一種結核分枝桿菌RNA/DNA同步定量分析方法與流程
本發明屬于生物工程檢測技術領域,具體涉及臨床標本中結核分枝桿菌的熒光定量檢測方法。
背景技術:
結核病(TB)是一種危害嚴重的慢性傳染病,全球有約20億人被感染,每年新增結核病患者達800-1000萬,每年因結核病死亡人數為200-300萬。我國結核病情況同樣較為嚴峻,年發病人數約為130萬,因結核病死亡人數每年達13萬,超過其它傳染病死亡人數的總和。我國是全球22個結核病流行嚴重的國家之一,同時也是全球27個耐多藥結核病流行嚴重的國家之一。我國結核病患病人數居世界第二位,僅次于印度。20世紀90年代以來,結核病群或高危人群的流動或移民以及結核病控制放松及艾滋病病毒(HIV)與結核分枝桿菌雙重感染蔓延等因素使結核病在全球的傳播有“死灰復燃”的趨勢。同時,耐藥、耐多藥(MDR)及超耐藥(XDR)結核病占結核病比率提高,導致多種一、二線抗結核藥物治療效果下降,診斷及治療成本上升,許多國家不同程度地出現了疫情下降緩慢或嚴重反彈的局面,發病率以每年1.1%的速度增長,結核病再次成為威脅人類健康的主要傳染病,帶來嚴重的公共衛生問題和重大的經濟社會問題。據世界衛生組織報道,目前全球有近1/3的人感染了結核桿菌,即20億人口感染了結核桿菌,其中活動性肺結核病人約2000萬,每年新增結核病人約800~1000萬,每年約有300萬人死于結核病。結核病已成為導致全世界成人因傳染病而死亡的主要疾病之一。我國是全球22個結核病高發國家之一,活動性肺結核病人數居世界第二位。結核病在我國流行的特點為“四高一低”:高患病率、高耐藥率、高死亡率、高感染率和低遞降率。據2000年全國結核病流行病學抽樣調查估計,全國現有活動性肺結核病人450萬,其中傳染性肺結核病人150萬。因此結核病防治是我國乃至全球疾病防治的重要課題。近年來世界各地結核分枝桿菌的多耐藥菌株逐漸增多,甚至引起暴發流行。結核分枝桿菌的耐藥可由自發突變產生(原發性耐藥)或由于用藥不當經突變選擇產生(繼發性耐藥)。目前濫用抗生素、過渡治療的現象很普遍,如何準確快速判斷藥物治療的藥效是目前急需解決的問題。臨床診斷肺結核的主要依據是檢測痰中結核分枝桿菌,其中,涂片法簡單易行,但陽性率較低;羅氏培養法雖然是金標準,但檢測周期長,均難以滿足臨床需要。因此提供敏感、特異、快速、可靠、簡便、適合臨床應用的實驗室檢查技術已成為國內外研究的重點。其中實時熒光PCR因其技術成熟,具有較高的靈敏性和特異性,已逐步應用到臨床中。實時熒光定量PCR技術,是指在PCR反應體系中加入一對引物的同時加入一個特異性熒光探針(TaqMan探針),目前常用的TaqMan探針5′端標記有報告基團(Reporter,R),如FAM、VIC等,3′端標記有熒光淬滅基團(Quencher,Q),如TAMRA等。對于完整的TaqMan探針,報告基團所發射的熒光能量被淬滅基團吸收,儀器檢測不到信號。隨著PCR的進行,Taq酶在鏈延伸過程中遇到與模板結合的探針,其3′→5′外切核酸酶活性就會將探針切斷,報告基團遠離淬滅基團,其能量不能被吸收,即產生熒光信號。每經過一個PCR循環,熒光信號隨著目的片段的擴增而指數增長,檢測每個循環結束后的熒光強度,整個反應結束后,便可得到一條擴增曲線,由已知濃度標準樣品的擴增曲線可以得到一條標準曲線,根據該標準曲線以及未知模板的擴增曲線可對未知模板進行定量分析。