具有增強的L-色氨酸生產力的埃希氏菌屬微生物以及使用其生產L-色氨酸的方法與流程
本發明涉及具有增強的L-色氨酸生產力的埃希氏菌屬的微生物,以及使用其生產L-色氨酸的方法。
背景技術:
必需氨基酸L-色氨酸已被廣泛用作飼料添加劑、用于醫療藥品諸如輸注溶液的原料、以及用于健康食品的材料,并已經通過化學合成、酶促反應、發酵等生產。近來,主要通過微生物發酵進行L-色氨酸的生產。在工業化初始階段,已經主要使用的是經化學突變獲得的類似物抗性菌株。然而,由于20世紀90年代基因重組技術的快速發展并在分子水平理解了調控機制,已經主要使用經基因工程技術獲得的重組大腸桿菌(E.coli)和棒狀桿菌(Corynebacterium)菌株。通過微生物生產色氨酸是從PEP(磷酸烯醇式丙酮酸(PhospoEnolPyruvate))與E4P(赤蘚糖-4-磷酸(Erythrose-4-Phosphate))的聚合產生的DAHP(3-脫氧D-阿拉伯庚酮糖酸-7-磷酸(3-DeoxyD-arabino-heptulosonate-7-phosphate))起始的,PEP是糖酵解(glycolysis)的中間產物,E4P是磷酸戊糖途徑的中間產物。然后,從分支酸(chorismate)經共同的芳香族生物合成途徑來生物合成色氨酸。具體地,色氨酸的合成通過trpE基因編碼的鄰氨基苯甲酸合酶(anthranilatesynthase,EC4.1.3.27),trpD基因編碼的鄰氨基苯甲酸合酶(anthranilatesynthase,EC4.1.3.27)和鄰氨基苯甲酸PRPP轉移酶(anthranilatePRPPtransferase,EC2.4.1.28),trpC基因編碼的吲哚-3-甘油磷酸合酶(indole-3-glycerolphosphatesynthase,EC4.1.1.48)和磷酸核糖基鄰氨基苯甲酸異構酶(phosphoribosylanthranilateisomerase,EC5.3.1.24),以及trpB基因和trpA基因編碼的色氨酸合酶(tryptophansynthase,EC4.2.1.20)。介導上述反應的基因簇trpEDCBA位于染色體中并具有含單一調控區的操縱子結構。色氨酸操縱子主動轉錄以產生細胞所需的足夠量的色氨酸。然而,如果細胞中的色氨酸水平高了,則阻遏物(repressor)就結合到色氨酸上,然后通過阻遏物結合到操縱子調控區而使色氨酸操縱子失活,從而抑制轉錄。此外,用于生物合成氨基酸(諸如蘇氨酸、苯丙氨酸、亮氨酸、色氨酸和組氨酸)的操縱子有另一種調控機制,被稱為弱化(attenuation)(JBacteriol.(1991)173,2328-2340)。如本領域關于弱化所已知的,在氨基酸不足的情況下,mRNA中與染色體上操縱子中啟動子與第一基因之間的特定序列區相對應的結構變成有利于翻譯過程的結構以促進生物合成基因的表達,但是在氨基酸豐富的情況下,短的轉錄mRNA形成稱為“發夾結構(hairpinstructure)”的三維結構以抑制翻譯過程(JBiolChem.,(1988)263:609-612)。在L-色氨酸生產菌株的初始開發階段,主要目的是通過增強酶活性來提高效率,或者通過解除由終產物色氨酸引起的對色氨酸生物合成途徑酶的反饋抑制,或者通過增加染色體上或者載體形式的色氨酸操縱子基因的拷貝數以增強色氨酸生物合成酶的表達(Appl.Environ.Microbiol.,(1982)43:289-297;Appl.Microbiol.Biotechnol.,(1993)40:301-305;TrendsBiotechnol.,(1996)14:250-256)。賦予微生物L-色氨酸生產能力的方法包括經化學突變賦予對色氨酸類似物或作為中間產物的鄰氨基苯甲酸抗性的方法,或者經基因工程修飾微生物的方法。化學突變方法的實例包括韓國專利注冊號1987-0001813、韓國專利注冊號0949312等中描述的那些方法,基于基因工程的修飾方法的實例包括使用以下菌株的各種方法,通過增強芳香族氨基酸生物合成途徑中編碼轉酮醇酶(transketolase)的tktA基因或編碼半乳糖透性酶(galactosepermease)的galP基因以增加E4P(赤蘚糖-4-磷酸(Erythrose4-phosphate)或PEP(磷酸烯醇式丙酮酸(phosphoenolpyruvate))的供應以及通過減少DAHP(3-脫氧-D-阿拉伯庚酮糖酸-7-磷酸(3-deoxy-D-arabino-heptulosonate-7-phosphate))的反饋抑制以增強芳香族生物合成途徑而獲得的菌株(TrendsBiotechnol.,(1996)14:250-256,MicrobialCellFactories(2009)8:19),或者通過另外引入色氨酸操縱子基因到載體或染色體中而獲得的菌株(Appl.Environ.Microbiol.,(1982)43:289-297,Appl.Microbiol.Biotechnol.,(1993)40:301-305)。然而,雖然引入色氨酸操縱子并解除生物合成酶的反饋抑制,但由于調控機制(例如轉錄水平的操縱子基因的抑制或弱化的緣故),這些方法未能實現色氨酸產量的增加。
