫瘤細胞的樣品中的PIK3CA基因為野生型抑或突變型;以及基于以上來選擇用具有化學式(I)的化合物治療的患者。該方法可以包括或不包括實際患者樣品分離步驟。因此,根據本發明的一個方面,提供一種用于選擇用具有化學式(I)的化合物治療的患者的方法,該方法包括確定先前從患者分離的包含腫瘤細胞的樣品中的PIK3CA基因為野生型抑或突變型;以及基于以上來選擇用具有化學式(I)的化合物治療的患者。在一個實施例中,如果腫瘤細胞DNA具有突變型PIK3CA基因,那么選擇該患者用具有化學式(I)的化合物治療。在其他實施例中,腫瘤細胞DNA具有野生型PIK3CA基因的患者不選擇用具有化學式(I)的化合物治療。根據本發明的另一個方面,提供一種用于預測患者對具有化學式(I)的化合物的反應的方法,該方法包括確定患者的腫瘤細胞中的PIK3CA基因為野生型抑或突變型以及基于以上,預測患者對用具有化學式(I)的化合物治療的反應。根據本發明的另一個方面,提供一種用于測定在罹患癌癥的人類患者中用具有化學式I的化合物治療的有效性的可能性的方法,該方法包括:測定患者的腫瘤細胞中的PIK3CA基因(多個)為野生型抑或突變型以及基于以上,預測患者對用具有化學式(I)的化合物治療的反應。為了本發明的目的,野生型的基因狀態打算表明基因的正常或恰當表達以及所編碼蛋白質的正常功能。相比之下,突變型狀態打算表明異常或不當基因表達,或蛋白質功能改變的表達,與癌癥中突變型PIK3CA的已知作用(如在此所述)一致。任何數目的遺傳或后生變化(包括(但不限于)突變、擴增、缺失、基因組重排或甲基化特征變化)可以導致突變體狀態。然而,如果這些變化仍然引起正常蛋白質或功能上等效變異體的恰當表達,那么該基因狀態被視為野生型。典型地將不產生功能突變型基因狀態的變異體實例包括同義編碼變異體和常見多態現象(同義或非同義)。如下文所討論,基因狀態可以通過功能分析來評估,或者它可以從與參考序列的檢測偏差的性質推斷。在某些實施例中,PIK3CA基因的野生型或突變型狀態通過這些基因中非同義核酸變異的存在或缺乏來確定。所觀察的非同義變異對應于未標注功能作用的已知常見多態現象,不促成突變型基因狀態。PIK3CA基因中意味著突變型狀態的其他變異包括剪接位點變異,這些變異在前體mRNA加工成mRNA期間降低內含子/外顯子接合點的識別。這可以導致外顯子跳躍或在剪接mRNA中包括通常內含的序列(內含子滯留或采用隱含的剪接接合點)。這可以轉而通過插入和/或缺失促使產生相對于正常蛋白質的異常蛋白質。因此,在其他實施例中,如果在內含子/外顯子接合點處存在改變剪接位點識別序列的變異體,那么該基因具有突變型狀態。另外,具有異常或失調PIK3CA或PI3K-α的腫瘤中的另外的基因(如Kras,一種潛在抗性標記)的突變狀態或激活狀態的測量可以有助于提高個人化藥物方法的預測。在我們于阿斯特拉捷利康進行的關于乳癌(基于COSMIC數據庫(韋爾科姆基金會桑格研究所(WelcomeTrustSangerInstitute),2011年9月)的調查中,從覆蓋>5千種人類腫瘤的整個數據集鑒別出PIK3CA基因中>55種不同突變。大部分突變以<1%頻率發生,3種以1%-3%頻率發生,但4種突變占總PIK3CA突變的約88%。這些突變為C端激酶結構域中的激酶結構域錯義突變H1047R(55%)和H1047L(5%),以及螺旋結構域殘基E545K(18%)和E542K(11%)。雖然并不打算為窮盡性的,但其他普遍的乳癌突變的更長清單涵蓋R38H、R38C、R88Q、N345K、C420R、E453Q、P539R、E542K、E545K、E545A、Q546K、Q546P、M1043I、M1043V、H1047R、H1047L、H1047Y。因此,診斷分析可以被構建成聚焦于檢測最常見突變,由此允許鑒別大部分PIK3CA突變。舉例來說,來自羅氏分子系統的Cobas(TM)PIK3CA突變測試經設計以檢測從福爾馬林固定的石蠟嵌埋的腫瘤樣品分離的DNA中PIK3CA基因的外顯子1、4、7、9以及20中的17種突變(E542K、E545A、E545G、E545K、E545D、Q546K、Q546R、Q546E、Q546L、N345K、C420R、R88Q、H1047L、H1047R、H1047Y、G1049R以及M1043I)。這一試劑盒能夠拾取ER陽性乳癌中的約95%突變。突變分布在其他腫瘤類型中不同并且診斷策略可以相應地調適。舉例來說,在子宮內膜癌中,相比于乳癌,擴散在整個PIK3CA基因編碼序列中的突變分布更均勻并且蛋白質N端區域中的突變數目更多(由醫學博士道格拉斯A.萊文(DouglasA.Levine,M.D)傳達,TCGA第2次年度研討會,2012年11月28日)。關于PIK3CA,參考序列可供用于基因(基因庫寄存編號:NG_012113)、mRNA(基因庫寄存編號:NM_006218)以及蛋白質(基因庫寄存編號:NP_006209或Swiss-Prot寄存號:P42336)。參考基因(基因組區域)序列包括上游序列的5000個堿基的下游序列的2000個堿基。PIK3CA內的突變眾所周知(COSMIC數據庫-韋爾科姆基金會桑格研究所),并且本領域普通技術人員將能夠基于與野生型比較DNA或蛋白質序列來確定PIK3CA基因狀態,即特定PIK3CA基因為野生型抑或突變型。關于PI3激酶α蛋白質序列的PIK3CA和p110α催化亞基所披露的基因和mRNA序列顯然各自為代表性序列。在正常個體中,每種基因存在兩個拷貝(母本和父本拷貝),這些拷貝將可能具有一些序列差異,此外在一個群體內將存在基因序列的許多等位基因變異體。視為野生型的其他序列包括具有以下的序列:核酸序列的一種或多種同義變化(這些變化不改變所編碼的蛋白質序列)、改變蛋白質序列但不影響蛋白質功能的非同義常見多態現象(例如生殖系多態現象)以及內含子非剪接位點序列變化。根據本發明的另一個方面,提供一種用于測定在罹患癌癥的人類患者中用具有化學式(I)的化合物治療的有效性的可能性的方法,該方法包括:檢測所述患者的PIK3CA基因中相對于野生型基因的至少一種非同義核酸變化的存在或缺乏,其中在PIK3CA基因中存在至少一種體細胞非同義核酸變化指示用具有化學式(I)的化合物治療可能為有效的。根據本發明的另一個方面,提供一種用于評估個體對用具有化學式(I)的化合物治療的易感性的方法,該方法包括:(i)確定個體的腫瘤細胞DNA中PIK3CA基因的非同義突變狀態;以及,(ii)通過參考腫瘤細胞中PIK3CA基因的非同義突變狀態來測定個體對用具有化學式(I)的化合物治療的可能易感性。存在本領域普通技術人員可獲得的以確定PIK3CA的基因狀態的許多技術。基因狀態可以通過測定核酸序列來確定。這可以經由對全長基因進行直接測序或分析該基因內的特異性位點,例如一般突變位點。用于確定PIK3CA基因為野生型抑或突變型的替代性手段為評估轉錄基因的功能。這一PIK3CA基因的功能突變產生脂質激酶活性增加的蛋白質,引起細胞中該路徑的下游信號傳導增加,包括(但不限于)Akt和S6激酶的激活。當PIK3CA變異體在細胞中表達時,用于評估它們功能狀態的分析包括(但不限于):(i)增加具有PIK3CA基因三磷酸磷脂酰肌醇(PI(3,4,5)P3)的激酶活性的產物的產生;(ii)增加磷酸化Akt或S6激酶的含量;(iii)增加用PIK3CA變異體轉染的NIH-3T3細胞的聚集和集落形成;(池上T(IkenoueT)等人,《癌癥研究》,200565,4562-4567)。樣品待測試基因狀態的患者樣品可以為從個體獲得或從個體可獲得的任何包含腫瘤組織或腫瘤細胞的樣品。測試樣品宜為從個體獲得的血液樣品、口腔抹片、活檢體或其他體液或組織。特定實例包括:循環腫瘤細胞、血漿或血清中的循環DNA、從卵巢癌患者的腹水液分離的細胞、具有腫瘤的患者肺內的肺痰、來自乳癌患者的細針抽出物、尿液、外周血液、細胞刮片、毛囊、皮膚鉆孔或面頰樣品。應了解,測試樣品同樣可以為對應于測試樣品中的序列的核酸序列,換句話說,樣品核酸中的所有或一部分區域可以在分析之前先使用任何便利技術(例如聚合酶鏈反應(PCR))擴增。核酸可以為基因組DNA或者部分分離的或全細胞RNA。在特定實施例中,RNA為全細胞RNA且直接用作使用隨機引物或polyA引物來標記一個第一鏈cDNA的模板。可以根據標準方法從測試樣品提取該樣品中的核酸或蛋白質(參見格林(Green)和薩姆布魯克(Sambrook)編,《分子克隆實驗指南》(MolecularCloning:ALaboratoryManual),(2012,第4版,第1-3卷,ISBN9781936113422),紐約州冷泉港的冷泉港實驗室出版社(ColdSpringHarborLaboratoryPress,ColdSpringHarbor,N.Y.))。本發明的診斷方法可以使用先前從個體或患者獲取的樣品進行。這些樣品可以通過冷凍或固定以及嵌埋于福爾馬林-石蠟或其他介質中來保存。可替代地,可以獲得且使用包含新鮮腫瘤細胞的樣品。可以使用來自任何腫瘤的細胞應用本發明方法。用具有化學式(I)的化合物治療的適合腫瘤已在上文中描述。在臨床腫瘤中廣泛發現PIK3CA突變,但每一基因突變的流行率隨腫瘤組織類型顯著不同。舉例來說,PIK3CA突變在乳癌中相對常見,但在腎臟腫瘤中相對罕見。表1.表1:臨床樣品中PIK3CA突變的流行率。PIK3CA信息來源為COSMIC數據庫(v62版)。本發明的患者選擇方法可以尤其有用于存在PIK3CA突變的高發生率的疾病(組織)區段(例如乳房、泌尿道、子宮內膜、大腸、子宮頸等)。如本領域普通技術人員將顯而易知,隨著新的且更全面的數據從如TCGA(癌癥基因組圖譜(TheCancerGenomeAtlas))和ICGC(國際癌癥基因組聯盟(InternationalCancerGenomeConsortium))的人類癌癥基因組分析協會(HumanCancerGenomeprofilingconsortia)出現,這一頻率數據持續地得到改進與更新。因此,可以鑒別具有PIK3CA依賴性的其他腫瘤類型且其適用于用在此所述的化合物治療。檢測核酸的方法在本發明的上下文中,可以采用突變型PIK3CA核酸的檢測來預測對藥物治療的反應。因為這些基因中的突變發生在DNA層面,所以本發明方法可以基于檢測基因組DNA的突變或變化,以及轉錄物和蛋白質本身。所希望的可以是通過分析轉錄物和/或多肽來確認基因組DNA突變,以便確保所檢測的突變實際上在該受試者中表達。本領域普通技術人員將清楚,存在可以用于檢測基因中一個或多個位置處變異體核苷酸的存在或缺乏的大量分析程序。一般來說,等位基因變異的檢測需要突變辨別技術、任選的擴增反應(如基于聚合酶鏈反應的擴增反應)以及任選的信號產生系統。在本領域中可獲得多種突變檢測技術并且這些突變檢測技術可以與信號產生系統組合使用,在本領域中可獲得許多信號產生系統。檢測等位基因變異的許多方法由諾勞(Nollau)等人,《臨床化學》(Clin.Chem.),1997,43,1114-1120;安德森SM.(AndersonSM.)《分子診斷學專家評論》(ExpertRevMolDiagn.),2011,11,635-642;邁耶森M.(MeyersonM.)等人,《自然綜述·遺傳學》,2010,11,685-696綜述;以及綜述于標準教科書中,例如《突變檢測的實驗室方案》(“LaboratoryProtocolsforMutationDetection”),由U.蘭德格倫(U.Landegren)編,牛津大學出版社(OxfordUniversityPress),1996和《PCR》(“PCR”),第2版,由牛頓(Newton)和格雷安(Graham)編,拜爾斯科學出版社有限公司(BIOSScientificPublishersLimited),1997。如上所指出,測定具有癌癥的患者中存在或缺乏PIK3CA基因的特定變化或多種變化可以按多種方式進行。