目前已有的檢測方法是單獨檢測樣本DNA或者RNA,但還沒有在同一管反應中同步檢測RNA和DNA的報道。在前期工作基礎上,本發明提出在同一管反應中同時檢測RNA和DNA的方法,具體為:分別以結核分枝桿菌復合群的16SrRNA基因和23SrRNA基因為靶目標,對16SrRNA設計一條逆轉錄引物、一對PCR引物以及一條Taqman探針,對23SrDNA設計另一對PCR引物以及另一條Taqman探針,在一個反應體系中同時進行RT-qPCR(16srRNA+16srDNA)和qPCR(23SrDNA)。分別測定總核酸(DNA+RNA)、以及DNA的絕對濃度,并算出(DNA+RNA)/DNA比值。具體地,通過分別比較16SrRNA和23SrDNA的Ct值以及5個絕對濃度已知的定量內參做出的標準曲線,可以求得待測標本中結核分枝桿菌(RNA+DNA)和DNA的絕對濃度,作為待測標本中結核分枝桿菌核酸定量檢測結果,判斷結核分枝桿菌的有無及豐度;通過比較16SrRNA總核酸與23SrDNA的Ct值(分別對應FAM和HEX檢測通道),可以算出(DNA+RNA)/DNA比值,作為判定標本中細菌生長活性的標準:當該比值大于200時,待測標本中結核分枝桿菌為活躍生長的活菌,當比值在2-200之間,為生長較慢的活菌,比值小于2時,為不生長的死菌。此方法的優勢:(1)可以比較結核分枝桿菌中RNA和DNA的相對量,從而判斷細菌的生長狀態;(2)樣本需求量變少,加上(DNA+RNA)的量一般比DNA量大數十倍甚至更多,可以提高檢測靈敏度;(3)方法更簡便,兩管PCR反應,合并到一管,節約成本和工作量;(4)由于在同一管中進行PCR,反應效率一致,DNA與RNA的比值更加精確。
技術實現要素:
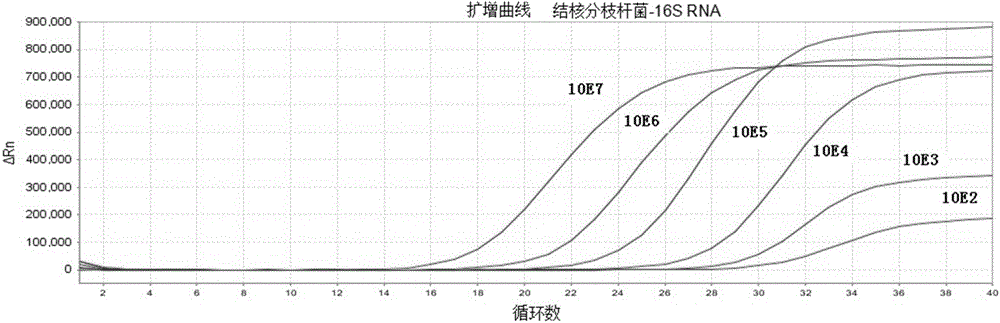
本發明的目的是克服現有技術中存在的缺陷,提供一種檢測靈敏度高、且操作簡單的結核分枝桿菌DNA與RNA同步檢測的熒光定量PCR分析方法。本發明提供的結核分枝桿菌的熒光定量PCR檢測方法,通過對RDP(RibosomalDatabaseProject)中結核分枝桿菌復合群的16SrRNA和23SrDNA全序列進行生物信息學比對分析,選取序列保守片段為靶目標,應用primerexpress軟件和primerpremier5.0軟件,設計一條逆轉錄引物和兩對PCR引物以及兩條Taqman探針。本發明方法所用的結核分枝桿菌復合群特異性定量檢測試劑包含一條逆轉錄引物和兩對特異性PCR引物以及兩條特異性熒光探針,擴增目的擴增片段長度分別為100bp(16SrRNA基因)和67bp(23SrDNA)。具體來說,本發明提供的結核分枝桿菌RNA/DNA同步定量分析方法,具體步驟如下:1、通過生物信息學分析,選擇結核分支桿菌的16SrRNA基因的保守片段為靶目標,其基因片段的擴增目標核苷酸序列為:GGGATGCATGTCTTGTGGTGGAAAGCGCTTTAGCGGTGTGGGATGAGCCCGCGGCCTATCAGCTTGTTGGTGGGGTGACGGCCTACCAAGGCGACGACGG(SEQIDNO.5)選擇結核分支桿菌的23SrRNA基因的保守片段為靶目標,其基因片段的擴增目標核苷酸序列為:TGGGTCCTGAGGCAACACTCGGACTTGTTCCAGGTGTTGTCCCACCGCCTTGGTGGTGGGGTGTGGT(SEQIDNO.9)2、應用primerexpress軟件和primerpremier5.0軟件,設計一條結核分支桿菌的逆轉錄引物和兩對PCR引物以及兩條Taqman探針,具體地:16SrRNA引物和探針序列為:逆轉錄引物MTBC-RT:5’-CCGTATCTCAGTCCCAGTGT-3’(SEQIDNO.1)上游引物:MTBC-16SF:5’-GGGATGCATGTCTTGTGGTG-3’(SEQIDNO.2)下游引物:MTBC-16SR:5’-CCGTCGTCGCCTTGGTAG-3’(SEQIDNO.3)Taqman探針序列MTBC-16SP:5'-FAM-CGGGCTCATCCCACACCGCTA-TAMRA-3’(SEQIDNO.4);23SrRNA引物和探針序列為:上游引物:MTBC-23SF:5’-CCTCTGCTGCCAAGAAAAGC-3’(SEQIDNO.6)下游引物:MTBC-23SR:5’-GTTATCTCGTACGCCTTGGTATGC-3’(SEQIDNO.7)Taqman探針序列MTBC-23SP:5'-HEX-TCTAGCGAGCACACACACGGCCC-TAMRA-3';(SEQIDNO.8);3、構建和制備質粒定量標準品根據本發明中的PCR產物,利用DNA重組技術將包含目的擴增片段的基因克隆到質粒載體pGEM-TEasyVector中,構建的重組質粒作為檢測結核桿菌RNA/DNA同步定量分析的定量標準品;4、分離樣本中結核分枝桿菌,提取總核酸,使提取的樣本中同時含有DNA與RNA成分;5、分別以結核分枝桿菌復合群的16SrRNA和23SrDNA為靶目標,對16SrRNA,使用設計的一條逆轉錄引物、一對PCR引物以及一條Taqman探針;對23SrDNA,使用設計的另一對PCR引物以及另一條Taqman探針,對提取的同一例樣本同時進行RT-qPCR,其中,RT(Reversetranscription)部分為單重的只針對16SrRNA的逆轉錄反應,qPCR(QuantitativePCR)部分為兩重(針對16SrRNA和23SrRNA)的實時定量PCR反應。通過比較16SrRNA和23SrDNA的Ct值以及5個絕對濃度已知的定量內參(105-101copies/ul)做出的標準曲線,求得待測標本中結核分枝桿菌DNA的絕對濃度,作為待測標本中結核分枝桿菌DNA定量檢測結果;通過比較16SrRNA與23SrDNA的Ct值(分別對應FAM和HEX檢測通道),算出(DNA+RNA)/DNA比值,作為判定標本中結核分枝桿菌生長活性的標準,即根據FAM通道或HEX通道的Ct值,計算FAM通道Ct值/HEX通道Ct值,也就是(16SrRNA+16SrDNA)/23SrDNA,代表了總核酸(RNA和DNA)與DNA的拷貝數比值;當該比值大于200時,待測標本中結核分枝桿菌為活躍生長的活菌,當比值在2-200之間,為生長較慢的活菌,比值小于2時,為不生長的死菌。結核分枝桿菌RNA/DNA同步定量分析方法進一步分析(1)結核分枝桿菌DNA、RNA同步抽提本發明利用新的核酸抽提方法,同時提取結核分支桿菌中的DNA與RNA,此類提取方法的難點是如何提高核酸的質與量。