技術實現要素:
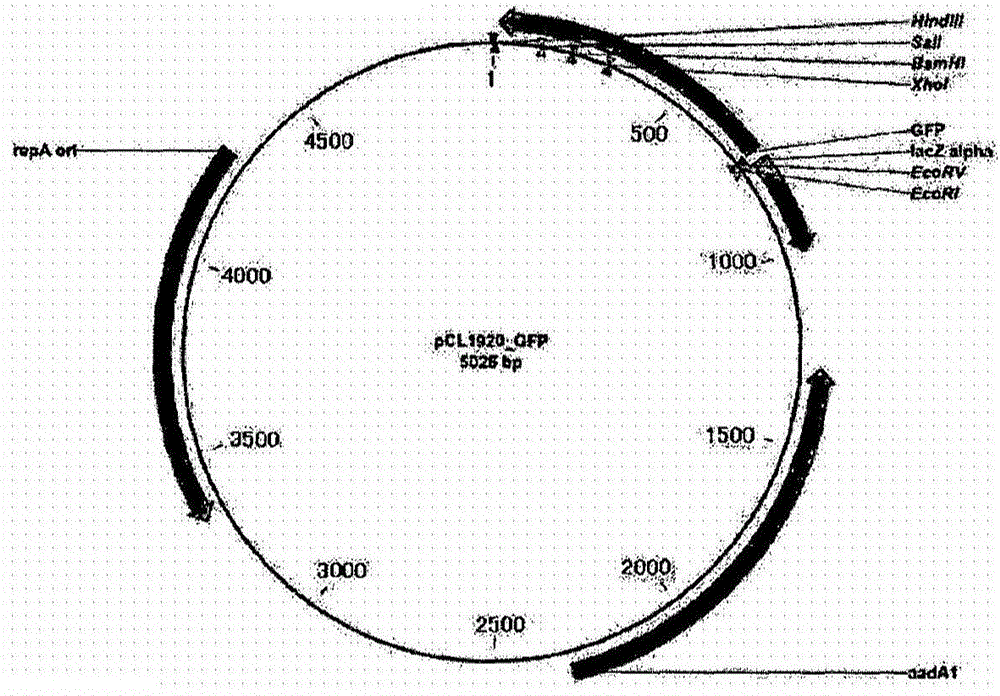
技術問題本發明人已開發了使L-色氨酸生產菌株在轉錄水平解除色氨酸操縱子基因的抑制或弱化的方法,以及能夠使用其增強色氨酸生物合成酶的方法。此外,為解決當增強色氨酸操縱子時由于鄰氨基苯甲酸積累而導致色氨酸生產菌株產量不增加的問題,本發明人已通過表達色氨酸操縱子基因中除編碼鄰氨基苯甲酸合酶(anthranilatesynthase,TrpE)的基因以外的基因簇,以對調控機制(諸如反饋抑制或抑制機制)脫敏的形式構建了具有增加的產量和低水平的鄰氨基苯甲酸積累的色氨酸生產菌株。本發明的一個目的是提供具有增強的L-色氨酸生產力的埃希氏菌(Escherichia)屬微生物,其通過修飾而對色氨酸操縱子的抑制或弱化脫敏并減少鄰氨基苯甲酸的積累。本發明的另一目的是提供使用所述埃希氏菌屬微生物生產L-色氨酸的方法。技術方案為實現以上目的,本發明的一個實施方案提供了具有增強的L-色氨酸生產力的埃希氏菌屬重組微生物,其已被修飾為缺失內源色氨酸操縱子的表達調控區中前導肽(leaderpeptide)的部分或全部,所述表達調控區具有SEQIDNO:1所示核苷酸序列,所述前導肽具有SEQIDNO:2所示核苷酸序列。本發明的另一實施方案還提供了生產L-色氨酸的方法,其包括培養上述埃希氏菌屬的重組微生物。有益效果根據本發明產生的重組微生物消除了其中鄰氨基苯甲酸的過度積累,并且可有利地用于以高產率生產L-色氨酸。附圖說明圖1示出了表示大腸桿菌染色體中的色氨酸操縱子基因、所述基因的調控區以及本發明所述的基因缺失形式的示意圖。A)大腸桿菌染色體中的色氨酸操縱子基因及其調控區(Ptrp);B)Ptrp形式;C)缺失了編碼前導肽的trpL基因的形式(DtrpL);以及D)缺失了編碼前導肽和弱化子的trpL基因的形式(Dtrpatt)。圖2示出了用于測量色氨酸操縱子表達調控區強度的pCL-GFP載體。圖3示出了用于將色氨酸生物合成基因trpDCBA引入到染色體中以增加基因拷貝數的載體pINT17E-Patt-trpDCBA。發明方式以下將詳細描述本發明。本發明的一個實施方案提供了具有增強的L-色氨酸生產力的埃希氏菌屬重組微生物,其已被修飾為缺失了內源色氨酸操縱子的表達調控區中前導肽的部分或全部,所述表達調控區具有SEQIDNO:1所示核苷酸序列,所述前導肽具有SEQIDNO:2所示核苷酸序列。本文使用的術語“色氨酸操縱子(tryptophanoperon)”或“Trp操縱子(Trpoperon)”意指包括所有trpEDCBA基因的整個操縱子。所述色氨酸操縱子具有SEQIDNO:9所示的核苷酸序列。本發明中可使用的L-色氨酸生產微生物可以是任何原核或真核微生物,只要它們具有L-色氨酸生產力。該微生物的實例可包括屬于埃希氏菌(Escherichia)屬、歐文氏菌(Erwinia)屬、沙雷菌(Serratia)屬、普羅威登斯菌(Providencia)屬、棒狀桿菌(Corynebacterium)屬以及短桿菌(Brevibacterium)屬的微生物。特別地,所述微生物是屬于埃希氏菌屬的微生物,更特別地是大腸桿菌。最特別地,本發明的大腸桿菌菌株可以是通過以下方式獲得的菌株:增強色氨酸生物合成酶(諸如鄰氨基苯甲酸合酶(TrpE)、鄰氨基苯甲酸PRPP轉移酶(TrpD)、磷酸核糖鄰氨基苯甲酸異構酶(TrpC)或色氨酸合酶(TrpA,TrpB))的活性同時維持解除了反饋抑制的3-脫氧-D-阿拉伯庚酮糖酸-7-磷酸合酶(aroG),增強芳香族生物合成途徑酶(諸如3-脫氫奎尼酸合酶(aroB)、莽草酸脫氫酶(aroE)、莽草酸激酶(aroL)、5-烯醇丙酮莽草酸-3-磷酸合酶(aroA)或分支酸合酶(aroC))的活性,增強磷酸甘油酸脫氫酶(serA)或轉酮醇酶(tktA)的活性以增強色氨酸生物合成途徑中間產物絲氨酸和PRPP的供應。此外,本發明的大腸桿菌菌株可以是通過失活芳香族生物合成途徑的預苯酸脫水酶和分支酸變位酶(pheA)的活性或者失活預苯酸脫氫酶、分支酸變位酶(tyrA),色氨酸酶(tnaA)和色氨酸轉運蛋白(tnaB,mtr)的活性而獲得的菌株。更特別地,本發明的重組微生物是重組大腸桿菌菌株CA04-2004(登記號KCCM11246P)。本文使用的術語內源色氨酸操縱子的“表達調控區(expressionregulatoryregion)”意指包括啟動子(promoter)、前導肽(leaderpeptide)和內源弱化子(attenuator)的區域。特別地,所述表達調控區具有SEQIDNO:1所示的核苷酸序列。本文使用的術語“前導肽(leaderpeptide)”意指由基因起始密碼子上游前導序列所編碼的低分子量肽。