新的羥基紅花黃色素藥用鹽的制作方法與工藝
本發明提供一種新的羥基紅花黃色素藥用鹽,具體地說,提供一種羥基紅花黃色素A藥用鹽及其制備方法、凍干粉針劑以及醫藥用途。屬于藥物化學領域。
背景技術:
中藥紅花為菊科植物CarthamustinctouiusL.的干燥花,是一種常見的活血化淤中藥,可用于冠心病、心絞痛等諸多血液循環障礙疾病的治療。羥基紅花黃色素A(hydroxysaffloryellowA)是具有單查爾酮苷類結構的化合物,是紅花藥理功效的最有效水溶性部位,可抑制血小板激活因子誘發的血小板聚集與釋放,可競爭性地抑制血小板激活因子與血小板受體的結合,是紅花黃色素的活血化瘀有效成分。研究表明,它具有多方面的心血管藥理作用,可抗凝、促進纖溶、抗血栓形成、改善微循環等。羥基紅花黃色素A作為紅花黃色素中含量最高的組分,其藥用價值在心血管的應用已得到證明,該機理也十分明確。現有技術中已經公開了大量的羥基紅花黃色素A生產工藝,包括以紅花為原料,經過水提取、大孔吸附樹脂分離、葡聚糖凝膠層析以及超濾等步驟,得到注射用的羥基紅花黃色素A。然而,現有的生產工藝所制得的羥基紅花黃色素A,產品純度不高,基本都存在10%以上的雜質,且這類雜質的結構性質均都未能定性,存在一定的質量不可控性,并且影響了產品,特別是注射用藥的穩定性和安全性。CN102675379A中公開了一種從紅花中提取精制羥基紅花黃色素A的方法,并具體公開了從中藥紅花中經過提取、弱堿性離子交換樹脂純化、中極性大孔吸附樹脂純化和非極性大孔吸附樹脂純化、冷凍干燥五個步驟,也僅僅得到含量為80%以上的羥基紅花黃色素A。
技術實現要素:
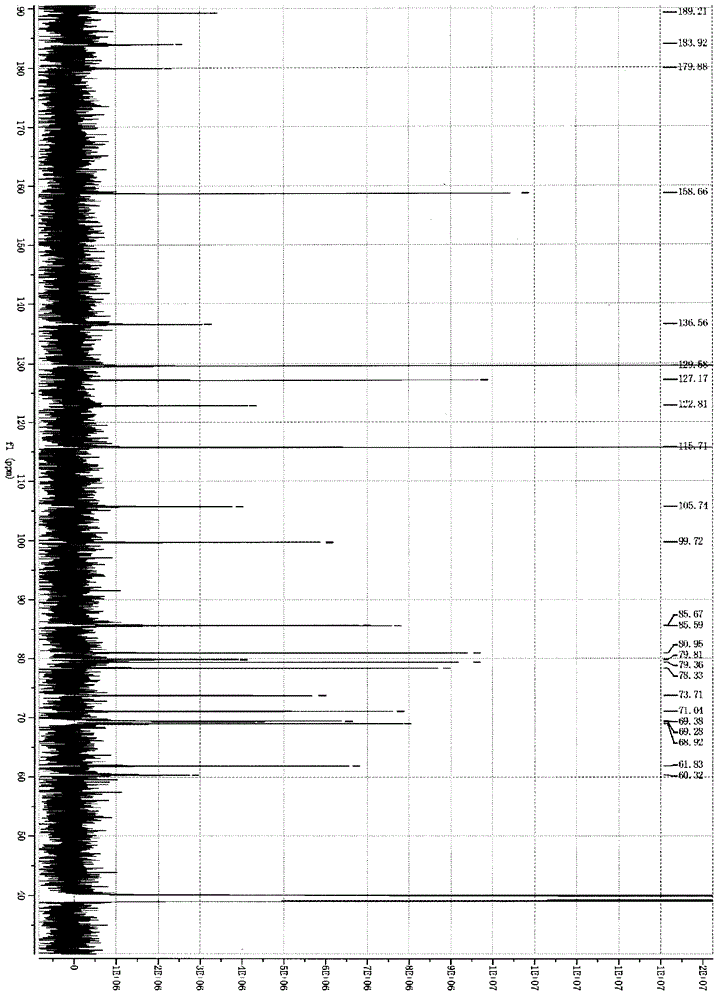
為克服現有技術的缺陷,本發明提供了一種羥基紅花黃色素A藥用鹽新化合物,其純度能保證達到98%以上,雜質個數控制在5個以下,將成為應用于冠心病、心絞痛、腦中風等諸多血液循環障礙疾病的治療上相較于羥基紅花黃色素A更安全有效、更穩定可控的新的單體化合物。本發明技術方案如下:本發明目的之一是提供一種如式(I)所示的羥基紅花黃色素A藥用鹽:其中,n為1或2,M選自Ca、Mg、K、NH4或其中,R1、R2、R3、R4分別相同或者不同,各自獨立選自氫、烷基。優選地,上述所述的羥基紅花黃色素A藥用鹽,其選自式(II)的鉀鹽、式(III)的銨鹽,式(IV)的鈣鹽或者式(V)的鎂鹽:作為本發明另一發明目的,還提供上述所述的羥基紅花黃色素A藥用鹽的制備方法,其包括紅花藥材的提取、強酸性H型陽離子交換樹脂的轉換、大孔吸附樹脂分離、葡聚糖凝膠層析分離和超濾的步驟,其特征在于:(1)紅花藥材的提取:紅花藥材為原料,水提取得到含有羥基紅花黃色素A的提取液;(2)強酸性H型陽離子交換樹脂轉換:將步驟(1)制得的提取液過強酸性H型陽離子交換樹脂柱,收集洗脫液,加入氫氧化鉀、氫氧化銨、烷基胺或烷基銨、氫氧化鎂、氫氧化鈣、碳酸鎂或者碳酸鈣,使羥基紅花黃色素A生成羥基紅花黃色素A藥用鹽,收集含羥基紅花黃色素A藥用鹽的洗脫液;(3)大孔吸附樹脂分離:將步驟(2)制得的含羥基紅花黃色素A藥用鹽的洗脫液用大孔吸附樹脂柱分離,以水為洗脫劑,收集洗脫液,減壓濃縮,得到羥基紅花黃色素A藥用鹽的粗品;(4)葡聚糖凝膠分離:將步驟(3)制得的羥基紅花黃色素A藥用鹽粗品用葡聚糖凝膠層析分離,以水為洗脫劑,收集含羥基紅花黃色素A藥用鹽洗脫液;(5)超濾:將步驟(4)所得含羥基紅花黃色素A藥用鹽洗脫液濃縮后經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液,干燥后即得羥基紅花黃色素A藥用鹽。其中,優選地,上述所述的制備方法,步驟(2)中所述的陽離子交換樹脂為強酸性H型陽離子交換樹脂,其選自001*7離子交換樹脂或者大孔HB-8交換樹脂。本發明所用的強酸性H型陽離子交換樹脂,可以采用市售的強酸性H型陽離子交換樹脂,例如001*7離子交換樹脂或者大孔HB-8交換樹脂均可購自上海華震科技有限公司,并且可以用HCl再生,重復使用。作為本發明另一目的,還提供了一種制備上述所述的羥基紅花黃色素A藥用鹽的方法,其包括紅花藥材的提取、大孔吸附樹脂分離、葡聚糖凝膠分離、超濾、酸化和轉鹽的步驟,其特征在于:(1)紅花藥材的提取:紅花藥材為原料,水提取得到含有紅花黃色素的提取液;(2)大孔吸附樹脂分離:將步驟(1)制得的含紅花黃色素提取液用大孔吸附樹脂柱分離,以水為洗脫劑,收集洗脫液,減壓濃縮,得到紅花黃色素的粗品;(3)葡聚糖凝膠分離:將步驟(2)制得的紅花黃色素粗品用葡聚糖凝膠層析分離,以水為洗脫劑,收集含紅花黃色素洗脫液;(4)超濾:將步驟(3)所得含紅花黃色素洗脫液濃縮后經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液,干燥后即得紅花黃色素粉末。(5)酸化:將步驟(4)得到的紅花黃色素粉末加水,加酸后放置冷處2~24小時,析出淡黃色固體羥基紅花黃色素A,濾去上層液體。(6)轉鹽:將步驟(5)得到的羥基紅花黃色素A,加水后加入氫氧化鉀、氫氧化銨、烷基胺或烷基銨、氫氧化鎂、氫氧化鈣、碳酸鎂或者碳酸鈣,使羥基紅花黃色素A生成羥基紅花黃色素A藥用鹽,再用超濾膜進行超濾,凍干制得所述的羥基紅花黃色素A藥用鹽。作為本發明另一目的,還提供一種藥物組合物,其包含治療有效量的上述所述的羥基紅花黃色素A藥用鹽為活性成分和藥學上可接受的載體為輔料。羥基紅花黃色素A藥用鹽,可制備成合適的制劑使用,例如凍干粉針劑或輸液劑。可以制成:(1)注射用羥基紅花黃色素A藥用鹽凍干粉針劑,每瓶含50mg-200mg,不加輔料或加入1∶0.5~1.5甘露醇;(2)羥基紅花黃色素A藥用鹽氯化鈉注射液,每100ml氯化鈉注射液含羥基紅花黃色素A藥用鹽50mg-200mg;(3)羥基紅花黃色素A藥用鹽葡萄糖注射液,每100ml葡萄糖注射液含羥基紅花黃色素A藥用鹽50mg-200mg。