為了保證提取過程中DNA與RNA的完整性,方法1中提取液同時含有異硫氰酸胍與酚:氯仿:異戊醇(25:24:1),方法2中提取液則含有PH值7.8的TE緩沖液。另外,上述兩種方法都利用了MiniBeadbeater-1研磨珠均質器對結核分支桿菌進行物理破壁,極大的提高了核酸抽提的量。分別以兩種方法提取相同菌量的總核酸作為模板進行R/P分析,方法1所得擴增曲線的Ct值較大,R/Pratio比值為0.6,說明模板中存在抑制劑。方法2所得擴增曲線的Ct值較小,R/Pratio比值為1.1,說明模板量損失少,提取效率較高。由于方法2通過純化試劑盒對提取的核酸進行了純化,故PCR反應抑制作用不明顯,完全可以保證模板總核酸的準確擴增。另外方法2的步驟簡單、需時少,成本較低、適合大樣本的處理。因此,在后續試驗中采用方法2進行模板總核酸的提取。(2)引物與探針的特異性分析NCBI數據庫的現有序列中,只有結核桿菌復合群的基因序列與提交的一對引物及一條探針具有高度同源性,其中正向引物與探針的特異性較反向引物好。與其他細菌及人的序列沒有同源性,引物與探針的特異性好。為了進一步驗證引物與探針在實際檢測中的特異性,運用本課題反應體系對人基因組DNA和大腸桿菌基因組DNA分別進行了檢測,結果均為陰性,其Ct值在36左右,與無模板陰性對照一致,PCR產物電泳未見100bp條帶。而結核桿菌復合群DNA標準品經上述方法檢測則顯示為陽性,其Ct值為22。將PCR產物電泳可以得到大小為100bp的目的條帶,且無引物二聚體。說明本課題引物和探針與人及大腸桿菌基因組DNA無交叉反應,特異性較強,擴增曲線見圖1。(3)熒光定量PCR反應條件的優化通過對反應體系中不同濃度的定量PCR引物、Taqman探針、以及退火溫度等實驗結果進行比較,最終選擇反應靈敏度高、本底熒光信號低、具有典型S型擴增熒光信號曲線、擴增效率接近于1的最優反應體系。引物濃度的優化:在反應體系中其他條件相同的情況下,將MTBC的PCR引物濃度從0.1umol/L至0.5umol/L以0.05umol/L遞增,均可行。通過試驗結果分析比較,確定較佳的引物終濃度為0.2-0.4umol/L,最佳的引物終濃度為0.3umol/L。探針濃度優化:在反應體系中其他條件相同的情況下,將MTBC的探針濃度分別從0.1umol/L至0.8umol/L做倍比連續稀釋后進行檢測,均可行。通過試驗結果分析比較,確定較佳的引物終濃度為0.3-0.6umol/L,最佳的引物終濃度為0.4umol/L。退火溫度優化:通過比較退火溫度的優化試驗結果后,選定較佳退火溫度為55℃-65℃,最佳退火溫度為60℃。(4)絕對定量標準曲線重組質粒標準品pGEM-TEasy-16srRNA首先運用紫外線分光光度計進行濃度測定,并根據其分子量計算出相關的拷貝數。然后將原始質粒標準品依次進行10倍梯度稀釋,分別生成101、102、103、104、105、106、107拷貝/微升等不同濃度的質粒標準品。每個濃度進行3個重復,并設陰性空白對照,進行實時熒光定量PCR(圖2,圖3)。利用平均CT值作圖可得到標準曲線(圖4)。從圖4分析可得出標準曲線的檢測極限是10拷貝/微升,相關系數是0.999,通過標準曲線的斜率(S=-3.39)可以計算出擴增效率E(E=10-1/斜率-1)是0.97。