特別地,所述前導肽具有SEQIDNO:2所示的核苷酸序列,由前導肽表達的多肽可以是SEQIDNO:4所示的氨基酸序列。該前導肽的功能是在色氨酸濃度高時形成發夾結構從而促進內源弱化子的結構形成以終止色氨酸生物合成基因的轉錄。本文使用的術語“缺失(delete)”意指從染色體上去除從靶基因起始密碼子到終止密碼子的核苷酸序列、或者其調控區的核苷酸序列的部分或全部。本發明的一個方面也提供了埃希氏菌屬的重組微生物,其已被進一步修飾為缺失了內源弱化子的(attenuator)部分或全部以增強其L-色氨酸生產能力,所述內源弱化子具有SEQIDNO:3所示核苷酸序列。本文使用的術語“內源弱化子(endogenousattenuator)”意指導致弱化機制的具有SEQIDNO:3所示核苷酸序列的區域,其不包括表達調控區的啟動子和前導肽。本發明的一個方面也提供了埃希氏菌屬的微生物,其已被進一步修飾為增強由色氨酸操縱子編碼的蛋白質的活性。本發明的一個方面也提供了埃希氏菌屬的微生物,其已被進一步修飾為增強由排除了編碼鄰氨基苯甲酸合酶的trpE基因的色氨酸生物合成基因簇trpDCBA所編碼的蛋白質的活性。本文使用的術語“色氨酸生物合成基因簇(tryptophanbiosyntheticgenecluster)”意指由色氨酸操縱子基因trpE、trpD、trpC、trpB和trpA的兩種或更多種的組合所組成的基因簇。特別地,色氨酸生物合成基因簇可以是具有SEQIDNO:10所示核苷酸序列的trpDCBA基因簇。其中,trpD基因編碼具有SEQIDNO:37所示氨基酸序列的蛋白質;trpC基因編碼具有SEQIDNO:38所示氨基酸序列的蛋白質;trpB基因編碼具有SEQIDNO:39所示氨基酸序列的蛋白質;而trpA基因編碼具有SEQIDNO:40所示氨基酸序列的蛋白質。對除了色氨酸操縱子中由trpE基因編碼的鄰氨基苯甲酸合酶以外的色氨酸生物合成基因簇的活性進行增強來解決當增強色氨酸操縱子時由于鄰氨基苯甲酸的積累導致色氨酸產量不增加的問題。增強所述基因之表達的方法包括:1)增加所述基因的染色體或細胞內拷貝數的方法;或2)用強的外源啟動子替換所述基因的染色體啟動子或者修飾染色體啟動子為強啟動子的方法。增加拷貝數的方法的實例包括將基因引入載體以增強基因表達的方法。本發明中可以使用的載體的實例包括質粒載體,例如pBR、pUC、pBluescriptII、pGEM、pTZ、pCL和pET型質粒。本發明可使用的載體不特別限定,可以使用任何已知的表達載體。特別地,可以使用pACYC177、pACYC184、pCL、pECCG117、pUC19、pBR322、pMW118或pCC1BAC載體。最特別地,可以使用pACYC177、pCL和pCC1BAC載體。同時,本發明可使用的外源啟動子的實例包括但不限于已知啟動子,諸如trc、lac和tac啟動子。此外,如上所述,可以通過缺失前導肽的部分或全部和/或進一步缺失內源弱化子的部分或全部來修飾染色體啟動子為強啟動子,但并不限于此。在本發明的一個特定實施方案中,埃希氏菌屬重組L-色氨酸生產微生物是通過缺失內源色氨酸操縱子上具有SEQIDNO:1所示核苷酸序列的表達調控區中具有SEQIDNO:2所示核苷酸序列的前導肽的部分或全部,并且缺失具有SEQIDNO:3所示核苷酸序列的內源弱化子的部分或全部以提高生產L-色氨酸的能力而產生。此外,通過進一步增強具有SEQIDNO:37、38、39和40所示氨基酸序列的蛋白質的活性產生重組L-色氨酸生產微生物,其由排除了編碼鄰氨基苯甲酸合酶的trpE基因的色氨酸生物合成基因簇trpDCBA所編碼。將所產生的重組微生物保藏為登記號KCCM11246P。本發明的另一實施方案還提供了生產L-色氨酸的方法,其包括培養具有增強的L-色氨酸生產力的埃希氏菌屬重組L-色氨酸生產微生物。在一個特定方面,本發明提供了一種方法,其包括培養具有增強的L-色氨酸生產力的埃希氏菌屬重組L-色氨酸生產微生物,其已被修飾為缺失了內源色氨酸操縱子上具有SEQIDNO:1所示核苷酸序列的表達調控區中具有SEQIDNO:2所示核苷酸序列的前導肽的部分或全部,并缺失了具有SEQIDNO:3所示核苷酸序列的內源弱化子的部分或全部以增強L-色氨酸生產能力,并且通過增強已排除編碼鄰氨基苯甲酸合酶的trpE基因的色氨酸生物合成基因簇trpDCBA的表達來進一步增強色氨酸操縱子所編碼的蛋白質的活性。本發明微生物培養中使用的培養基和培養條件可以是用于屬于埃希氏菌屬微生物培養的培養基和培養條件中任意那些,但是這些培養基和培養條件合適地滿足本發明微生物的需求。特別地,可以在含有合適的碳源、氮源、氨基酸、維生素等的常規培養基中于好氧條件下同時調節溫度、pH等培養本發明的微生物。可用于本發明的碳源包括碳水化合物,諸如葡萄糖、果糖、蔗糖、麥芽糖、甘露糖醇、山梨糖醇;醇類,諸如糖醇、甘油、丙酮酸、乳酸和檸檬酸;以及氨基酸,諸如有機酸、谷氨酸、甲硫氨酸和賴氨酸。