其中,優選上述所述的藥物組合物為凍干粉針劑,并用包括如下步驟的方法制備得到:(1)以紅花藥材為原料,加入溫度為50~100℃的水抽提,抽提是以水抽提2~3次,每次0.5~24小時,抽提用水量為紅花生藥重量的10~30倍,抽提后濾除藥渣,將提取液冷卻至5~30℃,靜置2~24小時;(2)將步驟(1)制得的提取液過強酸性H型陽離子交換樹脂,流速1~30ml/min,收集洗脫液,加入氫氧化鉀、氫氧化銨、烷基胺或烷基銨、氫氧化鎂、氫氧化鈣、碳酸鎂或者碳酸鈣,使羥基紅花黃色素A生成羥基紅花黃色素A藥用鹽,收集含羥基紅花黃色素A藥用鹽的洗脫液;(3)將步驟(2)制得的洗脫液用大孔吸附樹脂柱分離,以純化水為洗脫劑,洗脫流速10~30ml/min,收集洗脫液,減壓濃縮,得羥基紅花黃色素A藥用鹽濃縮液粗品;(4)將步驟(3)得到的羥基紅花黃色素A藥用鹽濃縮液粗品過濾或離心后用葡聚糖凝膠層析分離,以純化水為洗脫劑,洗脫流速控制線性流速為1~10cm/h,收集含羥基紅花黃色素A藥用鹽洗脫液,減壓濃縮得濃縮液;(5)將步驟(4)所得濃縮液經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液;(6)將步驟(5)所得超濾液經冷凍干燥,即得羥基紅花黃色素A藥用鹽精品;(7)將步驟(6)得到的羥基紅花黃色素A藥用鹽精品溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,即得到羥基紅花黃色素A藥用鹽凍干粉針。優選地,其中,上述所述的陽離子交換樹脂是用001*7離子交換樹脂或者大孔HB-8交換樹脂;所述的大孔吸附樹脂是用大孔吸附樹脂HZ801;所述的葡聚糖凝膠層析是用葡聚糖凝膠LH-20。本發明上述所述的方法,其中加入所述的藥用堿使羥基紅花黃色素A生成羥基紅花黃色素A藥用鹽,堿的加入量,優選為羥基紅花黃色素A的摩爾量的0.5~1倍。作為本發明一個具體實施方式,提供一種式(II)所述的羥基紅花黃色素A鉀及其凍干粉針劑的制備方法,其包括紅花藥材的提取、強酸性H型陽離子交換樹脂的轉換、大孔樹脂分離、葡聚糖凝膠層析分離和超濾的步驟,其特征在于:(1)以紅花藥材為原料,加入適量的溫度為50~100℃的水抽提,抽提是以水抽提2~3次,每次0.5~24小時,抽提用水量為紅花生藥重量的10~30倍。抽提后濾除藥渣,將提取液冷卻至5~30℃:,靜置2~24小時;(2)將提取液過強酸性H型陽離子交換樹脂,流速1~30ml/min,收集洗脫液,按羥基紅花黃色素A計,加入等摩爾的氫氧化鉀,得到含羥基紅花黃色素A鉀洗脫液;(3)大孔吸附樹脂分離:步驟(2)得到的洗脫液用大孔吸附樹脂HZ801柱分離,大孔吸附樹脂柱的柱內徑與柱高的比值為1∶8~15,以純化水為洗脫劑,洗脫流速10~30ml/min,收集洗脫液,減壓濃縮,得羥基紅花黃色素A鉀濃縮液粗品;(4)葡聚糖凝膠層析分離:步驟(3)得到的羥基紅花黃色素A鉀濃縮液粗品過濾或離心后用SephadexLH-20葡聚糖凝膠層析分離,層析柱的徑高比為1∶5~20,以純化水為洗脫劑,洗脫流速控制線性流速為1~10cm/h,收集含羥基紅花黃色素A鉀洗脫液,減壓濃縮得濃縮液;(5)超濾:步驟(4)所得濃縮液經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液;(6)凍干:步驟(5)所得超濾液經冷凍干燥,即得羥基紅花黃色素A鉀;如果需要,將步驟(6)得到的羥基紅花黃色素A鉀精品,溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,得到羥基紅花黃色素A鉀凍干粉針。作為本發明另一個具體實施方式,提供一種式(III)所述的羥基紅花黃色素A銨及其凍干粉針劑的制備方法,其包括紅花藥材的提取、強酸性陽離子交換樹脂的轉換、大孔樹脂分離、葡聚糖凝膠層析分離和超濾的步驟,其特征在于:(1)以紅花藥材為原料,加入適量的溫度為50~100℃的水抽提,抽提是以水抽提2~3次,每次0.5~24小時,抽提用水量為紅花生藥重量的10~30倍。抽提后濾除藥渣,將提取液冷卻至5~30℃:,靜置2~24小時;(2)將提取液過強酸性陽離子交換樹脂,流速1~30ml/min,收集的提取液,按羥基紅花黃色素A計,加入等摩爾的氫氧化銨(即氨水),得到含羥基紅花黃色素A銨的洗脫液;(3)大孔吸附樹脂分離:步驟(2)得到的洗脫液用大孔吸附樹脂HZ801柱分離,大孔吸附樹脂柱的柱內徑與柱高的比值為1∶8~15,以純化水為洗脫劑,洗脫流速10~30ml/min,收集洗脫液,減壓濃縮,得羥基紅花黃色素A銨鹽濃縮液粗品;(4)葡聚糖凝膠層析分離:步驟(3)得到的羥基紅花黃色素A銨鹽濃縮液粗品過濾或離心后用SephadexLH-20葡聚糖凝膠層析分離,層析柱的徑高比為1∶5~20,以純化水為洗脫劑,洗脫流速控制線性流速為1~10cm/h,收集含羥基紅花黃色素A銨鹽洗脫液,減壓濃縮得濃縮液;(5)超濾:步驟(4)所得濃縮液經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液;(6)凍干:步驟(5)所得超濾液經冷凍干燥,即得羥基紅花黃色素A銨鹽。如果需要,將步驟(6)得到的羥基紅花黃色素A銨鹽精品,溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,得到羥基紅花黃色素A銨鹽凍干粉針。作為本發明另一個具體實施方式,提供一種式(IV)所述的羥基紅花黃色素A鈣及其凍干粉針劑的制備方法,其包括紅花藥材的提取、強酸性H型陽離子交換樹脂的轉換、大孔樹脂分離、葡聚糖凝膠層析分離和超濾的步驟,其特征在于:(1)以紅花藥材為原料,加入適量的溫度為50~100℃的水抽提,抽提是以水抽提2~3次,每次0.5~24小時,抽提用水量為紅花生藥重量的10~30倍。