此類測試通常使用從生物樣品收集的DNA或RNA進行,這些生物樣品例如組織活檢體、尿液、大便、痰、血液、細胞、組織刮片、乳房抽出物或其他細胞物質,并且可以通過多種方法進行,這些方法包括(但不限于)PCR、與等位基因特異性探針雜交、酶突變檢測、錯配的化學裂解、質譜或DNA測序,包括微測序。適合的突變檢測技術包括擴增阻礙突變系統(ARMSTM)、擴增阻礙突變系統線性延伸(ALEXTM)、競爭性寡核苷酸引發系統(COPS)、塔克曼(Taqman)、分子信標(MolecularBeacons)、限制性片段長度多態現象(RFLP)以及基于限制位點的PCR以及熒光共振能量轉移(FRET)技術。在特定實施例中,用于測定生物標記基因內的核苷酸所用的方法是選自:等位基因特異性擴增(等位基因特異性PCR)(如擴增阻礙突變系統(ARMS))、測序、等位基因辨別分析、雜交、限制性片段長度多態現象(RFLP)或寡核苷酸連接分析(OLA)。在特定實施例中,與等位基因特異性探針雜交可以通過以下來進行:(1)溶液中結合到具有經標記樣品的固相(例如玻璃、硅、尼龍膜)的等位基因特異性寡核苷酸,例如,如同在許多DNA芯片應用中;或(2)溶液中的經結合樣品(通常為經克隆DNA或PCR擴增的DNA)和經標記寡核苷酸(或者等位基因特異性或者較短以便允許通過雜交來測序)。診斷測試可以涉及一組變化,通常在固體支撐物上,這使得能夠同時測定一種以上變化。這些雜交探針在本領域中為熟知的(參見例如格林和薩姆布魯克編,《分子克隆實驗指南》,(2012,第4版,第1-3卷,ISBN9781936113422),紐約州冷泉港的冷泉港實驗室出版社)并且可以跨越兩個或更多個變化位點。因此,在一個實施例中,檢測至少一種突變的存在或缺乏使包含假定突變位點的PIK3CA核酸與至少一個核酸探針接觸。探針優先在選擇性雜交條件下與包括變化位點且在該變化位點處包含互補核苷酸堿基的核酸序列雜交。雜交可以使用本領域普通技術人員已知的標記用可檢測標記來檢測。這些標記包括(但不限于)放射性、熒光、染料以及酶標記。在另一個實施例中,檢測至少一種突變的存在或缺乏使包含假定突變位點的PIK3CA核酸與至少一個核酸引物接觸。引物優選在選擇性雜交條件下與包括變化位點且在該變化位點處包含互補核苷酸堿基的核酸序列雜交。用作特異性擴增的引物的寡核苷酸可以攜帶與分子中間(因此擴增取決于差異性雜交;參見例如吉布斯(Gibbs)等人,1989,《核酸研究》(Nucl.AcidsRes.),17,2437-248)或在一個引物的3′遠端處的所關注的突變互補的核苷酸堿基,其中在恰當的條件下,可以防止錯配或減少聚合酶延伸(參見例如普羅森納(Prossner),1993,《蒂布技術》(Tibtech),11238)。在又另一個實施例中,檢測至少一種突變的存在或缺乏包括對至少一種核酸序列進行測序且比較所獲得的序列與已知野生型核酸序列。可替代地,至少一種突變的存在或缺乏包括質譜測定至少一種核酸序列。在一個實施例中,檢測至少一種核酸變化的存在或缺乏包括進行聚合酶鏈反應(PCR)。使包含假設變化的目標核酸序列擴增且測定經擴增的核酸的核苷酸序列。測定經擴增的核酸的核苷酸序列包括對至少一個核酸區段進行測序。可替代地,擴增產物可以使用能夠根據它們的大小分離擴增產物的任何方法來分析,包括自動化和手動凝膠電泳等。基因組核酸中的突變有利地通過基于擴增核酸片段中的活動位移的技術來檢測。舉例來說,陳(Chen)等人,《分析生物化學》(AnalBiochem)1996,239,61-9描述通過競爭性活動位移分析來檢測單堿基突變。此外,基于馬塞利諾(Marcelino)等人,《生物技術》(BioTechniques)1999,26,1134-1148的技術的分析可商購。在特定實例中,可以使用毛細管異雙螺旋分析以基于毛細管系統中雙螺旋核酸的活動位移來檢測突變的存在作為存在錯配的結果。從樣品產生用于分析的核酸總體上需要核酸擴增。許多擴增方法依賴于酶鏈反應(如聚合酶鏈反應、連接酶鏈反應或自持續序列復制)或來自已進行克隆的所有或一部分載體。優選地,根據本發明的擴增為指數擴增,如通過例如聚合酶鏈反應所展現。許多目標和信號擴增方法已描述于文獻中,例如這些方法的總體綜述描述于蘭德格倫U.(Landegren,U.)等人,《科學》,1988242,229-237和路易斯R.(Lewis,R.),《基因工程新聞》(GeneticEngineeringNews)1990,10,54-55中。這些擴增方法可以用于我們發明的方法中,并且包括聚合酶鏈反應(PCR)、原位PCR、連接酶擴增反應(LAR)、連接酶雜交、Qβ噬菌體復制酶、基于轉錄的擴增系統(TAS)、基因組擴增與轉錄物測序(GAWTS)、基于核酸序列的擴增(NASBA)以及原位雜交。適用于不同擴增技術的引物可以根據本領域中已知的方法制備。聚合酶鏈反應(PCR)PCR為核酸擴增方法,尤其描述于美國專利第4,683,195號和第4,683,202號中。PCR由DNA聚合酶產生的引物延伸反應的重復周期組成。目標DNA為熱變性的且使兩個寡核苷酸雜交,這兩個寡核苷酸支撐待擴增的DNA相對鏈上的目標序列。這些寡核苷酸變成引物用于DNA聚合酶。通過引物延伸復制DNA來制造兩個鏈的第二拷貝。通過重復熱變性、引物雜交以及延伸的周期,目標DNA可以在約二到四小時內擴增一百萬倍或更多。PCR為分子生物學工具,它必須與檢測技術結合用于測定擴增結果。PCR的優點為它通過將目標DNA的量在約4小時內擴增一百萬到十億倍來增加敏感性。PCR可以用于在診斷背景中擴增任何已知核酸(莫(Mok)等人,《婦科腫瘤學》(GynaecologicOncology),1994,52:247-252)。如擴增阻礙突變系統(ARMSTM)的等位基因特異性擴增技術(牛頓等人,《核酸研究》,1989,17,2503-2516)也可以用于檢測單堿基突變。在恰當的PCR擴增條件下,位于引物的3′末端的單堿基錯配對完全匹配的等位基因的優先擴增來說已足夠(牛頓等人,1989,前述),允許辨別密切相關的物質。使用上述引物的擴增系統的基礎為具有錯配3′-殘基的寡核苷酸在恰當的條件下將不充當PCR引物。這一擴增系統允許在瓊脂糖凝膠電泳之后僅通過檢查反應混合物來進行基因分型。擴增產物的分析可以使用能夠根據它們的大小分離擴增產物的任何方法來進行,包括自動化和手動凝膠電泳、質譜等。核酸分離、擴增以及分析方法對本領域普通技術人員來說為常規的并且方案的實例可以見于例如格林和薩姆布魯克編,《分子克隆實驗指南》,(2012,第4版,第1-3卷,ISBN9781936113422),紐約州冷泉港的冷泉港實驗室出版社)。PCR擴增中所用方法的尤其有用的方案來源為《PCR(基礎:從背景到實驗臺)》(PCR(Basics:FromBackgroundtoBench)),M.J.麥克弗森(M.J.McPherson),S.G.梅爾樂(S.G.Mailer),R.貝農(R.Beynon),C.豪(C.Howe),斯普林格出版社(SpringerVerlag);第1版(2000年10月15日),ISBN:0387916008。本發明還提供預測和診斷試劑盒,包括用于擴增PIK3CA基因中的目標核酸的簡并引物;以及說明書,包括擴增方案和結果分析。該試劑盒也可以可替代地包括用于進行擴增以及擴增產物的分析的緩沖劑、酶以及容器。該試劑盒也可以為包括其他工具(如DNA微陣列或其他支撐物)的篩選或診斷試劑盒的組件。優選地,該試劑盒還提供一個或多個對照模板,如從正常組織樣品分離的核酸;和/或一系列代表參考基因中的不同變化的樣品。在一個實施例中,該試劑盒提供兩個或更多個引物對,每一對均能夠擴增參考(PIK3CA)基因的不同區域(每一區域具有可能變化位點),由此提供用于分析生物樣品在一個反應或若干平行反應中的若干基因變化的表達的試劑盒。試劑盒中的引物可以經標記(例如熒光標記)以促進檢測擴增產物以及隨后分析核酸的變化。該試劑盒還可以允許在一個分析中檢測一種以上變化。組合試劑盒將因此包括能夠擴增參考基因的不同區段的引物。這些引物可以例如使用不同的熒光標記進行差示性標記,以便在這些變化之間進行區分。還披露用于檢測PIK3CA突變的′試劑盒的用途,包括(但不限于)由診斷公司(包括凱杰和羅氏分子系統)出售的PIK3CA突變檢測試劑盒。在另一個方面中,本發明提供一種治療罹患癌癥的患者的方法,該方法包括:確定患者的腫瘤細胞中的PIK3CA基因的突變型或野生型狀態,并且如果PIK3CA基因為突變型,那么向該患者給予有效量的具有化學式(I)的化合物。如在此所用,術語“有效”和“有效性”包括藥理學有效性和生理學安全性兩者。藥理學有效性是指在患者中產生所希望的生物作用的治療能力。生理學安全性是指由所給予治療在細胞、器官和/或生物體層面上造成的毒性程度或其他不良生理學作用(通常稱為副作用)。“更不有效”意指該治療產生治療學上顯著更低程度的藥理學有效性和/或治療學上更大程度的不良生理學作用。根據本發明的另一個方面,提供一種具有化學式(I)的化合物用于治療患有已經鑒別為具有突變型PIK3CA基因的腫瘤細胞的癌癥患者的用途。根據本發明的另一個方面,提供一種具有化學式(I)的化合物,用于治療具有已經鑒別為攜帶突變型PIK3CA基因的腫瘤細胞的癌癥。在另一個實施例中,本發明涉及包括具有化學式(I)的化合物的醫藥組合物,它用于預防和治療具有已經鑒別為攜帶突變型PIK3CA基因的腫瘤細胞的癌癥。對于所有以上方面,所測定/鑒別的PIK3CA的突變體形式處于基因中的所有位置處。對于所有以上方面,使用如乳癌的腫瘤作為一個實例,所測定/鑒別的PIK3CA的特定突變體形式為在R38、R88、N345、C420、E453、P539、E542K、E545K、Q546、M1043、M1043以及H1047R位置處者。對于所有以上方面,使用如乳癌的腫瘤作為一個實例,所測定/鑒別的PIK3CA的特定突變體形式為在E542、E545以及H1047位置處者。個人化醫療/個人化藥物實例腫瘤細胞系中的細胞增殖分析在標準增殖分析中測定人類癌細胞系池對化合物作用的靈敏度。分析方案細節根據以上生物分析(g)擷取。突變相關性分析方法從多種組織和從多種來源收集209種癌細胞系來獲得測量對用實例3治療起反應的細胞生長抑制的藥理學數據。將每一細胞系分類為敏感性(G150<=1.0μM)或抗性(G150>1.0μM)。每一細胞系中的基因突變狀態通過整合來自內部(阿斯特拉捷利康)和公共來源的結果獲得。公共數據包括來自以下的所有細胞系數據:癌癥項目中的藥物靈敏度的基因組學第3版(GenomicsofDrugSensitivityinCancerProjectrelease3)(伽尼特MJ(GarnettMJ)等人《自然》,2012年3月483,570-5)、癌細胞系百科全書項目(CancerCellLineEncyclopediaproject)(巴雷蒂那J(BarretinaJ)等人,《自然》2012,483,603-7)以及癌癥體細胞突變目錄(CatalogueofSomaticMutationsInCancer;COSMIC)數據庫(v61版;http://www.sanger.ac.uk/genetics/CGP/cosmic/;福布斯SA(ForbesSA)等人《核酸研究》,2011,39(數據庫發行號):D945-50;福布斯SA等人,《當前人類遺傳學方案》(CurrProtocHumGenet.)2008;第10章:第10.11單元)以及所選期刊文章。不包括沉默編碼區突變(同義變異體)和非同義多態現象,并且為了此分析的目的,忽略突變的接合子型式。對于細胞系和基因的每一組合,狀態概述為突變型(MUT)、野生型(WT)或不一致(INCON)。通過衡量內部觀察結果和COSMIC的癌細胞系項目(CancerCellLineProject;CCLP)子集的那些觀察結果或通過在手動回顧之后選擇狀態來解決一些最初不一致的情況(相同細胞系中相同基因的獨立WT和MUT觀察結果)。