此外,可以使用天然有機營養源,諸如淀粉水解物、糖蜜、赤糖糊、米糠、木薯、甘蔗渣和玉米漿。特別地,可以使用碳水化合物,諸如葡萄糖和滅菌預處理的糖蜜(即轉化為還原糖的糖蜜)。此外,可以不受限制地使用合適量的其它碳源。可用于本發明的氮源包括無機氮源,諸如氨、硫酸銨、氯化銨、乙酸銨、碳酸銨和硝酸銨;氨基酸,諸如谷氨酸、甲硫氨酸和谷氨酰胺;以及有機氮源,諸如蛋白胨、NZ胺、肉提取物、酵母提取物、麥芽提取物、玉米漿、酪蛋白水解物、魚粉或其消化產物、脫脂大豆餅或其消化產物等。可以單獨或組合使用這些氮源。培養基可含有磷酸二氫鉀、磷酸氫二鉀以及相應的含鈉鹽作為磷源。可用于本發明的無機化合物包括氯化鈉、氯化鈣、氯化鐵、硫酸鎂、硫酸鐵、硫酸錳和碳酸鈣。此外,培養基可含有氨基酸、維生素和合適的前體。這些來源或前體可以以分批或連續方式添加到培養基中。諸如氫氧化銨、氫氧化鉀、氨、磷酸和硫酸的化合物可以在培養期間以合適方式添加至培養基以調節培養基的pH。此外,在培養期間,可以使用消泡劑(諸如脂肪酸聚乙二醇酯)來抑制氣泡的形成。進一步地,為保持培養基的好氧狀態,可將氧氣或含氧氣體注入培養基中。此外,為保持培養基的厭氧或無氧狀態,不注入氣體,或者可以將氮氣、氫氣或二氧化碳氣體注入培養基中。通常將培養基保持在27℃至37℃,特別是30℃至35℃的溫度范圍。微生物的培養可持續直至獲得期望水平的有用物質。特別地,培養周期可以是10至100小時。本發明的方法可進一步包括純化或回收培養步驟中產生的L-氨基酸。純化或回收過程可以通過使用合適的方法(例如分批、連續或分批補料的培養方法)從培養基中純化或回收所述期望的L-氨基酸來進行。在下文中,將參照實施例進一步詳細描述本發明。然而能夠理解的是,這些實施例是用于說明目的,而非旨在限制本發明的范圍。實施例實施例1:包含GFP和去除前導肽的表達調控區的融合載體的構建以解除色氨酸生物合成的表達調控如圖1所示,在由啟動子(P)、前導肽(L)和弱化子(A)構成的色氨酸操縱子的表達調控區中,為擴增包含編碼前導肽(L)之trpL基因的缺失的表達調控區(該表達調控區在下文中稱為“DtrpL”,對應于圖1C),使用大腸桿菌W3110菌株(購自美國典型微生物菌種保藏中心(AmericanTypeCultureCollection):ATCC);GenBank登記號AC000091)的染色體DNA作為模板進行聚合酶鏈式反應(polymerasechainreaction)(在下文中稱為“PCR”)。具體地,使用引物1和2用Pfu聚合酶在以下條件下通過PCR擴增5’區具有KpnI限制性內切酶位點的155bp片段:30個循環,每個循環由94℃變性1分鐘、58℃退火30秒以及72℃延伸30秒組成。同時,使用引物3和4在上述條件下通過PCR擴增3’區具有EcoRV限制性內切酶位點的105bp片段。使用GeneAllRExpinTMGELSV試劑盒(韓國首爾)回收所獲得的DNA片段,然后用作交叉PCR的模板。為制備DtrpL,使用兩個DNA片段作為模板以及引物1和4進行交叉PCR。具體地,在上述條件下通過PCR擴增245bp片段(SEQIDNO:5)。用限制性內切酶KpnI和EcoRV處理擴增片段,然后與經同樣限制性內切酶處理的pCL1920GFP(SEQIDNO:8)連接(ligation),從而構建pCL-DtrpL_GFP。為擴增在色氨酸操縱子表達調控區中包含編碼前導肽(L)和弱化子(A)之基因的缺失的表達調控區(該表達調控區在下文中稱為“Dtrp_att”),使用大腸桿菌W3110菌株的染色體DNA作為模板以及引物1和5通過PCR擴增5’區具有KpnI限制性內切酶位點和3’區具有EcoRV限制性內切酶位點的148bp片段(SEQIDNO:6)。用限制性內切酶KpnI和EcoRV處理擴增片段,然后與經同樣限制性內切酶處理的pCL1920GFP連接,從而構建pCL-Dtrp_att-GFP。此外,為制備在后續實驗中用作對照的具有野生型表達調控區的載體,使用大腸桿菌W3110菌株的染色體DNA作為模板以及引物1和4通過PCR擴增5’區具有KpnI限制性內切酶位點和3’區具有EcoRV限制性內切酶位點的290bp片段。用限制性內切酶KpnI和EcoRV處理擴增片段,然后與經同樣限制性內切酶處理的pCL1920GFP連接,從而構建pCL-Ptrp-GFP。引物15′TTAGGTACCGGCGCACTCCCGTTCTGGATA3′(SEQIDNO11)引物25′ACTGCCCGTTGTCGATACCCTTTTTACGT3′(SEQIDNO12)引物35′TCGACAACGGGCAGTGTATTCACCATG3′(SEQIDNO13)引物45′AATGATATCTGTTATTCTCTAATTTTGTT3′(SEQIDNO14)引物55′AATGATATCACCCTTTTTACGTGAACTTG3′(SEQIDNO15)實施例2:GFP表達水平的測量將實施例1中制備的pCL-DtrpL_GFP、pCL-Dtrp_att-GFP和pCL-Ptrp_GFP的每一種轉化到野生型大腸桿菌W3110和色氨酸生產菌株大腸桿菌KCCM10812P中,然后測量菌株中GFP的強度。