抽提后濾除藥渣,將提取液冷卻至5~30℃:,靜置2~24小時;(2)將提取液過強酸性H型陽離子交換樹脂,流速1~30ml/min,收集洗脫液,按羥基紅花黃色素A計,加入一半摩爾的Ca(OH)2,得到含羥基紅花黃色素A鈣洗脫液;(3)大孔吸附樹脂分離:步驟(2)得到的洗脫液用大孔吸附樹脂HZ801柱分離,大孔吸附樹脂柱的柱內徑與柱高的比值為1∶8~15,以純化水為洗脫劑,洗脫流速10~30ml/min,收集洗脫液,減壓濃縮,得羥基紅花黃色素A鈣濃縮液粗品;(4)葡聚糖凝膠層析分離:步驟(3)得到的羥基紅花黃色素A鈣濃縮液粗品過濾或離心后用SephadexLH-20葡聚糖凝膠層析分離,層析柱的徑高比為1∶5~20,以純化水為洗脫劑,洗脫流速控制線性流速為1~10cm/h,收集含羥基紅花黃色素A鈣洗脫液,減壓濃縮得濃縮液;(5)超濾:步驟(4)所得濃縮液經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液;(6)凍干:步驟(5)所得超濾液經冷凍干燥,即得羥基紅花黃色素A鈣。如果需要,可以將步驟(6)得到的羥基紅花黃色素A鈣精品,溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,得到羥基紅花黃色素A鈣凍干粉針。作為本發明另一個具體實施方式,提供一種式(V)所述的羥基紅花黃色素A鎂及其凍干粉針劑的制備方法,其包括紅花藥材的提取、強酸性陽離子交換樹脂的轉換、大孔樹脂分離、葡聚糖凝膠層析分離和超濾的步驟,其特征在于:(1)以紅花藥材為原料,加入適量的溫度為50~100℃的水抽提,抽提是以水抽提2~3次,每次0.5~24小時,抽提用水量為紅花生藥重量的10~30倍。抽提后濾除藥渣,將提取液冷卻至5~30℃:,靜置2~24小時;(2)將提取液過強酸性陽離子交換樹脂,流速1~30ml/min,收集的提取液,按羥基紅花黃色素A計,加入一半摩爾的氫氧化鎂,得到含羥基紅花黃色素A鎂的洗脫液;(3)大孔吸附樹脂分離:步驟(2)得到的洗脫液用大孔吸附樹脂HZ801柱分離,大孔吸附樹脂柱的柱內徑與柱高的比值為1∶8~15,以純化水為洗脫劑,洗脫流速10~30ml/min,收集洗脫液,減壓濃縮,得羥基紅花黃色素A鎂鹽濃縮液粗品;(4)葡聚糖凝膠層析分離:步驟(3)得到的羥基紅花黃色素A鎂鹽濃縮液粗品過濾或離心后用SephadexLH-20葡聚糖凝膠層析分離,層析柱的徑高比為1∶5~20,以純化水為洗脫劑,洗脫流速控制線性流速為1~10cm/h,收集含羥基紅花黃色素A鎂鹽洗脫液,減壓濃縮得濃縮液;(5)超濾:步驟(4)所得濃縮液經過濾或離心后采用截留分子量8000-10000道爾頓的超濾膜進行超濾得到超濾液;(6)凍干:步驟(5)所得超濾液經冷凍干燥,即得羥基紅花黃色素A鎂鹽。如果需要,可以將步驟(6)得到的羥基紅花黃色素A鎂鹽精品,溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,得到羥基紅花黃色素A鎂鹽凍干粉針。其中,上述所述的離子交換樹脂是用001*7離子交換樹脂或者大孔HB-8交換樹脂;所述的大孔樹脂分離是用大孔吸附樹脂HZ801;所述的葡聚糖凝膠層析分離是用葡聚糖凝膠LH-20;所述的超濾是用截留分子量8000-10000道爾頓的超濾膜。作為本發明另一發明目的,還提供了上述所述的羥基紅花黃色素A藥用鹽在制備藥物中的應用,其中所述的藥物具有抗PAF或ADP誘導的血小板聚集作用,用于治療或者預防涉及心肌缺血、腦缺血、血栓形成所致損傷疾病。本發明的羥基紅花黃色素A藥用鹽的臨床應用劑量為50-200mg/每天。本發明以紅花藥材為原料,制備獲得了新型單體藥物羥基紅花黃色素A藥用鹽,純度能保證達到98%以上,雜質個數控制在5個以下,將成為應用于冠心病、心絞痛等諸多血液循環障礙疾病的治療上相較于羥基紅花黃色素A更安全有效、更穩定可控的單體化合物。本發明在研究中,通過反復試驗研究發現,紅花藥材提取液中的羥基紅花黃色素A是不以酸形式存在的,現有技術(例如CN101168539A、CN1895317A、CN101195647A)中通過紅花黃色素提取液直接加入氫氧化鈉等pH調節劑來調節pH值,羥基紅花黃色素A并不能轉化生成羥基紅花黃色素A藥用鹽,因此得不到羥基紅花黃色素A藥用鹽單體化合物。本發明令人意外發現,紅花黃色素提取液先通過用強酸性H型陽離子交換樹脂轉化后得到真正意義上的羥基紅花黃色素A,即以酸形式,然后通過不同的堿調節pH,轉化得到羥基紅花黃色素A相應的鹽,可以得到單一的羥基紅花黃色素A藥用鹽單體化合物。同樣地,本發明還發現,還可以通過將紅花黃色素精制品通過酸化先制得羥基紅花黃色素A,羥基紅花黃色素A再用氫氧化鉀、氫氧化銨、氫氧化鎂、氫氧化鈣、碳酸鎂或者碳酸鈣等成鹽,也可以高收率、高純度獲得本發明所述的羥基紅花黃色素A藥用鹽單體化合物。與羥基紅花黃色素A相比,本發明的羥基紅花黃色素A藥用鹽不僅純度更高,達到98%以上,雜質個數控制在5個以下,而且穩定性也更好。一、藥效學研究試驗藥效學研究試驗一:羥基紅花黃色素A鹽對大鼠急性心肌梗死的保護作用受試藥物注射用紅花黃色素(50mg/瓶),來源:浙江永寧藥業股份有限公司,含量:50mg/瓶含羥基紅花黃色素A42.5mg;羥基紅花黃色素A鉀(按照實施例1所制得);羥基紅花黃色素A鈣(按照實施例5所制得)。動物:雄性SD大鼠32只,體重250~350g.試驗分組及劑量設置:試驗方法1.大鼠急性心肌梗死模型的制備:以3%戊巴比妥鈉45mg/kg腹腔麻醉。右側股動、靜脈插管分別作血壓監測和靜脈給藥用;氣管插管行人工機械通氣,頻率60次/min、通氣量約10ml/kg。