在不可以解決不一致觀察結果的情況下,在分析期間保留INCON標記且基因狀態視為未知。突變狀態與反應之間的聯系通過構筑每一基因的列聯表且測定相應優勢比和雙尾費雪精確檢驗(two-tailedFisher′sexacttest)p值來鑒別。分類為反應邊緣的細胞系已從初始分析排除,從而鑒別候選生物標記。關于突變狀態,計數MUT或WT結果并且還排除少于4個WT或4個MUT細胞系的基因。結果與討論如方法中所述鑒別突變狀態與反應之間的聯系。細胞系對實例3的反應以及PIK3CA基因的相應遺傳狀態展示于表3中。表3.這項研究中所用的細胞系的PIK3CA基因的藥理學數據、反應分類以及突變狀態。突變與對實例3的敏感性最強烈相關的基因為PIK3CA。177個PIK3CAWT細胞系中僅12個(7.7%)對實例3敏感,而作為PIK3CA突變體的32個細胞系中的15個(46.9%)為敏感的,對應于12.1的優勢比和1.2×10-7的p值(參見表4)。表4.PIK3CA突變狀態和對實例3的反應的列聯表.如在此所表明,已報導具有異常或失調PIK3CA或PI3K-α的腫瘤中的另外的基因(如KRAS,一種潛在抗性標記)的突變狀態或激活狀態的測量可以有助于提高個人化藥物方法的預測。關于以上數據集,我們通過比較PIK3CA突變體細胞中的KRAS突變富集與該細胞系對抑制的反應來對此進行舉例說明。分析限于包含兩種基因的‘熱點’突變的細胞系(在PIK3CA的密碼子E542、E545以及H1047處和在KRAS的密碼子K12、13以及Q61處)。這證明了在PIK3CA突變體細胞系中,KRAS突變賦予對實例3抑制的抗性。-二十八個細胞系包含PIK3CA中的激活突變。-包含激活PIK3CA突變和野生型KRAS基因的19個細胞系中的6個(31.6%)對實例3具有抗性。-9個PIK3CA突變體細胞系中的7個(77.8%)包含共存KRAS突變且對實例3具有抗性。這轉化成7.5的優勢比和0.042的p值(參見表5)。表5.PIK3CA和KRAS突變狀態和對實例3的反應的列聯表。實例現將于下列實例中說明本發明,在這些實例中,總體上:(i)除非另行說明,否則在環境溫度(即在17℃到25℃范圍內)下和在如氮氣的惰性氣體的氣氛下進行操作;(ii)通過旋轉蒸發或利用Genevac設施在真空中進行蒸發且在通過過濾移除殘余固體之后進行處理程序;(iii)使用從德國達姆施塔特的默克公司(Merck,Darmstad,Germany)獲得的預填充Merck正相Si60二氧化硅濾筒(粒度計:15-40或40-63μm)在自動化ArmenGliderFlash:SpotIIUltimate(法國圣阿維的亞門儀器(ArmenInstrument,Saint-Ave,France))上進行快速色譜純化;(iv)使用水(包含0.2%碳酸銨)與乙腈的極性遞減混合物作為洗脫劑,在配備有ZMD或ZQESCi質譜儀和WatersX-Terra或WatersX-Bridge或WatersSunFire反相柱(C-18,5微米二氧化硅,19mm直徑,100mm長度,流速40毫升/分鐘)的Waters儀器(600/2700或2525)上進行制備色譜法;(v)產率(當存在時)并不必需為可得到的最大值;(vi)總體來講,具有化學式I的最終產物的結構通過核磁共振(NMR)光譜法確認;根據6量表測量NMR化學位移值[使用BrukerAvance500(500MHz)儀器測定質子磁共振光譜];除非另外說明,否則在環境溫度下進行測量;已使用以下縮寫:s,單峰;d,雙重峰;t,三重峰;q,四重峰;m,多重峰;dd,雙重峰的雙重峰;ddd,雙重峰的雙重峰的雙重峰;dt,三重峰的雙重峰;bs,寬信號;(vii)總體來講,具有化學式I的最終產物也通過液相色譜后的質譜(LCMS)表征;LCMS是使用配備有WatersZQESCi或ZMDESCi質譜儀和XBridge5μmC-18柱(2.1×50mm)的WatersAllianceHT(2790和2795)以2.4mL/min的流速,使用95%A+5%C到95%B+5%C的溶劑系統進行4分鐘,其中A=水,B=甲醇,C=1∶1甲醇∶水(包含0.2%碳酸銨);(viii)中間物總體上未經完全表征且純度通過薄層色譜、質譜、HPLC和/或NMR分析來評估;(ix)通過將結晶物質樣品安裝在Bruker單硅晶體(SSC)晶片支架上且借助于顯微鏡載片將樣品展布成薄層來測定(使用BrukerD4分析儀器)X射線粉末衍射光譜。使樣品以每分鐘30轉離心(以改良計數統計)且用由在40kV和40mA下操作的銅制長細聚焦管產生的具有1.5418埃的波長的X射線來照射。使準直X射線源穿過設定在V20下的自動可變發散狹縫且引導反射的輻射穿過5.89mm防散射狹縫和9.55mm檢測器狹縫。在2°到40°2θ范圍內以θ-θ模式每0.00570°2θ增量使樣品曝露0.03秒(連續掃描模式)。運作時間為3分鐘36秒。該儀器裝備有位置敏感性檢測器(聯凱(Lynxeye))。借助于用Diffract+軟件操作的DellOptiplex686NT4.0工作站進行控制和數據擷取。X射線粉末衍射領域的普通技術人員將認識到,峰的相對強度可以受例如大小在30微米以上的晶粒和非單一縱橫比影響,這可能影響樣品的分析。本領域普通技術人員也將認識到,反射位置可以受樣品在衍射計中所處的確切高度和衍射計的零點校正影響。樣品的表面平坦度也可能具有細微影響。因此,所呈現的衍射圖數據不應視為絕對值;(x)使用TA儀器(TAInstruments)Q1000DSC儀器進行差示掃描熱量測定。典型地,在25℃到300℃的溫度范圍內以10℃/min的恒定加熱速率加熱含于配備有蓋的標準鋁盤中的小于5mg的物質。以50mL/min的流速使用采用氮氣的凈化氣體;以及(xi)已使用以下縮寫:-aq.水性CDCl3氘化氯仿CHCl3氯仿DBU1,8-二氮雜雙環[5.4.0]十一-7-烯DCM二氯甲烷DEA二乙胺DIPEAN-乙基-N-異丙基丙-2-胺DMAN,N-二甲基乙酰胺DMFN,N-二甲基甲酰胺DMSO二甲亞砜DSC差示掃描熱量測定DTAD(E)-二氮烯-1,2-二甲酸二叔丁酯EDCI1-(3-二甲氨基丙基)-3-乙基碳化二亞胺鹽酸鹽Ether乙醚EtOH乙醇EtOAc乙酸乙酯%ee對映異構體過量%HOPO2-羥基-吡啶N-氧化物HPLC高效液相色譜IPA異丙醇MeCN乙腈MeOH甲醇MIBK甲基異丁基酮MTBE甲基叔丁醚NMP1-甲基-2-吡咯烷酮rt室溫sat.飽和sol.溶液THF四氫呋喃TEA三乙胺TBTU四氟硼酸2-(1H-苯并[d][1,2,3]三唑-1-基)-1,1,3,3-四甲基異脲v/v體積/體積TFA三氟乙酸實例11-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮將3-羥丙酸(30%v/v水溶液)(200μL,47.0mg,0.52mmol)蒸發到干燥,接著與甲苯共沸。將該酸溶解于NMP(1mL)中且添加分子篩(100mg,0.26mmol)、N-乙基-N-異丙基丙-2-胺(0.136mL,0.78mmol),接著添加四氟硼酸2-(1H-苯并[d][1,2,3]-三唑-1-基)-1,1,3,3-四甲基異脲(209mg,0.65mmol)。攪拌30分鐘后,添加3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(100mg,0.26mmol)且攪拌混合物2小時。通過制備型HPLC使用WatersX-Bridge反相柱(C-18,5微米二氧化硅,19mm直徑,100mm長度,流速40ml/min)和水(包含0.2%碳酸銨)與乙腈的極性遞減混合物作為洗脫劑來純化反應混合物。將包含所希望的化合物的洗脫份蒸發到干燥,得到呈透明黃色固體狀的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(45.0mg,37.9%):1HNMR光譜(CDCl3)1.52(9H,s),1.79-1.94(2H,m),2.07-2.15(2H,m),2.58(2H,t),2.84-2.94(1H,m,),3.00-3.10(1H,m),3.17-3.26(1H,m),3.53(1H,t),3.86-3.94(3H,m),4.30(3H,s),4.56-4.62(1H,m),9.02(1H,s);質譜[M+H]+=456。如下制備3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(實例1.1):在20℃下,在氮氣下經45分鐘的時間段,向三乙基氧鎓六氟磷酸鹽(V)(56.2g,226.47mmol)于二氯甲烷(500mL)中的攪拌溶液中逐滴添加于二氯甲烷(500mL)中的4-氨甲酰基哌啶-1-甲酸叔丁酯(47g,205.88mmol)。在20℃下攪拌所得溶液過夜。接著添加Na2CO3飽和水溶液直到獲得pH8。傾析各相且再用200mLCH2Cl2萃取水相,接著經MgSO4干燥有機相,過濾且濃縮,得到呈無色液體狀的4-(乙氧基(亞氨基)甲基)哌啶-1-甲酸叔丁酯(51.0g,97%):1HNMR光譜;(CDCl3)1.28(3H,t),1.46(9H,s),1.47(2H,m),1.79-1.93(2H,m),2.28(1H,m),2.73(2H,m),4.10(2H,q),4.13-4.18(2H,m);質譜[M+H]+=無質量離子。向4-(乙氧基(亞氨基)甲基)哌啶-1-甲酸叔丁酯(51g,198.95mmol)于二噁烷(500mL)中的攪拌溶液中添加甲酰肼(17.92g,298.43mmol)。使此溶液在40℃下在N2下攪拌過夜,產生白色固體沉淀(酰肼中間物)。接著將反應混合物加熱到80℃后持續6h,冷卻到室溫且濃縮。將殘余物溶解于500mLCH2Cl2中且添加300mL水。傾析各相且用鹽水洗滌有機相,經MgSO4干燥,過濾且濃縮,得到呈白色固體狀的4-(1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(46.0g,92%):1HNMR光譜;(CDCl3)1.47(9H,s),1.76(2H,m),1.98-2.11(2H,m),2.91(2H,s),2.97-3.08(m,1H),4.06-4.23(2H,m),8.05(1H,s);質譜[M+H]+=無質量離子。向4-(1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(22g,87.19mmol)于二氯甲烷(250mL)中的攪拌溶液中添加氫氧化鈉2N(131mL,261.58mmol)。通過機械攪拌劇烈攪拌反應混合物且接著逐滴添加三溴化苯甲基三甲基銨(37.4g,95.91mmol)于二氯甲烷(250mL)中的溶液,保持溫度在約15℃。使反應混合物在室溫下攪拌1h且添加2NHCl,得到pH5(保持溫度在約15℃)。傾析各相且用H2O(2×1L)洗滌有機相,經MgSO4干燥,過濾且濃縮,得到呈灰白色固體狀的4-(5-溴-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(25.00g,87%):1HNMR光譜;(CDCl3)1.46(9H,s),1.67-1.84(2H,m),1.90-2.13(2H,m),2.77-2.96(2H,m),2.98-3.10(1H,m),3.94-4.35(2H,m);質譜[M+H]+=無質量離子。在N2下,向4-(5-溴-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(26g,78.50mmol)于甲苯(200mL)和甲醇(50mL)中的攪拌懸浮液中逐滴添加(重氮甲基)三甲基硅烷2M己烷溶液(43.2mL,86.35mmol),保持溫度在約20℃:觀察到氣體逸出以及少量放熱。