本實施例中使用的親本菌株大腸桿菌KCCM10812P(韓國專利注冊號10-0792095)是源自具有L-苯丙氨酸生產力的大腸桿菌變體(KFCC10066,韓國專利公開號1985-0001232)。具體地,KCCM10812P是具有L-色氨酸生產力的重組大腸桿菌菌株,其中所述菌株已被修飾為恢復色氨酸營養缺陷,從而失活pheA、trpR、mtr和tnaAB基因并突變aroG和trpE基因。具體地,以1/100(v/v)體積比將每一株所述菌株接種到250ml燒瓶中的25mlM9培養基(含有0.5%葡萄糖+2g/L酵母提取物,對于KCCM10812還含有0.1g/L酪氨酸和0.1g/L苯丙氨酸)中,并在37℃下培養直至達到預定OD值。通過離心回收所培養的菌株,并用1×TE洗滌一次,使用SynergyHTMulti-ModeMicroplateReader(Biotek,USA)測量其中的GFP。測量結果示于下表1中。表1中的OD1和OD3指示在將每種培養產物稀釋到合適濃度后使用UVmini-1240分光光度計(Shimadzu)在600nm處測得的OD值。如圖1所示,在野生型W3110菌株中,如將Ptrp的相對強度定為1,則Dtrp_att(包含前導肽和弱化子的缺失)的相對強度在OD值為1(OD1)時是約7倍,在OD值為3(OD3)時是約10倍,而僅包含前導肽之缺失的DtrpL的相對強度比野生型調控區(Ptrp)的相對強度高約1.5至2倍。與此相比,在L-色氨酸生產菌株KCCM10812P中,如將Ptrp的相對強度定為1,Dtrp_att(包含前導肽和弱化子的缺失)的相對強度在OD值為1(OD1)時是約19倍,在OD值為3(OD3)時是約27倍,而僅包含前導肽缺失的DtrpL的相對強度比野生型調控區(Ptrp)的相對強度高約4倍。這樣的結果表明,前導肽或弱化子的缺失導致了表達的增加,盡管野生型菌株中的這種表達增加比L-色氨酸生產菌株中的表達增加要弱。[表1]實施例3:具有其表達調控區已被替換之色氨酸操縱子(trpEDCBA)的載體的構建基于實施例2的結果,為構建使用載體增強其色氨酸操縱子基因的大腸桿菌菌株,使用親本菌株大腸桿菌KCCM10812P的染色體DNA作為模板以及引物6和7在上述PCR條件下擴增6564bp片段(SEQIDNO:9)。使用GeneAllRExpinTMGELSV試劑盒(韓國首爾)回收擴增的DNA片段,然后用限制性內切酶EcoRV和HindIII處理。為使用所制備的DNA片段進行克隆,用EcoRV和HindIII處理pCL-Dtrp_att-GFP、pCL-DtrpL_GFP和pCL-Ptrp_GFP載體的每一個以去除GFP區域,從而獲得4291bp片段。用插入片段(insert)連接每種所制備載體,然后通過轉化將其引入到大腸桿菌DH5α中,從而構建pCL-Dtrp_att-trpEDCBA、pCL-DtrpL_trpEDCBA和pCL-Ptrp_trpEDCBA載體。引物65′CCCGATATCATGCAAACACAAAAACCGAC3′(SEQIDNO16)引物75′GGGAAGCTTAAAGGATCCGTGGGATTAACTGCGCGTCGCCGCTTT3′(SEQIDNO17)實施例4:其表達調控區被替換并且具有排除了trpE的色氨酸生物合成基因簇(trpDCBA)的載體的構建為通過用trpDCBA替換實施例1中制備的pCL-Dtrp_att-GFP、pCL-DtrpL-GFP和pCL-Ptrp_GFP載體的GFP區域構建載體,用EcoRV和HindIII處理pCLDtrp_att-GFP、pCL-DtrpL-GFP和pCL-Ptrp_GFP載體的每一種以去除GFP區域,從而獲得4291bp片段。然后,為構建使用載體增強其色氨酸操縱子trpDCBA基因的大腸桿菌菌株,使用親本菌株大腸桿菌KCCM10812P的染色體DNA作為模板以及引物7和8通過PCR擴增5002bp片段(SEQIDNO:10)。使用GeneAllRExpinTMGELSV試劑盒(韓國首爾)回收擴增的DNA片段,然后用限制性內切酶EcoRV和HindIII處理。將所制備的載體與插入片段互相連接,然后通過轉化將其引入到大腸桿菌DH5α中,從而構建pCL-Dtrp_att-trpDCBA、pCL-DtrpL-trpDCBA和pCL-Ptrp_trpDCBA載體。引物85′AAAGATATCATGGCTGACATTCTGCTGCT3′(SEQIDNO18)實施例5:具有低拷貝數的含多種表達調控區的色氨酸操縱子基因的載體的構建在大腸桿菌中以低拷貝數表達的典型載體是pCC1BAC(Epicentre,USA)。