四肢皮下置入針電極以記錄心電圖。于胸骨左緣外側約0.5cm處縱行切開皮膚,鈍性分離皮下組織,依次分離、結扎并剪斷胸大肌、胸小肌、肋間肌,在胸骨左緣第四肋間心尖博動最強外開胸,剪開心包,以止血鉗夾持心包于胸壁兩側,形成心包床,暴露左心室表面血管。6-0絲線穿過左心耳下約2mm處的淺層心肌(即冠狀動脈前降支)。穿線后穩定15min。如此期間出現心律失常或動脈收縮壓低于70mmHg(9.31KPa)持續超過5min則廢除該動物,穿線15min后靜脈給藥,給藥10min后結扎冠脈,結扎時間持續4小時。2.心律失常評分:對冠脈結扎后30min內出現心律失常嚴重程度進行評分。3.心肌酶學檢查:試驗結束取靜脈血約2ml,4℃下4000/min離心5min,取上清液作心肌酶學檢查。測定項目:LDH、AKP、CK。4.心肌梗死區面積測定:冠脈結扎后4h后于左心室前壁穿刺,注射碳素墨水后自處死動物,取出心臟,用生理鹽水洗凈,除去血污剔除血管、脂肪等非心肌組織,用吸水紙吸去水分,稱重。從心尖到心底部平行將心室切成約2mm左右薄片,將灌注區(墨水灌注部分)和缺血危險區(無墨水灌注部分)分離,將缺血危險區稱重并置入0.05%的NBT溶液中,37℃恒溫水浴箱中染色15min。NBT可使活組織內的基質、輔酶及脫氧酶等染成紫藍色,而壞死組織中這些代謝基質、酶等喪失,故不著色。可見到非梗死區域被NBT染成深藍色,梗死區不被染色。剪去各心肌中被染色的非梗死心肌,把末被染色的梗心心肌稱重,計算梗死心肌占危險區心肌的重量百分比(梗死區重量/危險區重量*100%)。統計學處理:試驗數據以均數±標準差表示,采用方差分析進行統計學檢驗,P<0.05表示差異顯著。一、對大鼠急性心肌缺血后心律失常的影響大鼠冠脈結扎后5min開始有心律失常出現,持續至30min,10min左右達到高峰。本試驗結果顯示紅花黃色素,羥基紅花黃色素A鉀與羥基紅花黃色素A鈣靜脈給藥都可降低心律失常的嚴重程度,但羥基紅花黃色素A鉀、羥基紅花黃色素A鈣的效果更佳。各實驗組心律失常評分*P<0.05vs組1二、比較紅花黃色素、羥基紅花黃色素A鉀與羥基紅花黃色素A鈣對大鼠心肌梗死范圍的影響各實驗組對心肌梗死范圍的影響*P<0.05vs組1三、注射用紅花黃色素對大鼠心肌梗死后血清LDH、AKP、CK的影響本試驗結果顯示注射用紅花黃色素、羥基紅花黃色素A鉀與羥基紅花黃色素A鈣靜脈給藥都可抑制大鼠血清LDH、CK的升高,但羥基紅花黃色素A鉀、羥基紅花黃色素A鈣效果更顯著。各實驗組血清LDH、AKP、CK值*p<0.05vs組1試驗結論1.注射用紅花黃色素、羥基紅花黃色素A鉀與羥基紅花黃色素A鈣均可明顯降低大鼠冠脈結扎后心律失常的嚴重程度,但羥基紅花黃色素A鉀、羥基紅花黃色素A鈣的效果更好;2.與注射用紅花黃色素比較,羥基紅花黃色素A鉀、羥基紅花黃色素A鈣降低大鼠心肌梗死范圍效果更好;3.與注射用紅花黃色素比較,羥基紅花黃色素A鉀、羥基紅花黃色素A鈣抑制大鼠血清LDH、CK的療效更明顯;4.羥基紅花黃色素A鉀與羥基紅花黃色素A鈣兩者藥效無顯著差別。藥效學研究試驗二:羥基紅花黃色素A鉀與羥基紅花黃色素A鈣靜脈注射給藥對急性腦缺血有防治作用。受試藥物注射用紅花黃色素(50mg/瓶),來源:浙江永寧藥業股份有限公司,含量:50mg/瓶含羥基紅花黃色素A42.5mg;羥基紅花黃色素A鉀(按照實施例1所制得);羥基紅花黃色素A鈣(按照實施例5所制得)。實驗內容如下:1.對犬離體心、腦血管的選擇性:該實驗將Beagle犬大腦基底動脈環和冠狀動脈環固定在離體血管測量裝置上,調整張力傳感器并向浴杯液中加入10-6mol/L的苯腎上腺素使血管維持適度張力,然后每間隔5min向浴杯液中按每毫升10mg的劑量加入注射用羥基紅花黃色素A鉀或羥基紅花黃色素A鈣,直到血管環反應很弱或不再出現反應為止(一般加藥次數為4-5次)。計算血管收縮或舒張變化值。實驗結果顯示:注射用羥基紅花黃色素A鉀對心臟血管環的舒張作用為31.6%,而對腦血管環的舒張作用為73.1%;注射用羥基紅花黃色素A鈣對心臟血管環的舒張作用為37.1%,而對腦血管環的舒張作用為69.7%。提示注射用羥基紅花黃色素A鉀與羥基紅花黃色素A鈣對腦血管均具有非常好的選擇性和舒張作用,兩者之間無顯著差別。2.對急性腦缺血的影響:實驗用SD大鼠,靜脈注射羥基紅花黃色素A鉀或羥基紅花黃色素A鈣后用常規的大腦中動脈栓塞(MCAO)線栓法制備急性腦缺血模型。飼養24h后先對其進行神經行為學評分,然后斷頭處死大鼠,取出大腦,放入模具中切成7片,予以TTC染色,存活腦組織被染成紅色,壞死腦組織不著色,用圖形分析軟件計算壞死腦組織占大腦半球的比例。試驗結果,腦梗死面積溶劑對照組為38%,尼莫地平陽性對照組為15.7%,注射用羥基紅花黃色素A鉀低中高三個劑量組分別為38.2%、27.6%和21.9%,注射用羥基紅花黃色素A鈣低中高三個劑量組分別為41.3%、25.7%和21.6%,與溶劑對照組比較,注射用羥基紅花黃色素A鉀與羥基紅花黃色素A鈣在中高劑量能夠顯著減輕急性腦缺血引起腦組織壞死,兩者之間無顯著差別。3.對大鼠腦血管通透性的影響:大鼠靜脈注射給藥,每天一次,連續7天,最后一次給藥后將大鼠麻醉,靜脈注射伊文思藍50mg/kg,5分鐘后結扎雙側頸總動脈,3小時后斷頭處死動物,取出大腦,稱重后浸泡于甲酰胺溶液中,置45℃恒溫箱中72h,此時腦血管中的伊文思藍能夠浸出到甲酰胺溶液中,用分光光度計檢測甲酰胺溶液中伊文思藍析出量的多少表示腦血管通透性的高低。實驗結果顯示:注射用羥基紅花黃色素A鉀及羥基紅花黃色素A鈣在中高劑量組有非常顯著的減少伊文思藍從腦血管中的溢出的作用,說明該藥對降低血管通透性有較好作用,兩者之間無顯著差別。4.