說明該反應的檢測結果可靠性高。(5)定量檢測結果判定標準1)檢測樣本中1×108copies/ml≥TBDNA(HEX通道)≥5×102copies/ml,測定結果有效,可直接報告相應的拷貝量;2)檢測樣本中TBDNA(HEX通道)≥1×108copies/ml,既可直接報告為≥1×108copies/ml,也可用生理鹽水按10倍梯度做相應稀釋,使其拷貝量落在1×105~5×107copies/ml范圍內再重新測定,測定結果應以稀釋倍數進行校正;3)檢測樣本TBDNA(HEX通道)的拷貝量為<5×102copies/ml時,報告拷貝數為<5×102copies/ml;4)Ct值不顯示時,報告該樣本TBDNA小于檢測極限。(6)死活菌檢測結果判定標準1)若DNA+RNA(FAM通道)及DNA(HEX通道)的CT值均≤35,且DNA+RNA(FAM通道)/DNA(HEX通道)的拷貝數比值>200,則判斷該檢測樣本含有大量活菌,該患者為活動性肺結核,有極大傳染性;2)若DNA+RNA(FAM通道)及DNA(HEX通道)的CT值均≤35,且DNA+RNA(FAM通道)/DNA(HEX通道)的拷貝數比值在2-200之間,則判斷該檢測樣本含有部分活菌或者可能為患者治療后體內的結核菌活力比正常低有一定傳染性;3)若DNA+RNA(FAM通道)及DNA(HEX通道)的CT值均≤35,且DNA+RNA(FAM通道)/DNA(VIC通道)的拷貝數比值在0-2之間,則判斷該檢測樣本是無活力的死菌,治療有效。本發明方法評價經過大量的反應條件優化、對比試驗和驗證試驗,并經過臨床樣本的檢測應用評估。對建立的方法進行靈敏度、穩定性、特異性以及對臨床樣本的有效性進行綜合評價:(1)靈敏度:用TE分別將質粒定量標準品pGEM-TEasy-23SrRNA稀釋至1010、109、108、107、106、105、104、103、102、101、100copies/ul,采用經優化后的體系進行檢測,驗證所建立的方法的靈敏度。本檢測方法在1010-102copies/reaction數量級范圍內具有良好的線性關系,相關系數R=0.9937,其檢測范圍可達9個數量級。(2)穩定性:從批間重復和批內重復兩個方面進行評價。批內重復性:選取同一份樣本設5個平行反應管同時進行反應,檢測其CT值及比值的變異系數。批間重復性:選取同一份樣本,一周內重復檢測5次,檢測其CT值及比值的變異系數。(3)特異性:本發明選取了5個人的基因組DNA樣本進行檢測,結果只有結核分支桿菌樣本呈陽性結果,5份人的基因組DNA均為陰性結果。本發明采用的熒光定量PCR技術巧妙的利用了PCR技術的高效擴增、探針技術的高特異性和光譜技術的敏感性及實時定量的優點,克服了常規PCR定性檢測的一些不足,極大地提高了檢測的敏感性和特異性,檢測靈敏度可達102數量級;并且縮短了實驗時間,簡化了實驗操作,而且熒光檢測的結果由電腦軟件分析,避免了產物后處理過程所致的污染,較常規PCR更為客觀、靈敏、準確。同時,本發明中采用的陽性對照是根據結核分支桿菌的保守序列設計構建的標準質粒,使得陽性標準品德來源可靠,避免了陽性毒株在每次實驗中的使用。本發明方法可進行大批量的樣本分析,并對結核分枝桿菌復合群進行定量,從而為結核分支桿菌的檢測和監控提供有效方法。附圖說明圖1.熒光定量PCR方法的特異性分析結果。