為使用該載體以低拷貝數表達色氨酸操縱子基因,用限制性內切酶HindIII消化實施例3和4中制備的pCL-Dtrp_att-trpEDCBA、pCL-DtrpL-trpEDCBA、pCL-Ptrp_trpEDCBA、pCL-Dtrp_att-trpDCBA、pCL-DtrpL-trpDCBA和pCL-Ptrp_trpDCBA。將所得到的DNA片段在瓊脂糖上電泳,然后根據它們的大小切下并使用GeneAllRExpinTMGELSV試劑盒(韓國首爾)回收。接下來,用pCC1BAC載體(在HindIII位點消化)連接每種片段,然后經轉化將其引入到大腸桿菌DH5α中。在LBCm固體培養基(LB+氯霉素瓊脂平板)上涂片每種經轉化菌株,選擇具有Cm抗性的菌株,從而構建pBAC-Dtrp_att-trpEDCBA、pBAC-DtrpL-trpEDCBA、pBAC-Ptrp_trpEDCBA、pBAC-Dtrp_att-trpDCBA、pBAC-DtrpL-trpDCBA和pBAC-Ptrp_trpDCBA載體。實施例6:其中pheA基因被失活的大腸桿菌菌株的構建為了從野生型大腸桿菌W3110菌株構建接近于色氨酸生產菌株的菌株,通過經同源重組的缺失失活編碼分支酸變位酶(chorismatemutase)/預苯酸脫水酶(prephenatedehydratase,CM-PDT)的pheA基因(NCBI基因ID:12934467)。CM-PDT是從分支酸生產苯丙氨酸的第一步中的酶,而pheA基因的缺失被用來抑制苯丙氨酸生物合成途徑。對于該缺失,使用一步失活方法(由DatsenkoKA等開發),其為使用λred重組酶(lambdaredrecombinase)的誘變技術(One-stepinactivationofchromosomalgenesinEscherichiacoliK-12usingPCRproducts,DatsenkoKA,WannerBL.,ProcNatlAcadSciUSA.2000年6月6日;97(12):6640-5)。使用pUCprmfmloxP的氯霉素抗性基因作為確認插入到基因中的標記(韓國專利特許公開,公開號2009-0075549)。使用載體pUCprmfmloxP作為模板以及引物9和10通過PCR擴增約1200bp的基因片段,其具有pUCprmfmloxP載體的pheA基因的一部分和氯霉素抗性基因核苷酸序列的一部分。引物95′-GGCCTCCCAAATCGGGGGGCCTTTTTTATTGATAACAAAAAGGCAACACTAGGTGACACTATAGAACGCG-3′(SEQIDNO19)引物105′-AACAGCCCAATACCTTCATTGAACGGGTGATTTCCCCTAACTCTTTCAATTAGTGGATCTGATGGGTACC-3′(SEQIDNO20)將通過PCR擴增所獲得的DNA片段在0.8%瓊脂糖凝膠上電泳,然后洗脫并用作二次PCR的模板。進行二次PCR以使得初次DNA片段的5’和3’區具有20bp互補核苷酸堿基對。此外,使用洗脫的初次PCR產物作為模板以及引物11和12通過PCR擴增約1300bp的基因片段,其包含pheA基因的5’和3’區。將所得到的DNA片段在0.8%瓊脂糖凝膠上電泳,然后洗脫并用于重組。引物115′-GAATGGGAGGCGTTTCGTCGTGTGAAACAGAATGCGAAGACGAACAATAAGGCCTCCCAAATCGGGGGGC-3′(SEQIDNO21)引物125-GGCACCTTTTCATCAGGTTGGATCAACAGGCACTACGTTCTCACTTGGGTAACAGCCCAATACCTTCATT-3′(SEQIDNO22)根據DatsenkoKA等開發的方法,將用pKD46載體轉化的W3110大腸桿菌菌株制備為感受態,然后用經PCR獲得的1300bp基因片段轉化。在LB培養基上選擇具有氯霉素抗性的菌株。使用引物13和14進行PCR,PCR擴增產物具有約2500bp的大小,表明菌株中的pheA基因被缺失了。引物135′-TTGAGTGTATCGCCAACGCG-3′(SEQIDNO23)引物145′-AAAGCCGCGTGTTATTGCGT-3′(SEQIDNO24)從具有氯霉素抗性的初次重組菌株中去除pKD46載體,然后將pJW168載體引入到菌株中,并從菌株中去除氯霉素標記基因(Gene,(2000)247,255-264)。所得菌株使用引物13和14經PCR獲得約500bp的擴增產物,表明實現了靶基因的缺失。將所構建的菌株命名為“大腸桿菌W3110trpΔ1”。實施例7:tnaAB基因從其被失活的大腸桿菌菌株的構建經同源重組從實施例6所構建的大腸桿菌W3110trpΔ1菌株中缺失tnaAB操縱子(NCBI基因ID:12933600,12933602),該操縱子由編碼色氨酸酶的tnaA基因和編碼色氨酸輸入蛋白(importer)的tnaB基因構成。由于該缺失,色氨酸產生后的降解途徑可以被封閉,并且分泌到培養基中的色氨酸向細胞的流入可以被避免,從而賦予了色氨酸生產菌株的特性。