對犬腦血流量的影響:實驗動物用Beagle犬,犬用戊巴比妥鈉麻醉后手術分離出一側頸外靜脈、頸內靜脈和椎動脈,結扎頸外靜脈,放置流量探頭在頸內靜脈和椎動脈,兩處探頭記錄的血流量相加乘2代表全腦供血量,實驗結束取出大腦稱重計算每100g大腦組織血流量。試驗結果,注射用羥基紅花黃色素A鉀及羥基紅花黃色素A鈣在靜脈給藥后中高劑量均有明顯增加血流量的作用,但兩者血流量增加維持時間較短(約15min),該實驗提示今后在臨床用于治療急性腦缺血時應選用靜脈滴注的方法給藥,兩者之間無顯著差別。5.對急性腦缺氧的影響:實驗用昆明種小鼠和SD大鼠進行,將動物放入缺氧環境中,記錄存活時間,了解動物用藥后是否能增加對急性缺氧的耐受性。實驗發現:小鼠在密閉的容器中,溶劑對照組存活時間為32min,而注射用羥基紅花黃色素A鉀低中高三個劑量組的小鼠存活時間分別為36、37、36min;注射用羥基紅花黃色素A鈣低中高三個劑量組的小鼠存活時間分別為35、38、37min,經統計學處理與溶劑對照組比較有顯著差異(P<0.05~0.01)。大鼠實驗在含97%的氮氣和3%氧氣的環境中進行,動物放入容器后到呼吸停止,各組的生存時間為,溶劑對照組平均3分43秒;陽性對照組(尼莫地平)為5分38秒,兩者間比較差別非常顯著;注射用羥基紅花黃色素A鉀低中高三個劑量組分別為3分20秒、4分30秒和4分9秒;注射用羥基紅花黃色素A鈣低中高三個劑量組分別為3分31秒、4分35秒和4分21秒,和溶劑對照組比較中注射用羥基紅花黃色素A鉀及注射用羥基紅花黃色素A鈣的高劑量組存活時間顯著增加,兩者之間無顯著差別。6.抗血小板聚積:實驗分為用儀器檢測和活體實驗兩部分,1)儀器檢測方法:家兔靜脈注射羥基紅花黃色素A鉀,每天一次連續5天,最后一次給藥結束2小時內從心臟取血4ml,血液經低速離心得到富含血小板的血清;經高速離心得到貧血小板血清,血小板聚積誘導劑選用二磷酸腺苷(adenosinediphosphate;ADP)和血小板聚集活化因子(Platelet-ActivatingFactorPAF)兩種,用血小板聚集儀進行測試。在羥基紅花黃色素A鉀注射液抗ADP和PAF誘導的血小板聚集實驗中,均顯示有非常好的抗血小板聚集作用,在三個劑量之間顯示出有良好的量-效關系存在。2)活體方法:用乳膠管在大鼠動靜脈之間形成短路,乳膠管內固定有手術用絲線,利用血小板粘附的特點,開放短路使血液經乳膠管跨動靜脈流動15分鐘,取出絲線稱重,減去絲線干重即是粘附在絲線上的血小板重量。實驗結果,在絲線上粘附的血小板重量溶劑對照組為14.8±1.57mg;陽性對照組為8.62±2.79mg;注射用羥基紅花黃色素A鉀低中高三個劑量組的粘附血小板重量分別為13.6±1.89mg,9.90±1.53mg和8.91±1.34mg;注射用羥基紅花黃色素A鈣低中高三個劑量組的粘附血小板重量分別為13.9±1.54mg,10.26±1.15mg和8.73±1.79mg;注射用羥基紅花黃色素A鉀及羥基紅花黃色素A鈣的中高劑量組和溶劑對照組比較相差非常明顯,說明注射用羥基紅花黃色素A鉀或羥基紅花黃色素A鈣均有非常好的抗大鼠血小板聚集的作用,兩者之間無顯著差別。7.對血液粘滯度的影響:家兔靜脈給藥,每天一次,連續5天,最后一次給藥2小時心臟取血抗凝后直接用血液流變儀檢測。試驗結果,與溶劑對照組比較,羥基紅花黃色素A鉀及羥基紅花黃色素A鈣治療組隨著用藥劑量的增加,血液粘滯度低切、中切和高切三個指標均隨之降低,降低幅度隨劑量增加而增加,兩者高劑量組效果均優于陽性對照藥尼莫地平注射用液組,說明注射用羥基紅花黃色素A鉀及羥基紅花黃色素A鈣對降低血液粘滯度均有顯著作用,兩者之間無顯著差別。二、穩定性試驗和理化數據(一)觀察羥基紅花黃色素A鹽與羥基紅花黃色素A穩定性受試藥物注:含量是以羥基紅花黃色素A計。試驗結果表明,本發明的羥基紅花黃色素A藥用鹽單體化合物與羥基紅花黃色素A的穩定性比較試驗,說明羥基紅花黃色素A藥用鹽的穩定性更好。(二)觀察羥基紅花黃色素A鹽與羥基紅花黃色素A理化數據。羥基紅花黃色素A藥用鹽理化數據結果試驗結果表明,本發明的羥基紅花黃色素A藥用鹽單體化合物與羥基紅花黃色素A的理化數據比較,說明羥基紅花黃色素A藥用鹽的耐受性更好。附圖說明圖1:式(IV)的羥基紅花黃色素A鈣核磁共振1H-NMR圖2:式(IV)的羥基紅花黃色素A鈣核磁共振13C-NMR圖3:式(V)的羥基紅花黃色素A鎂核磁共振1H-NMR圖4:式(V)的羥基紅花黃色素A鎂核磁共振13C-NMR具體實施方式實施例1:羥基紅花黃色素A鉀(本發明式II化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的001*7強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的氫氧化鉀,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A鉀粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A鉀粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A鉀部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A鉀精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A鉀精品粉末,純度為98.5%,收率按紅花計為0.55%左右。羥基紅花黃色素A鉀的紅外光譜(IR)、質譜MS、核磁共振1H-NMR、13C-NMR數據如下:1.紅外吸收光譜儀器型號:BrukerVECTOR-22型紅外吸收光譜儀IR(溴化鉀壓片)2.