從左至右,依次為結核分枝桿菌質粒標準品、陰性對照、人基因組DNA、大腸桿菌DNA。圖2.結核分枝桿菌16SrRNA基因不同濃度的標準品實時熒光定量PCR的擴增曲線。圖3.結核分枝桿菌23SrRNA基因不同濃度的標準品實時熒光定量PCR的擴增曲線。圖4.絕對定量標準曲線,橫坐標是DNA拷貝數,縱坐標是平均CT值。圖5.病人的R/P比值與治療天數之間的關系,病人1(A),2(B),3(C),4(D)。橫坐標是治療天數,縱坐標是R/P比值。具體實施方式下面結合具體實施例詳細說明本發明,但不以任何形式限制本發明要求保護的范圍。實施例1:肺內臨床標本的檢測總共檢測了207份肺內標本,在總的207份肺內標本中,30份是培養陽性標本(其中24份是涂片陽性,6份涂片陰性),另外177份則是培養陰性(其中27份涂片陽性,150份涂片陰性)。24份培養和涂片同時陽性的標本,R/P檢測結果都為陽性;其余6份培養陽性涂片陰性的樣本中,只有4份樣本R/P檢測結果陽性;而177份培養陰性樣本中,其中156份樣本R/P檢測結果陰性,只有21份樣本R/P檢測結果陽性。在所有肺內樣本中,一共有23份樣本的培養和R/P結果之間是存在分歧的:其中21份樣本是R/P陽性而培養陰性的,另外2份樣本則是R/P陰性而培養陽性(如表1)。基于表1,計算出肺內標本的靈敏度、特異度、陽性預測值、陰性預測值分別為93.3%、88.1%、57.1%、98.7%。為了更好的解決上述23例分歧標本,我們回顧性的查看了病人的臨床證據,發現17份分歧樣本可以得到解決。另外4份未得到解決的樣本,我們則進行了樣本重新檢測。調整后的肺內標本檢測結果(如表2),其中45份樣本是真陽性,157份樣本是真陰性,4份樣本是假陽性,1份樣本是假陰性。基于表2,計算得出臨床靈敏度、特異度、陽性預測值、陰性預測值分別為97.8%、97.5%、91.8%、99.4%。實施例2:評價肺外標本的結核分枝桿菌的活動性在總的28份肺外標本中,4份樣本同時涂片和培養陽性,2份樣本涂片陰性而培養陽性。另外的22份培養陰性樣本中,有5份涂片陽性,17份涂片陰性。4份培養和涂片同時陽性的標本,R/P檢測結果都為陽性;其余2份培養陽性涂片陰性的樣本中,R/P檢測結果也都為陽性。而22份培養陰性樣本中,18份樣本R/P檢測結果陰性,另外4份樣本R/P檢測結果陽性,即4份分歧樣本(如表1)。基于這些數據,計算出總的肺外標本的靈敏度為100%,特異度為81.8%,陽性預測值為60%,陰性預測值為100%。通過查看分歧樣本的臨床證據之后,其中10份樣本是真陽性,17份樣本是真陰性,1份樣本是假陽性。基于表2,計算得出臨床肺外標本靈敏度、特異度、陽性預測值、陰性預測值則分別為100%、94.4%、91.0%、100%。表1.運用R/P分析檢測臨床樣本MTBC的結果+:陽性;—:陰性;n:樣本量。表2.調整R/P分析與培養結果之間分歧數據之后的臨床樣本MTBC檢測結果n:樣本量。在總的235份臨床樣本中,當分歧樣本解決后,以培養結果和臨床評估為參考標準,R/P分析中55份樣本是真陽性,1份樣本是假陰性,5份樣本是假陽性,174份樣本是真陰性。而熒光定量PCR分析中,47份樣本是真陽性,9份樣本是假陰性,18份樣本是假陽性,161份樣本是真陰性。