在室溫下攪拌所獲得的黃色溶液1h。蒸發溶劑且經二氧化硅純化所得油,用40%EtOAc/石油醚洗脫,得到呈油狀物的4-(5-溴-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(15.00g,55.3%):1HNMR光譜;(CDCl3)1.46(9H,s),1.65-1.78(2H,m),1.90-2.01(2H,m),2.68-3.02(3H,m),3.83(3H,s),3.94-4.31(2H,m);質譜[M+H]+=無質量離子。在室溫下,向3-氨基吡嗪-2-甲酸甲酯(21.3g,139.09mmol)于乙醇(65mL)中的攪拌懸浮液中逐份添加單水合肼(34mL,1094.95mmol)。在60℃下攪拌所得漿料2小時,冷卻到室溫且過濾。用冷乙醇(2×25ml)洗滌固體且干燥到恒重,得到呈米色固體狀的3-氨基吡嗪-2-甲酰肼(20.75g,97%):1HNMR光譜;(DMSO-d6)4.49(2H,d),7.46(2H,brs),7.78(1H,d),8.17(1H,d),9.79(1H,t);質譜[M+H]+=154。經15分鐘向N-乙基-N-異丙基丙-2-胺(70.6mL,405.51mmol)、特戊酸(17.08mL,148.69mmol)以及3-氨基吡嗪-2-甲酰肼(20.7g,135.17mmol)于乙腈(350mL)中的攪拌懸浮液中逐份添加四氟硼酸2-(1H-苯并[d][1,2,3]三唑-1-基)-1,1,3,3-四甲基異脲(47.7g,148.69mmol),并且在80℃下攪拌反應混合物20分鐘(獲得溶液)。將反應混合物冷卻到0℃且經15分鐘的時間段相繼添加N-乙基-N-異丙基丙-2-胺(70.6mL,405.51mmol)、4-甲苯-1-磺酰氯(77g,405.51mmol)。使反應混合物(黃色懸浮液)回流(溶解),且接著允許在室溫下攪拌14小時,得到暗橙色溶液。濃縮該溶液。殘余物用二氯甲烷稀釋,用水、鹽水洗滌,經硫酸鎂干燥且濃縮。用0%到40%乙酸乙酯/二氯甲烷洗脫,通過硅膠快速色譜純化粗產物。蒸發溶劑到干燥。所得混合物用乙醚(100mL)濕磨,過濾,用極少的乙醚洗滌且干燥,得到呈淺黃色固體狀的3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(20.8g,70.2%):1HNMR光譜;(CDCl3)1.53(9H,s),1.58-1.68(2H,m),6.67(2H,s),8.13(2H,dt);質譜[M+H]+=220。向3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(20.8g,94.87mmol)于THF(320mL)中的溶液中逐份添加1-溴吡咯烷-2,5-二酮(18.57g,104.36mmol)且在室溫下攪拌該溶液16小時。濃縮反應混合物且將殘余物溶解于二氯甲烷(300mL)中,用水(2×150mL)、鹽水洗滌,經硫酸鎂干燥且濃縮。蒸發溶劑且用0%到10%乙酸乙酯/二氯甲烷洗脫,通過硅膠快速色譜純化粗產物。蒸發溶劑到干燥,得到呈米色固體狀的5-溴-3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(25.5g,90%):1HNMR光譜;(CDCl3)1.52(9H,s),8.23(1H,s);質譜[M+H]+=300。向5-溴-3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(45g,150.94mmol)于甲苯(450mL)中的懸浮液中添加1,1,1,2,2,2-六甲基二錫烷(37.6mL,181.12mmol)和氯化雙(三苯基膦)鈀(II)(5.30g,7.55mmol)。用氬氣使反應混合物脫氣且在80℃下加熱2小時。(一旦加熱即溶解,橙色溶液接著再沉淀且變為黑色,表明反應完全。)使反應混合物冷卻下來,濃縮且將殘余物溶解于CH2Cl2中并經Decalite過濾以移除不溶性雜質。濃縮濾液且用0%到10%EtOAc/CH2Cl2洗脫,經二氧化硅純化。濃縮溶劑,得到呈橙色固體狀的3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(三甲基錫烷基)吡嗪-2-胺(22.63g,39.2%):1HNMR光譜;(CDCl3)0.38(9H,s),1.53(9H,s),6.49(2H,brs),8.13(1H,s);質譜[M+H]+=384。向4-(5-溴-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(2700mg,7.82mmol)和3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(三甲基錫烷基)吡嗪-2-胺(2988mg,7.82mmol)于4-甲基-2-戊醇(28mL)中的攪拌懸浮液中添加氯化鋰(995mg,23.46mmol)和氯化雙(三苯基膦)鈀(II)(220mg,0.31mmol)。用氬氣使混合物脫氣且在140℃下加熱2h。使反應冷卻下來且通過過濾收集所得沉淀,用異丙醇(25mL)、水(25mL)洗滌且在抽吸下干燥。濃縮異丙醇有機洗脫份且收集所形成的沉淀并與主要沉淀合并,得到4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(3.0g,79%):1HNMR光譜:(DMSO-d6)1.41(9H,s),1.45(9H,s),1.50-1.68(2H,m),1.95(3H,dd),2.78-3.05(1H,m),3.96(3H,d),4.21(3H,s),8.86(1H,s);質譜[M+H]+=484。在25℃下攪拌4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(3g,6.20mmol)于TFA(15mL)和CH2Cl2(15mL)中的溶液1小時。使混合物與甲苯共沸,添加氨于甲醇和二氯甲烷中的7N溶液且經硅膠吸附混合物。相繼用0%到8%甲醇/二氯甲烷、0%到10%甲醇氨/二氯甲烷洗脫,通過硅膠快速色譜純化粗產物。蒸發溶劑到干燥,得到呈黃色結晶固體狀的3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(2.040g,86%):1HNMR光譜:(DMSO-d6)1.45(9H,s),1.55-1.66(2H,m),1.86(2H,dd),2.52-2.61(2H,m),2.69-2.78(1H,m),2.95-3.02(2H,m),4.20(3H,s),8.86(1H,s);質譜[M+H]+=384。分離實例1的單一晶形經以上分離的物質的X射線粉末衍射譜顯示該物質為結晶但為多晶型形式的混合物。此物質具有226.4℃(開始)的熔點。形式A物質通過使初始物質在25℃下在乙腈中制成漿料而產生。將約20mg初始物質置放于具有磁性攪拌棒的小瓶中,且添加約2mL乙腈,接著用帽蓋緊密地密封小瓶且使其在磁性攪拌板上攪拌。約5天后,從該板移出樣品,脫去帽蓋且使漿料在環境條件下干燥,隨后通過XRPD和DSC對其進行分析。此形式(形式A)通過XRPD測定為結晶。此物質具有227.2℃(開始)的熔點。相同晶形可以通過在室溫下在乙腈中攪拌粗物質過夜,接著過濾所得固體,用冷乙腈洗滌且干燥來制造。在本發明的一個方面中,提供一種通過使化合物樣品在乙腈中制成漿料來形成實例1的晶形(形式A)的方法。十個X射線粉末衍射峰顯示于下表中:實例1形式A的十個X射線粉末衍射峰實例1形式A的X射線粉末衍射譜顯示于圖1中。實例1形式A的DSC分析顯示開始于227.2℃的熔融吸熱和在228.6℃處的峰(圖2)。因此,DSC分析顯示實例1形式A為在約227.2℃開始熔融且具有在228.6℃處的峰的高熔點固體。實例1形式的DSC顯示于圖2中。X射線粉末衍射分析儀器:BrukerD4。通過將結晶物質樣品安裝在Bruker單硅晶體(SSC)晶片支架上且借助于顯微鏡載片將樣品展布成薄層來測定X射線粉末衍射圖。使樣品以每分鐘30轉離心(以改良計數統計)且用由在40kV和40mA下操作的銅制長細聚焦管產生的具有1.5418埃的波長的X射線來照射。使準直X射線源穿過設定在V20下的自動可變發散狹縫且引導反射的輻射穿過5.89mm防散射狹縫和9.55mm檢測器狹縫。在2°到40°2θ范圍內以θ-θ模式每0.00570°2θ增量使樣品曝露0.03秒(連續掃描模式)。運作時間為3分鐘36秒。該儀器裝備有位置敏感性檢測器(聯凱)。借助于用Diffract+軟件操作的DellOptiplex686NT4.0工作站進行控制和數據擷取。X射線粉末衍射領域的普通技術人員將認識到,峰的相對強度可以受例如大小在30微米以上的晶粒和非單一縱橫比影響,這可能影響樣品的分析。本領域普通技術人員也將認識到,反射位置可以受樣品在衍射計中所處的確切高度和衍射計的零點校正影響。樣品的表面平坦度也可能具有細微影響。因此,所呈現的衍射圖數據不應視為絕對值。差示掃描熱量測定分析儀器:TA儀器Q1000DSC。典型地,在25℃到300℃的溫度范圍內以10℃/min的恒定加熱速率加熱含于配備有蓋的標準鋁盤中的小于5mg的物質。以50mL/min的流速使用采用氮氣的凈化氣體。實例1化合物的替代性合成在下文中提供為實例2。實例2:1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮在氮氣下,向1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(37g,68.57mmol)于甲醇(275mL)中的懸浮液中添加吡啶4-甲基苯磺酸鹽(3.58g,14.25mmol)。于60℃下攪拌混合物1.5小時。5分鐘后混合物溶解。使混合物在50℃下靜置過夜,在此期間形成沉淀。將反應混合物溶解于二氯甲烷(400mL)中,用水(300mL)和鹽水(100mL)洗滌。水性萃取物用DCM(100mL)回洗且經MgSO4干燥經合并的有機層且濃縮。用100%乙酸乙酯到10∶50∶40甲醇/乙酸乙酯/DCM洗脫,通過硅膠快速色譜純化粗產物。將包含洗脫份的產物蒸發到干燥,得到米色固體(24.5g)。使固體在乙腈(500mL)中制成漿料過夜,過濾且在高真空下干燥,得到呈奶白色固體狀的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(實例2)(24g,78%):1HNMR光譜:(DMSO-d6)1.51(9H,s),1.55-1.68(1H,m),1.68-1.84(1H,m),1.96-2.13(2H,m),2.78-2.93(1H,m),2.98-3.1(1H,m),3.19-3.3(1H,m),3.71(2H,q),3.93-4.04(1H,m),4.27(3H,s),4.35-4.48(1H,m),4.54(1H,t),7.96(2H,s),8.92(1H,s);質譜[M+H]+=456。(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(實例2.1)制備如下:向3-氨基-6-溴吡嗪-2-甲酸甲酯(100g,418.04mmol)于EtOH(2L)中的攪拌混合物中逐滴添加水合肼(23.59mL,480.75mmol)。在50℃下在氮氣下加熱混合物。在50℃下攪拌所得稠懸浮液16小時。一次性添加另一份肼(2.5mL)且將懸浮液在50℃下再攪拌24小時。向稠反應混合物中饋入乙醇(500mL)且使混合物冷卻到室溫。