質譜儀器型號:美國FINNIGAN公司液質聯用多級離子阱質譜系(LCQ-DECAXP)測試條件:ESI質譜MS+cESI651.06(M)+-cESI611.24(M-K)-3.核磁共振氫譜和碳譜儀器型號:BRUCKERAVANCEIII500型超導核磁共振儀測試條件:溶劑:DMSO,內標:TMS羥基紅花黃色素A鉀的1H一NMR數據羥基紅花黃色素A鉀鹽的13C一NMR數據將所制得的羥基紅花黃色素A鉀精品,溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,得到羥基紅花黃色素A鉀凍干粉針。實施例2:羥基紅花黃色素A鉀(本發明式II化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的HB-8大孔強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的氫氧化鉀,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A鉀粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A鉀粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A鉀部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A鉀精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A鉀精品粉末,純度為98.6%,收率按紅花計為0.50%左右。實施例3:式III的羥基紅花黃色素A銨(本發明式III化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的001*7強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的氨水,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A銨粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A銨粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A銨部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A銨精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A銨精品粉末,純度為99.3%,收率按紅花計為0.45%左右。羥基紅花黃色素A銨的紅外光譜(IR)、質譜MS、核磁共振1H-NMR、13C-NMR數據如下:1.紅外吸收光譜儀器型號:BrukerVECTOR-22型紅外吸收光譜儀IR(溴化鉀壓片)2.質譜儀器型號:美國FINNIGAN公司液質聯用多級離子阱質譜系(LCQ-DECAXP)測試條件:ESI質譜MS+cESI613.17(M)+-cESI611.22(M-H)-3.核磁共振氫譜和碳譜儀器型號:BRUCKERAVANCEIII500型超導核磁共振儀測試條件:溶劑:DMSO,內標:TMS核磁共振羥基紅花黃色素A銨的1H一NMR數據羥基紅花黃色素A銨的13C一NMR數據實施例4:羥基紅花黃色素A銨(即本發明式III化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的HB-8大孔強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的氨水,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A銨粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A銨粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A銨部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A銨精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A銨精品粉末,純度為99.4%,收率按紅花計為0.50%左右。實施例5:羥基紅花黃色素A鈣(式IV化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的001*7強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入一半摩爾的Ca(OH)2,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A鈣粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A鈣粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A鈣部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A鈣精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A鈣精品粉末,純度為98.8%,收率按紅花計為0.50%左右。羥基紅花黃色素A鈣的質譜MS、核磁共振1H-NMR、13C-NMR數據如下:1.質譜儀器型號:美國FINNIGAN公司液質聯用多級離子阱質譜系(LCQ-DECAXP)測試條件:ESI質譜MS+cESI1263.08(M)+-cESI611.29(M)-2.