對于該缺失,使用載體pUCprmfmloxP作為模板與引物15和16一起以實施例6所述同樣的方式通過PCR擴增約1200bp的基因片段,其具有pUCprmfmloxP載體tnaAB基因的一部分以及氯霉素抗性基因核苷酸序列的一部分。此外,將由PCR擴增獲得的DNA片段使用引物17和18以實施例6所述同樣方式經PCR進一步擴增,從而獲得1300bp的基因片段。引物155′-TTAGCCAAATTTAGGTAACACGTTAAAGACGTTGCCGAACCAGCACAAAAAGGTGACACTATAGAACGCG-3′(SEQIDNO25)引物165′-ATGAAGGATTATGTAATGGAAAACTTTAAACATCTCCCTGAACCGTTCCGTAGTGGATCTGATGGGTACC-3′(SEQIDNO26)引物175′-TGATTTCCTGAGAGGCAAGAAGCCAGCGAATGGCTGGCTTCTTGAAGGATTTAGCCAAATTTAGGTAACA-3′(SEQIDNO27)引物185′-AATCGGTATAGCAGATGTAATATTCACAGGGATCACTGTAATTAAAATAAATGAAGGATTATGTAATGGA-3′(SEQIDNO28)為缺失tnaAB基因,以實施例6所述同樣方式構建引入了載體pKD46的大腸桿菌菌株W3110trpΔ1并使其成為感受態,然后將由PCR獲得的1300bp基因片段轉化到大腸桿菌菌株中。在LB培養基上選擇具有氯霉素抗性的菌株。使用引物19和20進行PCR,PCR擴增產物具有約5400bp的大小,表明菌株中的tnaAB基因已被缺失。引物195′-CGGGATAAAGTAAAACCAGG-3′(SEQIDNO29)引物205′-CGGCGAAGGTAAGTTGATGA-3′(SEQIDNO30)以實施例6所述同樣方式從具有氯霉素抗性的初次重組菌株中去除pKD46載體,然后從菌株中去除氯霉素標記基因。所得菌株使用引物19和20經PCR獲得約550bp的擴增產物,表明實現了所期望基因的缺失。所構建的菌株命名為“大腸桿菌W3110trpΔ2”。實施例8:鑒定具有含多種表達模式的色氨酸操縱子的菌株的L-色氨酸生產力用根據實施例3、4和5中所述方法制備的載體轉化大腸桿菌菌株。使用實施例6和7中制備的W3110trpΔ2作為親本菌株評估大腸桿菌變體的影響,其碳源是葡萄糖。為評估效價,通過接種環接種每個菌株并在LB固體培養基上過夜培養。然后,將一接種環的每個菌株接種到25mL含葡萄糖的培養基中,所述培養基的組成示于下表2中。接種后,每個菌株在37℃和200rpm孵育48小時。結果示于下表3中。所有結果是以三個燒瓶的結果的平均值記錄的。[表2]組成濃度(每升)葡萄糖2gKH2PO41g(NH4)2SO412gNaCl1gNa2HPO4·H2O5gMgSO4·H2O1gMnSO4·H2O15mgCuSO4·H2O3mgZnSO4·H2O30mg檸檬酸鈉1g酵母提取物1g苯丙氨酸0.15gpH6.8[表3]從上表3的結果可見,在用多種載體的組合轉化親本菌株大腸桿菌W3110trpΔ2的情況下,如果只有色氨酸操縱子持續地被增強,則不會在鄰氨基苯甲酸積累的同時出現對色氨酸產量的正面影響。相反,與僅增強色氨酸操縱子的菌株相比,被修飾為增強了Trp操縱子和trpDCBA的菌株顯示了對色氨酸產量的正面影響以及鄰氨基苯甲酸積累的減少。因此,確認了生產色氨酸的菌株中鄰氨基苯甲酸積累的減少是增加L-色氨酸產量的有效方式。實施例9:鑒定具有含多種表達模式的色氨酸操縱子的菌株的L-色氨酸生產力根據下表5所示的組合,將根據實施例3、4和5中所述方法構建的載體引入到生產L-色氨酸的的親本菌株大腸桿菌KCCM10812P中。使用葡萄糖作為碳源評估菌株的效價。結果,看起來不僅trpDCBA的增強是重要的,而且色氨酸操縱子的增強也是重要的,與實施例8的結果類似。由此評估了對色氨酸生產菌株的影響。為評估效價,通過接種環接種每個菌株并在LB固體培養基上過夜培養。然后,將一接種環的每個菌株接種到25mL含葡萄糖的培養基中,所述培養基的組成示于下表4中。接種后,每個菌株在37℃和200rpm孵育48小時。結果示于下表5中。所有結果是以三個燒瓶的結果的平均值記錄的。[表4]組成濃度(每升)葡萄糖60gK2HPO41g(NH4)2SO410gNaCl1gMgSO4·H2O1g檸檬酸鈉5g酵母提取物2g碳酸鈣40g檸檬酸鈉5g苯丙氨酸0.15g酪氨酸0.1gpH6.8[表5]*在33小時測量**在48小時測量從上表5的結果可見,在用多種載體的組合轉化親本菌株大腸桿菌KCCM10812P的情況下,如果只有色氨酸操縱子持續地被增強,則不會在鄰氨基苯甲酸積累的同時出現對色氨酸產量的正面影響。相反,與僅增強色氨酸操縱子的菌株相比,看起來通過使用pCL載體增強操縱子并且使用pBAC載體增強trpDCBA來修飾菌株顯示了對色氨酸產量的正面影響以及鄰氨基苯甲酸積累的減少。