過濾所得懸浮液且用乙醇(1L)洗滌固體且在真空中干燥,得到呈奶白色固體狀的3-氨基-6-溴吡嗪-2-甲酰肼(98g,定量):1HNMR光譜;(DMSO-d6)4.52(2H,s),7.59(2H,s),8.30(1H,s),9.74(1H,s);質譜[M+H]+=232。向3-氨基-6-溴吡嗪-2-甲酰肼(172g,741.26mmol)于乙腈(1.8L)中的攪拌混合物中添加特戊酸酐(165mL,815.38mmol)且在80℃下加熱混合物1小時。攪拌反應物16小時。通過過濾分離所需黃色固體物質。將濾液分配于EtOAc(2L)與碳酸氫鈉水溶液(2L)之間。用飽和鹽水洗滌有機層并經MgSO4干燥。過濾且濃縮溶液,得到橙色粘性固體,用MTBE(250mL)對其進行濕磨。通過過濾分離不溶性黃色固體且此物質顯示與第一固體相同。在真空烘箱中在50℃下干燥經合并的固體3天,得到呈黃色固體狀的3-氨基-6-溴-N′-特戊酰基吡嗪-2-甲酰肼(224g,96%):1HNMR光譜:(DMSO-d6)1.17(9H,s),7.62(2H,s),8.37(1H,s),9.42-9.56(1H,m),10.09-10.23(1H,m);質譜[M+H]+=318。向3-氨基-6-溴-N′-特戊酰基吡嗪-2-甲酰肼(227g,718.00mmol)和N,N-二異丙基乙胺(297mL,1795.01mmol)于乙腈(2200mL)中的懸浮液中分批添加對甲苯磺酰氯(164g,861.60mmol)。在70℃下攪拌混合物2小時。使反應物冷卻到室溫過夜。將反應混合物分配于乙酸乙酯(2L)與碳酸氫鈉溶液(2L)之間。用飽和鹽水洗滌有機層,經硫酸鎂干燥,過濾且在減壓下濃縮。所得棕色/米色固體用熱MTBE(1000mL)濕磨且通過過濾分離并干燥,得到呈黃色固體狀的5-溴-3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(187g,87%)。將母液蒸發到干燥。用MTBE(500mL)濕磨粗固體,過濾且用100mLMTBE洗滌。風干所得固體過夜,得到第二批5-溴-3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(36g,17%):1HNMR光譜:(DMSO-d6)1.43(9H,s),7.70(2H,s),8.39(1H,s);質譜[M+H]+=298。在一個替代性制備中,在50℃下經30min的時間向于MeCN(10.8L)中的3-氨基-6-溴-N′-特戊酰基吡嗪-2-甲酰肼(2301g,7.28mol)中逐份(約280g×6)添加DIPEA(3.044L,17.48mol)和對甲苯磺酰氯(1665g,8.73mol)。通過控制添加速率將反應溫度維持在65℃-70℃之間。在添加完成之后,在70℃下攪拌反應混合物1h。將混合物冷卻到室溫且用5%NaHCO3(水溶液,24.2L)淬滅。攪拌所得懸浮液30min,接著過濾。產物用水(14.8L)洗滌,吸干且在50℃下干燥16h。將產物溶解于DCM(12L)中且分離各相。將有機相裝載到二氧化硅墊(6kg)上,且用20%EtOAc/DCM(8×10L)洗脫產物。包含產物的洗脫份的濃度得到根據HPLC純度為99.8%的1987g(92%產率)。使氮氣流穿過5-溴-3-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-胺(89.35g,239.75mmol)于DMA(1.2L)中的溶液,持續20分鐘。向攪拌混合物中依次添加二環己基(2′,4′,6′-三異丙基聯苯-2-基)膦(11.43g,23.98mmol)、三(二苯亞甲基丙酮)二鈀(0)(5.49g,5.99mmol)、鋅(1.568g,23.98mmol)以及二氰基鋅(16.89g,143.85mmol)。將混合物加熱到100℃且攪拌1小時。將混合物冷卻且分配于DCM(3L)與水(1L)之間。經由硅藻土過濾黑色混合物且分離有機層。相繼用水、鹽水洗滌溶液。經硫酸鎂干燥溶液且在減壓下濃縮。殘余物用MTBE濕磨且通過過濾分離,用MTBE洗滌。在真空中干燥濾餅,得到呈淺橙色固體狀的5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-甲腈(55.7g,95%):1HNMR光譜:(DMSO-d6)1.46(9H,s),6.02(1H,s),8.38(2H,s);質譜[M-H]242。產物可以在庚烷中制成漿料,接著過濾且干燥,作為用MTBE濕磨的一個替代方案。向于IPA(200mL)中的5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-甲腈(55g,225.18mmol)中添加水合肼(82mL,1.69mol)且在50℃下在氮氣下加熱混合物16小時。在冰浴中冷卻混合物。通過過濾來收集所得沉淀,用IPA和乙醚洗滌且干燥到恒重,得到呈黃色固體狀的(Z)-5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-甲腙酰胺(49.2g,79%):1HNMR光譜:(DMSO-d6)1.45(9H,s),5.26(2H,s),5.58(2H,s),7.56(2H,s),8.75(1H,s);質譜[M+H]+=277。向N-Boc-異哌啶甲酸(41.1g,179.15mmol)和4-甲基嗎啉(35.9mL,325.74mmol)于DMA(800mL)中的溶液中添加六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲脲(74.3g,195.44mmol)。攪拌混合物10分鐘,接著向該溶液中一次性添加(Z)-5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-甲腙酰胺(45g,162.87mmol)(從22℃到27℃觀察到放熱)。數分鐘后,從反應混合物結晶出產物。從容器中移出反應混合物且經由燒結物過濾。從容器側面向洗滌產物中再添加DMA(150mL)且將此傾到濾餅上。向容器中添加異丙醇(600mL)且使用劇烈攪拌將容器中產物的剩余部分懸浮于此溶劑中。在DMA已通過抽吸移除之后,使用異丙醇懸浮液洗滌濾餅。吸干濾餅,接著用MTBE洗滌且再次吸干。在真空中干燥濾餅,得到呈黃色固體狀的(Z)-4-(2-(氨基(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)亞甲基)肼羰基)哌啶-1-甲酸叔丁酯(76g,95%):1HNMR光譜:(DMSO-d6)1.40(9H,s),1.46(9H,s),1.63-1.9(2H,m),2.33-2.6(2H,m,因DMSO信號而模糊),2.63-3.03(2H,m),3.18-3.48(4H,m,因水信號而模糊),3.88-4.11(2H,m),6.43(2H,s),7.76(2H,br),8.84(0.5H,s),8.87(0.5H,s),9.85(1H,s);質譜[M+H]+=488在一個替代性制備中,可以如下原位制得N-Boc-異哌啶甲酸:將異哌啶甲酸(858g,3.74mol)溶解于DMA(25.3L)中,且添加4-甲基嗎啉(393mL,3.74mol)。攪拌5min,且添加氯甲酸異丁酯(489mL,3.74mol)。將反應混合物在25℃下攪拌2h,且冷卻到15℃,隨后經10min逐份添加(Z)-5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-甲腙酰胺(940g,3.4mol)。在15℃下攪拌反應混合物1-2h。經1h逐份添加水(20.5L),且再攪拌1h,隨后過濾。濾餅接著用水(4×4L)洗滌,且在過濾器上吸干,隨后在真空烘箱中在50℃下干燥直到無水,得到所希望的產物。向3L固定雙夾套容器中的二噁烷(500mL)中添加乙酸(200mL)且將該溶液在氮氣下加熱到70℃。向溫熱的混合物中逐份添加(Z)-4-(2-(氨基(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)亞甲基)肼羰基)-哌啶-1-甲酸叔丁酯(74.5g,152.80mmol)。10分鐘后,升高溫度到100℃(略微回流)。在100℃下攪拌反應混合物1.5小時(懸浮液),接著在80℃下靜置過夜(在靜置過夜之后形成溶液)。在減壓下濃縮所得溶液,接著用甲苯稀釋,蒸發到干燥,用甲苯吸收且再次濃縮。剩余油與一些乙酸乙酯混合且濃縮到干燥。從溶液結晶的固體用MTBE(200mL)濕磨且通過過濾分離。用水和MTBE洗滌濾餅,得到呈灰色固體狀的4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(50g,70%)。在減壓下濃縮濾液,得到黃色固體。此物質用MTBE濕磨且過濾。相繼用乙酸乙酯、MTBE洗滌濾餅,得到呈淺黃色固體狀的第二批(4.93g,7%)。此物質與第一批相同:1HNMR光譜:(DMSO-d6)1.17(9H,s),1.22(9H,s),1.29-1.47(2H,m),1.67-1.78(2H,m),2.57-2.87(3H,m),3.57-3.92(2H,m),7.56(2H,br),8.56(1H,s),13.47(2H,brs);質譜[M+H]+=470。向4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(48g,102.23mmol)于2-甲基THF(300mL)中的懸浮液中添加1,8-二氮雜雙環[5.4.0]十一-7-烯(19.87mL,132.90mmol)。5分鐘后獲得暗溶液,該溶液用木炭處理且經由硅藻土墊過濾,再用2-甲基THF(100mL)洗滌木炭和木炭。在-5℃下在3L夾套固定容器中在氮氣氛下用空氣攪拌器攪拌濾液。添加2-甲基THF(100mL)來幫助攪拌黃色懸浮液。經15分鐘逐滴添加碘甲烷(7.96mL,127.78mmol)。攪拌混合物2小時且將反應混合物溫到室溫。16小時后,再添加碘甲烷(6mL)和DBU(20mL)且繼續攪拌16小時。將混合物傾入水中且攪拌5分鐘。分離出呈米色固體狀的不溶性物質且在真空中干燥,得到4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(24.77g,50.1%)。在減壓下濃縮母液且使用MTBE作為洗脫劑,通過二氧化硅快速柱色譜純化殘余物。由此獲得呈黃色固體狀的第二批所希望的產物(13.04g,26%):1HNMR光譜:(DMSO-d6)1.47(9H,s),1.51(9H,s),1.57-1.76(2H,m),1.94-2.1(2H,m),2.87-3.09(3H,m),3.9-4.08(2H,m),4.26(3H,s),7.97(2H,br,s),8.92(1H,s);質譜[M+H]+=484向2,2,2-三氟乙酸(100mL,1305.87mmol)于DCM(100mL)中的溶液中添加4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(36.81g,76.12mmol)。在室溫下攪拌混合物3小時。在減壓下濃縮混合物。將殘余物溶解于DCM(1.5L)中且添加到劇烈攪拌濃縮的氨(150mL)于水(400mL)中。用DCM(400mL)洗滌水溶液且經硫酸鎂干燥經合并的有機物溶液,過濾且濃縮到干燥,得到呈黃色固體狀的3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(30.0g,103%):1HNMR光譜:(DMSO-d6)1.44(9H,s),1.54-1.69(2H,m),1.8-1.92(2H,m),2.53-2.63(2H,m),2.68-2.83(1H,m),2.93-3.05(2H,m),4.19(3H,s),7.89(2H,br),8.85(1H,s);質譜[M+H]+=384。