磁共振氫譜和碳譜儀器型號:BRUCKERAVANCEIII500型超導核磁共振儀測試條件:溶劑:DMSO,內標:TMS結果見圖1和2。羥基紅花黃色素A鈣的1H一NMR數據歸屬(由于結構對稱,編號一致)化學位移δ(ppm)3.01~4.15歸屬于糖部分氫G1~G6和G’1~G’6;4.29~4.88歸屬于糖上羥基氫;7.33(1H)7.38(1H)歸屬于8和9;6.77~7.47(4H)歸屬于11~15;18.74(1H)歸屬于3-OH;4.72(1H)歸屬于4-OH;9.82(1H)歸屬于13-OH。羥基紅花黃色素A鈣的13C一NMR數據歸屬化學位移δ(ppm)60.32(1C)、61.83(1C)(仲碳)歸屬于糖部分碳G6、G’6;68.92、69.28、69.38、71.04、73.71、78.33、79.36、79.81、80.95、85.67(10C)(叔碳)歸屬于糖部分碳G1~G5和G’1~G'5;115.71(2C)(叔碳)歸屬于12和14;129.58(2C)(叔碳)歸屬于11和15;122.81(1C)136.56(1C)(叔碳)歸屬于8和9;化學位移δ(ppm)189.21、105.74、85.59、183.92、99.72(5C)歸屬于1~2、4~6;179.88(1C)、127.17(1C)、158.66(1C)分別歸屬于7、10、13。將所制得的羥基紅花黃色素A鈣精品,溶解于注射用水,經采用0.22μm的微孔濾膜或者截留分子量8000-10000道爾頓的超濾膜過濾后分裝于瓶中,冷凍干燥后,得到羥基紅花黃色素A鈣凍干粉針。實施例6:羥基紅花黃色素A鈣(式IV化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的HB-8大孔強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入一半摩爾的Ca(OH)2,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A鈣粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A鈣粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A鈣部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A鈣精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A鈣精品粉末,純度為98.6%,收率按紅花計為0.50%左右。實施例7:羥基紅花黃色素A鈣(本發明式IV化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得紅花黃色素粗品濃縮液。以紅花計每公斤紅花得濃縮液100ml。紅花黃色素粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,以純化水為洗脫劑,洗脫流速為每分鐘5ml,收集含紅花黃色素部分。收集液經60℃減壓濃縮后,得紅花黃色素濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的紅花黃色素粉末,純度約為90%。紅花黃色素粉末加水溶解后用HCl酸化,放置冷處2~24小時析出羥基紅花黃色素A固體。固體過濾后加水溶解,按羥基紅花黃色素A計,再加入一半摩爾的氫氧化鈣,經冷凍干燥即得淡黃色的羥基紅花黃色素A鈣精品,純度為99.2%,收率按紅花計為0.7%左右。實施例8:式III的羥基紅花黃色素A鎂(本發明式V化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的001*7強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入一半摩爾的氫氧化鎂,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A鎂粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A鎂粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A鎂部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A鎂精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A鎂精品粉末,純度為99.0%,收率按紅花計為0.5%左右。羥基紅花黃色素A鎂的質譜MS、核磁共振1H-NMR、13C-NMR數據如下:1.質譜儀器型號:美國FINNIGAN公司液質聯用多級離子阱質譜系(LCQ-DECAXP)測試條件:ESI質譜MS+cESI1247.01(M)+-cESI611.35(M-H)-2.核磁共振氫譜和碳譜儀器型號:BRUCKERAVANCEIII500型超導核磁共振儀測試條件:溶劑:DMSO,內標:TMS結果見圖3和4。羥基紅花黃色素A鎂的1H一NM和13C一NMR,跟案例(埃索美拉唑鈉與埃索美拉唑鎂)類似,由于鎂的存在,會干擾樣品的測試。只能得到部分的氫譜和碳譜。由于工藝和骨架結構都與羥基紅花黃色A鈣一致,故可作為參考。羥基紅花黃色素A鎂的1H一NMR數據歸屬(由于結構對稱,編號一致)化學位移δ(ppm)2.85~4.11歸屬于糖部分氫G1~G6和G’1~G’6;4.38~4.81歸屬于糖上羥基氫;7.30(1H)7.38(1H)歸屬于8和9;6.74~7.41(4H)歸屬于11~15;18.68(1H)歸屬于3-OH;4.72(1H)歸屬于4-OH;9.80(1H)歸屬于13-OH。