R/P分析的靈敏度(98.2%vs83.9%,p<0.05)和特異度(97.2%vs89.9%,p<0.05)都比熒光定量PCR高,且具有統計學差異(如表3)。R/P分析的陽性預測值和陰性預測值分別為91.7%,99.4%,比熒光定量PCR的好很多(91.7%vs72.3%,p<0.05;99.4%vs94.7%,p<0.05)(如表3)。表3.基于分歧數據調整之后,R/P分析與熒光定量PCR之間的比較*:有統計學差異(p<0.05);n:樣本量。實施例3:新藥臨床試驗治療效果的監測為了對某種新型的抗結核分枝桿菌的藥物臨床效果進行評估,我們收集了4例結核病患者多份臨床樣本(標本數≥2)。這4例病人我們一直監測其治療效果,直至他們的樣本培養陰性為止,樣本的R/P結果和培養及臨床證據結果始終一致,但是熒光定量PCR的檢測結果則一直保持陽性(如表4)。表4.運用培養、涂片、熒光定量PCR、R/P分析等方法監測結核患者治療效果的比較這4例結核患者初始抗結核治療(0天)的R/P分析比值,比治療一段時間之后的比值要高很多。經過抗結核治療之后,病人1的R/P分析比值從第0天的6.2降低到第75天的1.23;病人2的R/P分析比值從第0天的4降低到第50天的1.4;病人3的R/P分析比值從第0天的4.5降低到第63天的0.9;病人4的R/P分析比值從第0天的3.8降低到第40天的1.2。4例結核病人經過治療之后的R/P比值都小于1.51,即都轉為陰性(如圖5)。SEQIDNO.1:ccgtatctcagtcccagtgtSEQIDNO.2:gggatgcatgtcttgtggtgSEQIDNO.3:ccgtcgtcgccttggtagSEQIDNO.4:cgggctcatcccacaccgctaSEQIDNO.5:gggatgcatgtcttgtggtggaaagcgctttagcggtgtgggatgagcccgcggcctatcagcttgttggtggggtgacggcctaccaaggcgacgacggSEQIDNO.6:CCTCTGCTGCCAAGAAAAGCSEQIDNO.7:GTTATCTCGTACGCCTTGGTATGCSEQIDNO.8:TCTAGCGAGCACACACACGGCCCSEQIDNO.9:TGGGTCCTGAGGCAACACTCGGACTTGTTCCAGGTGTTGTCCCACCGCCTTGGTGGTGGGGTGTGGT
相關技術
網友詢問留言
已有0條留言
- 還沒有人留言評論。精彩留言會獲得點贊!
1
- 一種敲除c?di?AMP分解酶的重組恥垢分枝桿菌株及其應用的制造方法與工藝
- 檢測結核分枝桿菌感染的抗原多肽池及其應用的制造方法與工藝
- 一種獼猴桃分枝定位器的制造方法與工藝
- 一種xanthone類化合物及其單晶的制備方法與作為抗海分枝桿菌藥物的應用與制造工藝
- 一種糖肽類抗生素耐藥基因檢測試劑盒的制造方法與工藝
- 分枝桿菌快速分離培養方法與制造工藝
- 一種用于檢測潛在結核的特異性標志物及其應用的制造方法與工藝
- 結核分枝桿菌抗原蛋白Rv2941及其T細胞表位肽的應用的制造方法與工藝
- 一種微流控芯片上基于熒光量子點磁性富集并分離結核分枝桿菌TB的方法及裝置與制造工藝
- 二肽meso?DAP?D?Ala和/或四肽L?Ala?D?Glu?L?Lys?D?Ala的新用途的制造方法與工藝