在25℃下,向溶解于乙腈(200mL)中的3-(四氫-2H-吡喃-2-基氧基)丙酸(12.67g,72.76mmol)和N-乙基-N-異丙基丙-2-胺(25.3mL,145.52mmol)的攪拌溶液中逐份添加六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲脲(30.4g,80.04mmol)。在25℃下攪拌所得溶液20分鐘,接著逐份添加3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(30g,72.76mmol),將最終部分在乙腈(100mL)中洗滌成呈漿料狀的混合物。攪拌1小時后,通過過濾來收集沉淀,用乙腈洗滌且在真空中干燥,得到呈米色固體狀的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(35.0g,89%)。濾液用DCM(600mL)稀釋,用水洗滌,經硫酸鎂干燥且濃縮。用2%到2.5%7N氨/甲醇與二氯甲烷溶液梯度洗脫,通過硅膠快速色譜純化殘余物。還獲得呈奶白色固體狀的第二批產物(3.31g,6.13mmol,8.43%)。合并兩個樣品,得到米色固體:1HNMR光譜:(DMSO-d6)1.44(9H,s),1.52-1.79(4H,m),1.88-2.04(2H,m),2.53-2.73(2H,m),2.73-2.87(1H,m),2.91-3.05(1H,m),3.13-3.24(1H,m),3.37-3.47(1H,m),3.53-3.65(1H,m),3.7-3.8(1H,m),3.81-3.89(1H,m),3.89-3.99(1H,m),4.20(3H,s),4.29-4.4(1H,m),4.54-4.61(1H,m),7.60-8.20(2H,br),8.85(1H,s);質譜[M+H]+=540。實例31-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮在氮氣下,向1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(128g,231.19mmol)于甲醇(1L)中的懸浮液中添加吡啶4-甲基苯磺酸鹽(11.62g,46.24mmol)。于60℃下攪拌混合物1.5小時。5分鐘后混合物溶解。使混合物在50℃下靜置過夜,在此期間形成沉淀。固體物質通過過濾來分離且用水和乙腈洗滌。此物質仍包含來自先前階段的少量雜質且需要進一步純化。將該物質溶解于二氯甲烷中且通過硅膠快速色譜(0%甲醇/DCM到10%甲醇/DCM)來純化。由此分離出呈奶白色固體(形式A)狀的所希望的產物1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(實例3)(92g,85%):1HNMR光譜:(DMSO-d6)1.4-1.51(12H,m),1.51-1.78(2H,m),1.89-2.05(2H,m),2.72-2.86(1H,m),2.91-3.05(1H,m),3.12-3.24(1H,m),3.64(2H,q),3.83-4.01(1H,m),4.29-4.41(1H,m),4.47(1H,t),4.58(2H,q),8.26(2H,s),8.85(1H,s);質譜[M+H]+=470。1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(實例3.1)制備如下:向4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(150g,319.46mmol)于2-甲基THF(1.2L)中的懸浮液中添加1,8-二氮雜雙環[5.4.0]十一-7-烯(76mL,511.14mmol)。添加碘乙烷(46mL,575.03mmol)且在35℃下攪拌混合物16小時。再添加碘乙烷(46mL,575.03mmol)和1,8-二氮雜雙環[5.4.0]十一-7-烯(76mL,511.14mmol)且在35℃下繼續攪拌24小時。將混合物傾入水中且通過過濾來分離不溶性物質,用水和MTBE洗滌且在真空中干燥,得到呈黃色固體狀的4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(116g,73.0%)。用DCM萃取濾液且有機溶液經硫酸鎂干燥,過濾且在減壓下濃縮。使用梯度洗脫(30%MTBE/庚烷到100%MTBE),通過二氧化硅快速柱色譜純化殘余物。由此分離出呈黃色固體狀的第二批所希望的產物(12g,24.12mmol,7.55%),隨后將其與第一批合并:1HNMR光譜:(DMSO-d6)1.41(9H,s),1.44(9H,s),1.48(3H,t),1.52-1.69(2H,m),1.87-2.04(2H,m),2.79-3.03(3H,m),3.86-4.03(2H,m),4.59(2H,q),7.89(2H,s),8.85(1H,s);質譜[M+H]+=498。THF也可以為以上反應的適合溶劑。向4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(126g,253.22mmol)于DCM(400mL)中的溶液中逐份添加TFA(400mL)。在室溫下攪拌混合物16小時。在減壓下濃縮混合物,將其溶解于DCM(1L)中且在0℃下緩慢添加到濃氨水(500mL)于水中的劇烈攪拌溶液中。從水溶液分離出有機溶液且在減壓下濃縮*,得到呈黃色固體狀的3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-乙基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(101g,100%):1HNMR光譜:(DMSO-d6)1.4-1.52(12H,m),1.57-1.73(2H,m),1.83-1.93(2H,m),2.57-2.7(2H,m),2.71-2.84(1H,m),2.96-3.09(2H,m),4.58(2H,q),8.06(2H,s),8.84(1H,s);質譜[M+H]+=398。*在類似規模(約170g起始物質)的另一個實驗中,使用以下分離程序:分離各層且過濾頂層(含固體的乳液)。用DCM(0.5L)洗滌固體且將濾液轉移到分液漏斗。分離各層且用DCM(0.5L)萃取水層。有機層經MgSO4干燥,過濾并濃縮。在50℃下使產物干燥過夜(81.75g)。使來自萃取的固體在水(200mL)中在室溫下制成漿料,持續30min且過濾。在50℃下在真空中干燥產物(61.8g)。另一個變化形式如下;將4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-甲酸叔丁酯(3009.5g,6.05mol)于DCM(9L)中的懸浮液在N2下冷卻到5℃-10℃。向懸浮液中逐份添加TFA(9L),同時維持溫度<30℃。在室溫下攪拌反應混合物1h。濃縮混合物,將所得殘余物溶解于水(30L)中,且在0℃-5℃下緩慢添加到35%氨水溶液(12L)中。將懸浮液攪拌30min,接著過濾出產物且用水(2×6L)洗滌。在50℃下在真空中干燥產物2天(2496g)。在25℃下,向溶解于乙腈(600mL)中的3-(四氫-2H-吡喃-2-基氧基)丙酸(44.3g,254.10mmol)和N-乙基-N-異丙基丙-2-胺(89mL,508.21mmol)的攪拌溶液中逐份添加六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲脲(HATU,106g,279.51mmol)。在25℃下攪拌所得溶液20min,接著逐份添加3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-乙基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(101g,254.10mmol),將最終部分在乙腈(300mL)中洗滌成呈漿料狀的混合物。攪拌1小時后,通過過濾來收集沉淀,用乙腈洗滌且在真空中干燥,得到呈米色固體狀的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(128g,91%)。濾液用DCM(600ml)稀釋,用水洗滌,經硫酸鎂干燥且濃縮。用2%到2.5%7N氨/甲醇與二氯甲烷溶液梯度洗脫,通過硅膠快速色譜純化殘余物。獲得呈奶白色固體狀的第二批所希望的產物(40g,72.2mmol,28.4%),將其與第一批合并:1HNMR光譜:(DMSO-d6)1.29-1.48(16H,m),1.48-1.75(4H,m),1.83-1.99(2H,m),2.48-2.68(2H,m),2.68-2.79(1H,m),2.87-2.99(1H,m),3.07-3.19(1H,m),3.32-3.42(1H,m),3.47-3.6(1H,m),3.64-3.75(1H,m),3.75-3.84(1H,m),3.84-3.95(1H,m),4.24-4.39(1H,m),4.47-4.6(3H,m),7.84(2H,s),8.79(1H,s);質譜[M+Na]+=577。替代性制備:在室溫下在氮氣下,向3-(四氫-2H-吡喃-2-基氧基)丙酸(48.80g,0.2774mol)和N-乙基-N-異丙基丙-2-胺(86.96mL,0.4993mol)于THF(552mL)中的溶液中逐份添加六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲脲(115.73g,0.3051mol)。攪拌所得混合物20min,接著經1h逐份添加3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-乙基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(122.5g(110.25g有效),0.2774mol)。3.5h后,濃縮混合物且使殘余物在室溫下在MeCN(275mL)中制成漿料,持續15min。過濾出產物,用MeCN(3×110mL)洗滌且在50℃下在真空中干燥過夜。由此得到1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(131.9g,96%)。在另一個替代性制備中,HBTU(六氟磷酸O-(苯并三唑-1-基)-N,N,N′,N′-四甲脲)于THF中可以用作偶合劑代替HATU。替代性制備實例3在N2下,向1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-(四氫-2H-吡喃-2-基氧基)丙-1-酮(131.9g,0.2382mol)于甲醇(1045mL)中的懸浮液中添加對甲苯磺酸吡啶鎓(11.97g,47.7mmol)。在60℃下攪拌反應混合物5.5h,接著在50℃下過夜。將反應混合物冷卻到0℃且過濾出固體。使產物在水(250mL)中在室溫下制成漿料,持續20分鐘,過濾,用水(3×40mL)洗滌且在50℃下在真空中干燥。由此得到呈形式A(參見以下)的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(21.4g)。濃縮甲醇液體且使所得固體在水(0.6L)中在室溫下制成漿料,持續20分鐘。通過過濾分離固體,且用水(3×100mL)洗滌。濾餅在水(0.5L)中第二次制成漿料,再持續20分鐘。產物通過過濾分離,用水(100mL)洗滌且在50℃下在真空中干燥。