羥基紅花黃色素A鎂的13C一NMR數據歸屬化學位移δ(ppm)61.26(1C)、62.26(1C)(仲碳)歸屬于糖部分碳G6、G’6;69.24、70.03、71.51、74.13、79.70、80.54、81.33、85.91(8C)(叔碳)歸屬于糖部分碳G1~G5和G’1~G'5部分;116.09(2C)(叔碳)歸屬于12和14;129.90(2C)(叔碳)歸屬于11和15;122.81(1C)136.56(1C)(叔碳)歸屬于8和9;化學位移δ(ppm)195.38(1C)歸屬于3;179.83(1C)、127.62(1C)、159.01(1C)分別歸屬于7、10、13。實施例9:羥基紅花黃色素A鎂(即本發明式V化合物)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得紅花黃色素粗品濃縮液。以紅花計每公斤紅花得濃縮液100ml。紅花黃色素粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,以純化水為洗脫劑,洗脫流速為每分鐘5ml,收集含紅花黃色素部分。收集液經60℃減壓濃縮后,得紅花黃色素濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的紅花黃色素粉末,純度約為90%。紅花黃色素粉末加水溶解后用HCl酸化,放置冷處2~24小時析出羥基紅花黃色素A固體。固體過濾后加水溶解,按羥基紅花黃色素A計,再加入一半摩爾的氫氧化鎂,經冷凍干燥即得淡黃色的羥基紅花黃色素A鎂精品,純度為99.2%,收率按紅花計為0.7%左右。實施例10:羥基紅花黃色素A三乙胺(即式I中,n為1,R1為氫,R2、R3、R4為乙基)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的001*7強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的三乙胺,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A三乙胺粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A三乙胺粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A三乙胺部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A三乙胺精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A三乙胺精品粉末,純度為99.0%,收率按紅花計為0.45%左右。實施例11:羥基紅花黃色素A三乙胺(即式I中,n為1,R1為氫,R2、R3、R4為乙基)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的HB-8大孔強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的三乙胺,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A三乙胺粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A三乙胺粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A三乙胺部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A三乙胺精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A三乙胺精品粉末,純度為99.0%,收率按紅花計為0.47%左右。實施例12:羥基紅花黃色素A四甲基銨(即式I中,n為1,R1、R2、R3、R4為甲基)稱取紅花,加12.5倍藥材重量的去離子水,于100℃提取20-25分鐘,過濾,濾渣加10倍藥材重量的去離子水再按上述條件重復提取一次,過濾。合并二次提取液,冷卻至室溫,用離心機離心后,取離心液備用。將上述離心液緩緩加到已處理平衡好的001*7強酸性H型陽離子交換樹脂,柱徑高比為1∶10,柱體積為500ml,流速為3ml/min,收集流出液,按羥基紅花黃色素A計,加入等摩爾的四甲基氫氧化銨,然后緩緩加到大孔吸附樹脂分離柱中,柱徑高比為1∶12,上樣流速為每分鐘10ml。上樣結束后,用常溫的去離子水以每分鐘20ml的流速洗脫。洗脫液于60℃減壓濃縮,得羥基紅花黃色素A四甲基銨粗品濃縮液。以紅花計,每公斤紅花得濃縮液100ml。羥基紅花黃色素A四甲基銨粗品濃縮液上凝膠LH-20柱,柱的徑高比為1∶5,上樣量為柱床體積的10%,洗脫流速為每分鐘5ml,收集含羥基紅花黃色素A四甲基銨部分。收集液經60℃減壓濃縮后,得羥基紅花黃色素A四甲基銨精品濃縮液,以紅花計,每公斤紅花得濃縮液35~50ml,經冷凍干燥,即得淡黃色的羥基紅花黃色素A四甲基銨精品粉末,純度為99.0%,收率按紅花計為0.45%左右。羥基紅花黃色素A四甲基銨的質譜MS、核磁共振1H-NMR數據如下:1.質譜儀器型號:美國FINNIGAN公司液質聯用多級離子阱質譜系(LCQ-DECAXP)測試條件:ESI質譜MS-cESI611.22(M-H)-2.核磁共振氫譜儀器型號:BRUCKERAVANCEIII500型超導核磁共振儀測試條件:溶劑:DMSO,內標:TMS核磁共振羥基紅花黃色素A四甲基銨的1H一NMR數據
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1