因此,確認了色氨酸產生菌株中鄰氨基苯甲酸積累的減少是增加L-色氨酸產量的有效方式。實施例10:菌株的構建,其中染色體中色氨酸生物合成基因簇trpDCBA的拷貝數增加并且鄰氨基苯甲酸的積累減少基于實施例9的結果,構建載體以增加染色體中色氨酸生物合成基因簇trpDCBA的拷貝數。具體地,用限制性內切酶EcoRI和BamHI切割實施例5中所述的pCL-Dtrp_att-trpDCBA以獲得Dtrp_att-trpDCBA,然后與經同樣限制性內切酶處理的pINT17E連接,從而獲得pINT17E-Patt-trpDCBA。為將pINT17E-Patt-trpDCBA引入到生產色氨酸的親本菌株大腸桿菌KCCM10812P中以增加色氨酸生物合成基因簇trpDCBA的拷貝數,根據實施例6在一步失活方法(由DatsenkoKA等開發)中使用pKD46,所述方法為使用λred重組酶的誘變技術。使用pUCprmfmloxP的氯霉素抗性基因作為確認插入到基因中的標記。具體地,用pINT17E-Patt-trpDCBA轉化已引入pKD46的親本菌株,然后在37℃培養1至2天以獲得菌落。為確認pINT17E-Patt-trpDCBA是否已正確地插入到所獲得菌落的染色體中,使用引物21和22通過PCR擴增約2000-bp的片段。引物215′TATTTGCTGTCACGAGCAGG3′(SEQIDNO31)引物225′AGTTCCGGCATACAACCGGCTT3′(SEQIDNO32)從具有氯霉素抗性的初次重組菌株中去除pKD46,然后將pJW168質粒引入以從菌株中去除氯霉素標記基因(Gene,(2000)247,255-264)。使用引物23和24經PCR獲得約5000bp的擴增產物,并使用引物25和26經PCR獲得約6500bp的擴增產物,這證明trpDCBA是連續地位于色氨酸操縱子之后,所述色氨酸操縱子內源位于染色體上。將該菌株命名為“KCCM10812P/trpDCBA”。引物235′TAATACGACTCACTATAGGG3′(SEQIDNO33)引物245′CTGTTGGGCGGAAAAATGAC3′(SEQIDNO34)引物255′TGATCGCCAGGGTGCCGACG3′(SEQIDNO35)引物265′CCCTATAGTGAGTCGTATTA3′(SEQIDNO36)為另外插入一個拷貝到以上制備的已增加trpDCBA拷貝數的菌株中,將pKD46引入到以上制備的KCCM10812P/trpDCBA菌株中。然后將pINT17E-Patt-trpDCBA載體引入到KCCM10812P/trpDCBA/pKD46中,從而構建具有兩個trpDCBA拷貝插入到染色體中的菌株。將該構建的菌株命名為“KCCM10812P/2trpDCBA”。將該菌株于2011年12月29日保藏在國際保藏單位韓國微生物培養中心(361-221,Hongje1-dong,Seodaemun-gu,Seoul,Korea),登記號為KCCM11246P。實施例11:L-色氨酸生產菌株效果的檢測,所述菌株具有增加的由色氨酸生物合成基因簇trpDCBA編碼的蛋白質活性根據實施例10中描述的方法,使用葡萄糖作為碳源評估KCCM10812P/trpDCBA的效價。KCCM10812P/trpDCBA的獲得是通過進一步將trpDCBA引入到色氨酸生產菌株大腸桿菌KCCM10812P中以增強色氨酸生物合成途徑一些酶的活性。為評估效價,通過接種環接種菌株并在LB固體培養基上過夜培養。然后,將一接種環的菌株培養物接種到每燒瓶25mL的效價培養基中,培養基的組成示于上表4中。接種后,將菌株在37℃和200rpm孵育48小時。結果示于下表6中。所有結果是以三個燒瓶的結果的平均值記錄的。[表6]*在33小時測量**在48小時測量如從上表6中可見的,當在染色體中插入一個拷貝的色氨酸生物合成基因簇trpDCBA時,與親本菌株相比,鄰氨基苯甲酸的濃度降低了39%。然而在染色體中插入兩個拷貝,與親本菌株相比,鄰氨基苯甲酸的濃度降低了69%。此外,兩種菌株中L-色氨酸濃度分別增加了10%和13%。如上表6所示,當trpDCBA的拷貝數增加時,葡萄糖消耗速率在一些情況下輕微降低,但是色氨酸生物合成基因簇的增強對L-色氨酸濃度的增加和鄰氨基苯甲酸濃度的降低具有正面影響。盡管已經參照特定說明性實施方案描述了本發明,但是本發明所屬領域技術人員能夠理解,本發明可以在不偏離本發明技術精神或實質特征的情況下以其它特定方式實施。因此,上述實施方案在所有方面都被認為是說明性的而非限制性的。此外,本發明的范圍是由所附權利要求書而不是具體說明書限定的,應當理解的是,源自本發明涵義和范圍的所有改變或變化及其等同形式都被包括在所附權利要求書的范圍內。登記號:保藏單位:韓國微生物培養中心(國際)登記號:KCCM11246P保藏日期:2011年12月29日
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1