由此得到81.9g呈形式A的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(81.9g)。合并兩批(103.3g),用形式B(16.68g)接種且在MeCN(826mL)中在室溫下制成漿料過夜。由此得到117.4g呈淺黃色固體狀的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(117.4g),即形式B(參見下文)。這一物質通過在庚烷(7.5相對體積)中制成漿料持續1小時來進一步純化。過濾混合物,在過濾器上吸干,且在50℃下在真空烘箱中干燥過夜,得到呈形式B的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(102.5g)。形式B也可以通過在不接種的情況下使形式A在MeCN中制成漿料來制造。形式A或B也可以如下轉化成形式C:在回流下加熱1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(例如通過以上概述的方法制得的形式B)于IPA(12體積)中的懸浮液,直到固體溶解。熱過濾該溶液,接著冷卻到室溫。由此得到呈淺黃色固體狀的1-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-羥基丙-1-酮(99.3g,97%)作為形式C。形式C也可以如下轉化成形式B:在10L法蘭燒瓶中,將形式C(377.8克份1)于MIBK(7900mL)中加熱到110℃-115℃,得到溶液。使溶液冷卻到97℃-103℃,且立即精制過濾到在-15℃下攪拌的包含形式B(0.8g)晶種于乙腈(8220mL)中的50L容器中。在添加期間,借助于夾套冷卻將50L容器中的溫度維持在-15℃與25℃之間。通過類似方法再添加三份溶解于MIBK中的該化合物。向所得漿料中添加形式B的晶種(0.8g),且接著在10℃-20℃下攪拌混合物過夜。過程內分析證實所希望的形式(形式B),不具有形式C或可見非晶形。過濾混合物,且用乙腈(3340mL)洗滌。將固體烘箱干燥2天(固體在干燥期間破碎成粉末以及大小為約1mm到約3-4mm的小團塊的混合物),直到獲得恒重。產率=1532.8g(93.5%)。3-(四氫-2H-吡喃-2-基氧基)丙酸制備如下:在0℃下在氮氣下,向甲醇(2.4L)和濃硫酸(44.4mL,832.61mmol)的攪拌溶液中逐滴添加β-丙內酯(175mL,2.78mol)。在室溫下攪拌此溶液2天。將反應混合物冷卻到10℃,隨后逐份添加碳酸氫鈉(145g,1.72mol),在室溫下攪拌所得懸浮液75分鐘。過濾此溶液,用甲醇(800mL)洗滌濾餅。將濾液蒸發為油,將其再溶解于二氯甲烷(1.2L)中且攪拌60分鐘,隨后再過濾。過濾此溶液,隨后蒸發,得到呈油狀物的3-羥基丙酸甲酯(219g,76%)。1HNMR光譜:(CDCl3)2.50(2H,t),3.63(3H,s),3.78(2H,t)。在室溫下在氮氣下,向3-羥基丙酸甲酯(63.4g,609.00mmol)和3,4-二氫-2H-吡喃(78mL,852.60mmol)于二氯甲烷(650mL)中的澄清溶液中添加對甲苯磺酸吡啶鎓(7.65g,30.45mmol),得到混濁溶液。在室溫下攪拌此溶液過夜。反應混合物用水(250mL)和鹽水(250mL)洗滌,隨后干燥(MgSO4)且蒸發成油。通過快速二氧化硅色譜,15%到30%EtOAc/庚烷的洗脫梯度來純化此粗產物。蒸發純洗脫份到干燥,得到呈無色油狀物的3-(四氫-2H-吡喃-2-基氧基)丙酸甲酯(67.7g,59.0%):1HNMR光譜:(CDCl3)1.47(4H,dddd),1.55-1.84(2H,m),2.55(2H,t),3.33-3.53(1H,m),3.53-3.7(4H,m),3.78(1H,ddd),3.93(1H,dt),4.42-4.72(1H,m);質譜[MH]+=189。在室溫下,向3-(四氫-2H-吡喃-2-基氧基)丙酸甲酯(67.68g,359.58mmol)于THF(680mL)中的溶液中添加氫氧化鈉(2M,349mL,697.58mmol)。在室溫下攪拌反應混合物3小時。在真空中移除THF,接著用乙酸乙酯(260mL)洗滌水層,隨后冷卻到0℃且通過添加鹽酸(2M)小心酸化到pH5。用乙酸乙酯(3×250mL)萃取產物,隨后干燥(MgSO4)且蒸發,得到呈透明油狀物的3-(四氫-2H-吡喃-2-基氧基)丙酸(57.0g,91%)。將此物質溶解于乙酸乙酯(750mL)中,接著用水(3×250mL)和鹽水(250mL)洗滌以移除剩余乙酸。干燥(MgSO4)且蒸發有機溶液,得到呈無色油狀物的3-(四氫-2H-吡喃-2-基氧基)丙酸(45.67g,72.9%):1HNMR光譜:1HNMR(CDCl3)1.43-1.67(4H,m),1.65-1.95(2H,m),2.68(2H,t),3.48-3.58(1H,m),3.73(1H,dt),3.88(1H,ddd),4.02(1H,dt),4.59-4.7(1H,m);質譜[M-H]-=173。如以上所分離出的實例3為呈三種不同晶形的結晶固體,在此描述為形式A、B以及C。實例3的形式A的晶體結構可以通過XRPD和DSC表征。進行這些技術的方法如關于實例1所述。實例3形式A的十個X射線粉末衍射峰實例3形式A的XRPD顯示于圖3中。實例3形式A的DSC分析顯示開始于27.0℃的初始吸熱和在63.0℃處的峰,另外的吸熱位移可見在以下溫度處的開始和峰:166.5℃和168.7℃、172.2℃和173.2℃以及在174.8℃處的最終熔融物和在175.7℃處的峰(圖4)。因此,DSC分析顯示實例3形式A為在約27.0℃下開始去溶劑化且具有在約63.0℃處的峰的溶劑化物質。實例3(形式A)的X射線粉末衍射譜顯示該物質為結晶。此物質具有28.0℃(開始)的去溶劑化點。實例3也可以按替代性多晶型形式存在,在此稱作形式B。形式B的制備描述于上文中。此物質具有172.5℃(開始)的熔點。在本發明的另一個方面中,提供一種通過使實例3樣品在乙腈中制成漿料來制造實例3的形式B的方法。在本發明的另一個方面中,提供一種用于從實例3的形式C于MIBK中的溶液制造實例3的形式B的方法。實例3形式B的十個X射線粉末衍射峰實例3形式B的XRPD顯示于圖5中。實例3形式B的DSC分析顯示開始于172.5℃的熔融吸熱和在174.2℃處的峰(圖6)。因此,DSC分析顯示實例3B為在約172.5℃下開始熔融且具有在約174.2℃處的峰的高熔點固體。實例3也可以按第三晶形存在,在此稱作形式C。通過從異丙醇(IPA)結晶,從例如形式B物質制造形式C物質的方法描述于上文中。因此,在本發明的另一個方面中,提供一種通過從IPA結晶出實例3來制造實例3的形式C的方法。實例3形式C的特征為提供以下使用CuKa輻射所測量的2θ值中的至少一者:6.9和12.3。實例3形式C的特征為提供實質上如圖A中所示的X射線粉末衍射圖。十個X射線粉末衍射峰顯示如下:實例3形式C的十個X射線粉末衍射峰實例3形式C的DSC分析顯示開始于183.0℃的熔融吸熱和在185.6℃處的峰(圖B)。因此,DSC分析顯示實例3形式C為在約183.0℃下開始熔融且具有在約185.6℃處的峰的高熔點固體。用于形式C分析的技術細節X射線粉末衍射分析儀器:PanalyticalCubix。通過將結晶物質樣品安裝在Panalytical單硅晶體(SSC)晶片支架上且借助于顯微鏡載片將樣品展布成薄層來測定X射線粉末衍射圖。使樣品以每分鐘30轉離心(以改良計數統計)且用由在45kV和40mA下操作的銅制長細聚焦管產生的具有1.5418埃的波長的X射線來照射。使X射線光束穿過0.04拉德索勒(soller)狹縫,接著穿過設定在20mm下的自動可變發散狹縫和最后穿過20mm光束遮罩。引導反射的輻射穿過20mm防散射狹縫和0.04拉德索勒狹縫。在2°到40°2θ范圍內以θ-θ模式每0.0025067°2θ增量使樣品曝露1.905秒(連續掃描模式)。該儀器裝備有X-Celerator檢測器。借助于用X′PertIndustry軟件操作的DellPentium4HT工作站進行控制和數據擷取。X射線粉末衍射領域的普通技術人員將認識到,峰的相對強度可以受例如大小在30微米以上的晶粒和非單一縱橫比影響,這可能影響樣品的分析。本領域普通技術人員也將認識到,反射位置可以受樣品在衍射計中所處的確切高度和衍射計的零點校正影響。樣品的表面平坦度也可能具有細微影響。因此,所呈現的衍射圖數據不應視為絕對值。差示掃描熱量測定分析儀器:TA儀器Q1000DSC。典型地,在25℃到300℃的溫度范圍內以10℃/min的恒定加熱速率加熱含于配備有蓋的標準鋁盤中的小于5mg的物質。以50mL/min的流速使用采用氮氣的凈化氣體。實例4(3R)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-丁-1-酮向3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(200mg,0.52mmol,描述于實例1中)、N-乙基-N-異丙基丙-2-胺(0.273mL,1.56mmol)以及(R)-3-羥基丁酸(65.2mg,0.63mmol)于N,N-二甲基甲酰胺(3mL)中的攪拌懸浮液中添加四氟硼酸2-(1H-苯并[d][1,2,3]三唑-1-基)-1,1,3,3-四甲基異脲(201mg,0.63mmol)。在室溫下攪拌所得懸浮液2小時。使用WatersX-Bridge反相柱(C-18,5微米二氧化硅,30mm直徑,150mm長度,流速60ml/min),使用31%乙腈于水(包含碳酸銨(2g/L)中的等位溶劑混合物,通過制備型HPLC來純化所得混合物。將包含所希望的化合物的洗脫份蒸發到干燥,得到淺黃色固體。在乙腈(3mL)中在室溫下攪拌此固體。過濾所得固體,用冷乙腈洗滌且干燥,得到呈淺黃色固體狀的標題化合物(125mg,51.0%)。1HNMR光譜:(CDCl3)1.24(3H,d),1.52(9H,s),1.85(2H,m),2.10(2H,m),2.35(1H,dd),2.55(1H,d),2.90(1H,m),3.05(1H,m),3.20(1H,m),3.90(1H,m),4.25(1H,m),4.31(3H,s),4.6(1H,m),9.03(1H,s);質譜[M+H]+=470。實例5(3S)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-丁-1-酮使用與實例4類似的程序,使3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺與(S)-3-羥基丁酸反應,得到呈淺黃色固體狀的標題化合物(167mg,68.2%)。1HNMR光譜:(CDCl3)1.24(3H,d),1.52(9H,s),1.85(2H,m),2.10(2H,m),2.35(1H,dd),2.55(1H,d),2.90(1H,m),3.05(1H,m),3.20(1H,m),3.90(1H,m),4.25(1H,m),4.31(3H,s),4.6(1H,m),9.03(1H,s);質譜[M+H]+=470。實例6(2R)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-羥基-2-甲基-丙-1-酮使用與實例4類似的程序,使3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺與(R)-3-羥基-2-甲基丙酸反應,得到呈淺黃色固體狀的標題化合物(87mg,47.4%)。1HNMR光譜:(CDCl3)1.55(9H,s),1.61(3H,sbr),1.8-2.0(2H,m),2.10-2.25(2H,m),2.90(2H,m),3.10(1H,m),3.3(2H,m),3.77(2H,m),4.33(3H,s),4.6(1H,m),9.05(1H,s);質譜[M+H]+=470。實例71-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-2-羥基-2-甲基-丙-1-酮在氬氣下,向溶解于NMP(1.2mL)中的2-羥基-2-甲基丙酸(38.0mg,0.37mmol)、3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(100mg,0.26mmol)以及2-羥基-吡啶N-氧化物(57.9mg,0.52mmol)中一次性添加1-(3-二甲氨基丙基)-3-乙基碳化二亞胺鹽酸鹽(100mg,0.52mmol)。在25℃下攪拌所得溶液3小時。添加吡啶(100μL,1.24mmol)且攪拌混合物18小時。再添加2-羥基吡啶1-氧化物(57.9mg,0.52mmol)和1-(3-二甲氨基丙基)-3-乙基碳化二亞胺鹽酸鹽(100mg,0.52mmol)。接著加熱混合物到70℃,持續48小時,再添加2-羥基-2-甲基丙酸(15mg,0.14mmol)、1-(3-二甲氨基丙基)-3-乙基碳化二亞胺鹽酸鹽(50.0mg,0.26mmol)以及2-羥基吡啶1-氧化物(25.0mg,0.23mmol),且接著保持混合物在70℃,持續8小時。使用WatersX-Bridge反相柱(C-18,5微米二氧化硅,19mm直徑,100mm長度,流速40ml/min)和水(包含0.2%碳酸銨)與乙腈的極性遞減混合物作為洗脫劑,通過制備型HPLC來純化溶液,得到呈淺黃色固體狀的標題化合物(71mg,58%)。1HNMR光譜:(CDCl3)1.55(15H,sbr),1.90(2H,m),2.15(2H,m),3.05-3.3(4H,m),4.32(3H,s),4.6(1H,m),9.03(1H,s);質譜[M+H]+=470。實例83-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-甲基-1,2,4-三唑-3-基]-1-哌啶基]-3-氧代-丙酸在0℃下在氮氣下,經2分鐘的時間段,向溶解于CH2Cl2(1.5mL)中的3-(5-叔丁基-1,3,4-噁二唑-2-基)-5-(1-甲基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(100mg,0.26mmol)和三乙胺(0.047mL,0.34mmol)的攪拌溶液中逐滴添加3-氯-3-氧代丙酸乙酯(0.037mL,0.29mmol)。在0℃下攪拌混合物10分鐘,接著使其溫至室溫且攪拌1小時。蒸發混合物,使其溶解于DMF中;過濾出白色固體且使用WatersX-Terra反相柱,用水(包含0.2%碳酸銨)與乙腈的混合物洗脫,通過制備型HPLC來純化濾液,得到呈黃色固體狀的3-(4-(5-(5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-甲基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-氧代丙酸乙酯(80mg,61.7%)。將此物質懸浮于THF(2mL)中。添加2N氫氧化鈉(0.235mL,0.47mmol)和水(0.5ml)。在室溫下攪拌混合物過夜。向該混合物中添加2N鹽酸(230μL)。蒸發溶劑。用CH2Cl2(30mL)和水(5mL)稀釋殘余物。有機相用鹽水洗滌且經MgSO4干燥。蒸發溶劑。在乙醚中濕磨所得泡沫。過濾所得黃色固體,干燥,在乙腈(3mL)中濕磨。通過過濾來收集黃色固體,在40℃下干燥,得到呈黃色固體狀的標題化合物(50mg,68%)。1HNMR光譜:(DMSO-d6)1.46(9H,s),1.58(1H,m),1.74(1H,m),1.98(2H,m),2.84(1H,m),3.0(1H,m),3.21(1H,m),3.46(2H,m),3.83(1H,m),4.22(3H,s),4.34(1H,m),7.8-8.2(1H,m),8.87(1H,s);質譜[M+H]+=470。實例9:3-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-乙基-1,2,4-三唑-3-基]-1-哌啶基]-3-氧代-丙酸在50℃下,向溶解于DMF(20mL)中的3-乙氧基-3-氧代丙酸(150mg,1.13mmol)、N-乙基-N-異丙基丙-2-胺(0.394mL,2.26mmol)以及3-(5-(叔丁基)-1,3,4-噁二唑-2-基)-5-(1-乙基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(450mg,1.13mmol)的攪拌溶液中經30秒逐份添加六氟磷酸O-(7-氮雜苯并三唑-1-基)-N,N,N′,N′-四甲脲(474mg,1.25mmol)。1分鐘后(完全反應)對所得溶液進行取樣且使其即刻冷卻到環境溫度。濃縮反應混合物且用EtOAc(100mL)稀釋,依次用水(20mL)和飽和鹽水(20mL)洗滌。經MgSO4干燥有機層,過濾且蒸發,得到粗3-(4-(5-(5-氨基-6-(5-(叔丁基)-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-氧代丙酸乙酯(850mg)。將該物質中的一些(780mg)溶解于THF(20ml)中。向此溶液中添加2N氫氧化鈉水溶液(2.3ml,4.57mmol)和水(5ml),隨后添加甲醇(5ml),得到澄清溶液。于室溫下攪拌混合物3小時。蒸發THF。用2N鹽酸水溶液(2.5ml)將水層酸化到pH3。添加二氯甲烷(50ml)且萃取有機相。用鹽水(10ml)洗滌有機相且經MgSO4干燥。蒸發溶劑。使用水(包含1%氨)與乙腈的極性遞減混合物作為洗脫劑,通過制備型HPLC(WatersX-BridgePrepC18OBD柱,5μ二氧化硅,50mm直徑,100mm長度)來純化所得膠狀物。將包含所希望的化合物的洗脫份蒸發到干燥,得到純銨鹽。將此銨鹽溶解在水中且用2N鹽酸(約0.3ml)酸化到pH3。添加二氯甲烷(50ml)且分離出有機相,用鹽水(5ml)洗滌且經MgSO4干燥。在過濾之后,將所得溶液蒸發到干燥且用乙醚(5ml)濕磨殘余物并過濾,得到呈奶白色固體狀的3-(4-(5-(5-氨基-6-(5-(叔丁基)-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)-3-氧代丙酸(195mg,26.5%)。1HNMR光譜:(DMSO-d6)1.45(9H,s),1.48(3H,m),1.55-1.62(1H,m),1.70-1.80(1H,m),1.95-2.05(2H,m),2.80-2.90(1H,m),2.95-3.05(1H,m),3.15-3.25(1H,m),3.45(2H,s),3.78-3.85(1H,m),4.30-4.40(1H,m),4.55-4.65(2H,m),7.80-8.00(2H,brs),8.88(1H,s),12.60(1H,s);質譜[M+H]+=484實例10:(2S)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-乙基-1,2,4-三唑-3-基]-1-哌啶基]-2,3-二羥基-丙-1-酮向3-(5-(叔丁基)-1,3,4-噁二唑-2-基)-5-(1-乙基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺(257mg,0.50mmol,TFA鹽)、(S)-2,2-二甲基-1,3-二氧戊環-4-甲酸鉀(101mg,0.55mmol)以及EDCI(105mg,0.55mmol)于DCM(5mL)中的混合物中添加水合1-羥基-1H-苯并三唑(85mg,0.56mmol)和DIPEA(194mg,1.50mmol)。在室溫下攪拌混合物16小時。向混合物中添加水,且用DCM萃取混合物。有機層用鹽水洗滌且經Na2SO4干燥,過濾且濃縮,得到(S)-(4-(5-(5-氨基-6-(5-(叔丁基)-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)(2,2-二甲基-1,3-二氧戊環-4-基)甲酮(320mg)。質譜[M+H]+=526。在室溫下向(S)-(4-(5-(5-氨基-6-(5-(叔丁基)-1,3,4-噁二唑-2-基)吡嗪-2-基)-1-乙基-1H-1,2,4-三唑-3-基)哌啶-1-基)(2,2-二甲基-1,3-二氧戊環-4-基)甲酮(320mg)于DCM(10mL)中的混合物中逐滴添加TFA(1.6ml,20.77mmol)。在室溫下攪拌混合物16h,濃縮,且使用水(包含0.1%NH3)與MeCN的極性遞減混合物作為洗脫劑,通過制備型HPLC(WatersXBridgePrepC18OBD柱,5μ二氧化硅,19mm直徑,100mm長度)來純化。將包含所希望的化合物的洗脫份蒸發到干燥,得到呈白色固體狀的標題化合物(142mg,48%)。1HNMR光譜(400Hz,DMSO-d6,30℃):1.45(12H,m),1.56(1H,m),1.70(1H,m),1.98(2H,m),2.85(1H,m),3.00(1H,m),3.20(1H,m),3.45(1H,s),3.55(1H,s),4.05(1H,m),4.35(2H,m),4.60(2H,m),4.70(1H,m),4.85(1H,m),7.90(2H,m),8.85(1H,s);質譜[M+H]+=486。實例11:(2R)-1-[4-[5-[5-氨基-6-(5-叔丁基-1,3,4-噁二唑-2-基)吡嗪-2-基]-1-乙基-1,2,4-三唑-3-基]-1-哌啶基]-2,3-二羥基-丙-1-酮使用與實例10中所述類似的程序,使3-(5-(叔丁基)-1,3,4-噁二唑-2-基)-5-(1-乙基-3-(哌啶-4-基)-1H-1,2,4-三唑-5-基)吡嗪-2-胺與(R)-2,2-二甲基-1,3-二氧戊環-4-甲酸鉀反應,獲得呈固體狀的標題化合物(0.145g,40%)。1HNMR光譜(400Hz,DMSO-d6,30℃):1.45(12H,m),1.60(2H,m),1.98(2H,m),2.85(1H,m),3.00(1H,m),3.17(1H,m),3.45(1H,s),3.55(1H,s),4.05(1H,m),4.35(2H,m),4.60(2H,m),4.70(1H,m),4.85(1H,m),7.90(2H,m),8.85(1H,s);質譜[M+H]+=486。附圖簡要說明圖1顯示實例1形式A的X射線粉末衍射圖。圖2顯示實例1形式A的DSC熱譜圖。圖3顯示實例3形式A的X射線粉末衍射圖。圖4顯示實例3形式A的DSC熱譜圖。圖5顯示實例3形式B的X射線粉末衍射圖。圖6顯示實例3形式B的DSC熱譜圖。圖7顯示實例3形式C的X射線粉末衍射圖。圖8顯示實例3形式C的DSC熱譜圖。圖9顯示通過實例3與AKT抑制劑(AZD5363)的組合依序給藥的腫瘤生長抑制圖10顯示通過實例3與AKT抑制劑(AZD5363)的組合共同給藥的腫瘤生長抑制圖11顯示在BT474異種移植模型中通過實例3與PARP抑制劑(奧拉帕尼)的組合的腫瘤生長抑制圖12顯示在MCF7異種移植模型中通過實例3與PARP抑制劑(奧拉帕尼)的組合的腫瘤生長抑制圖13顯示通過實例3與(AZD8186)的組合的腫瘤生長抑制。
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1